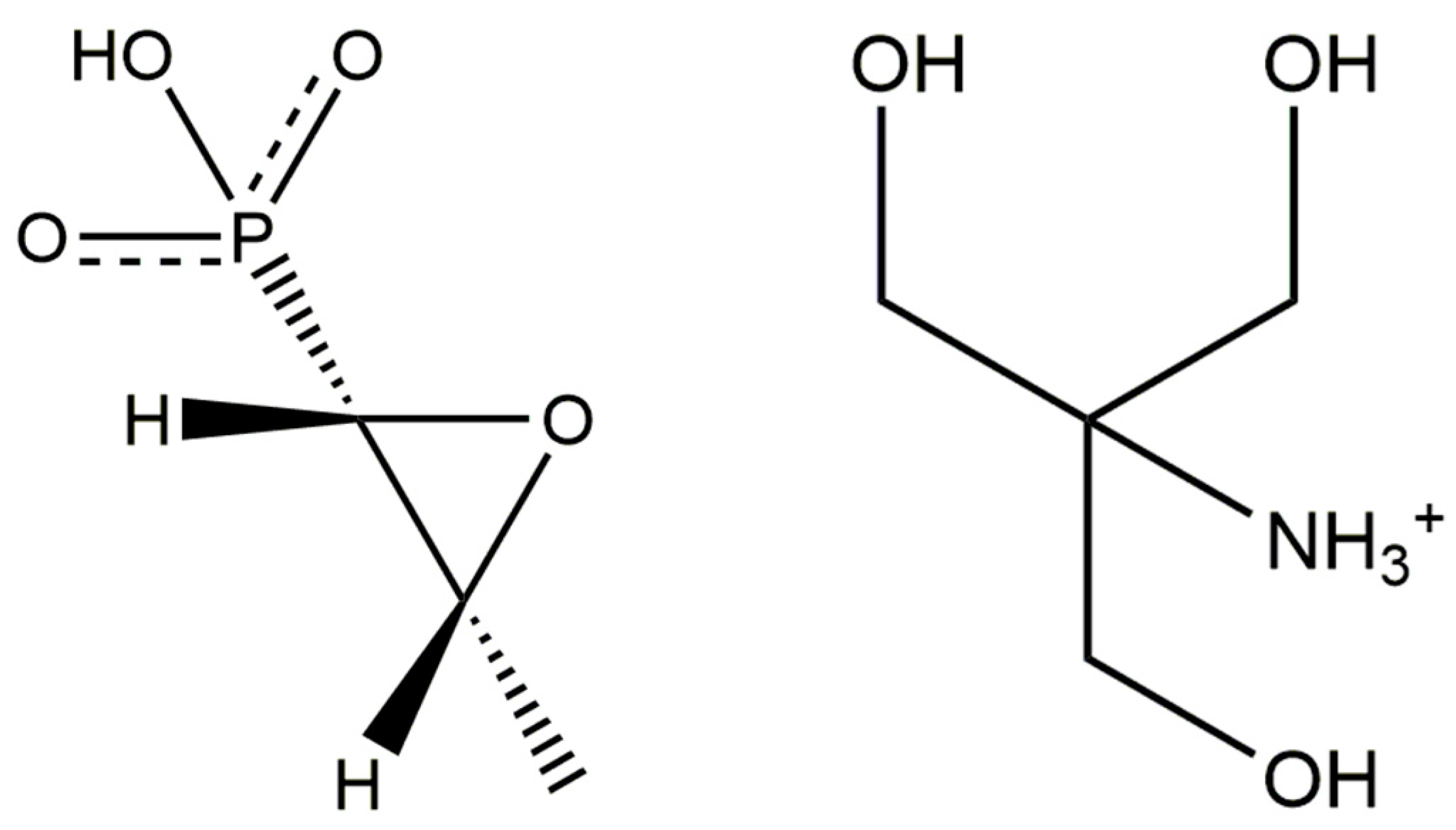
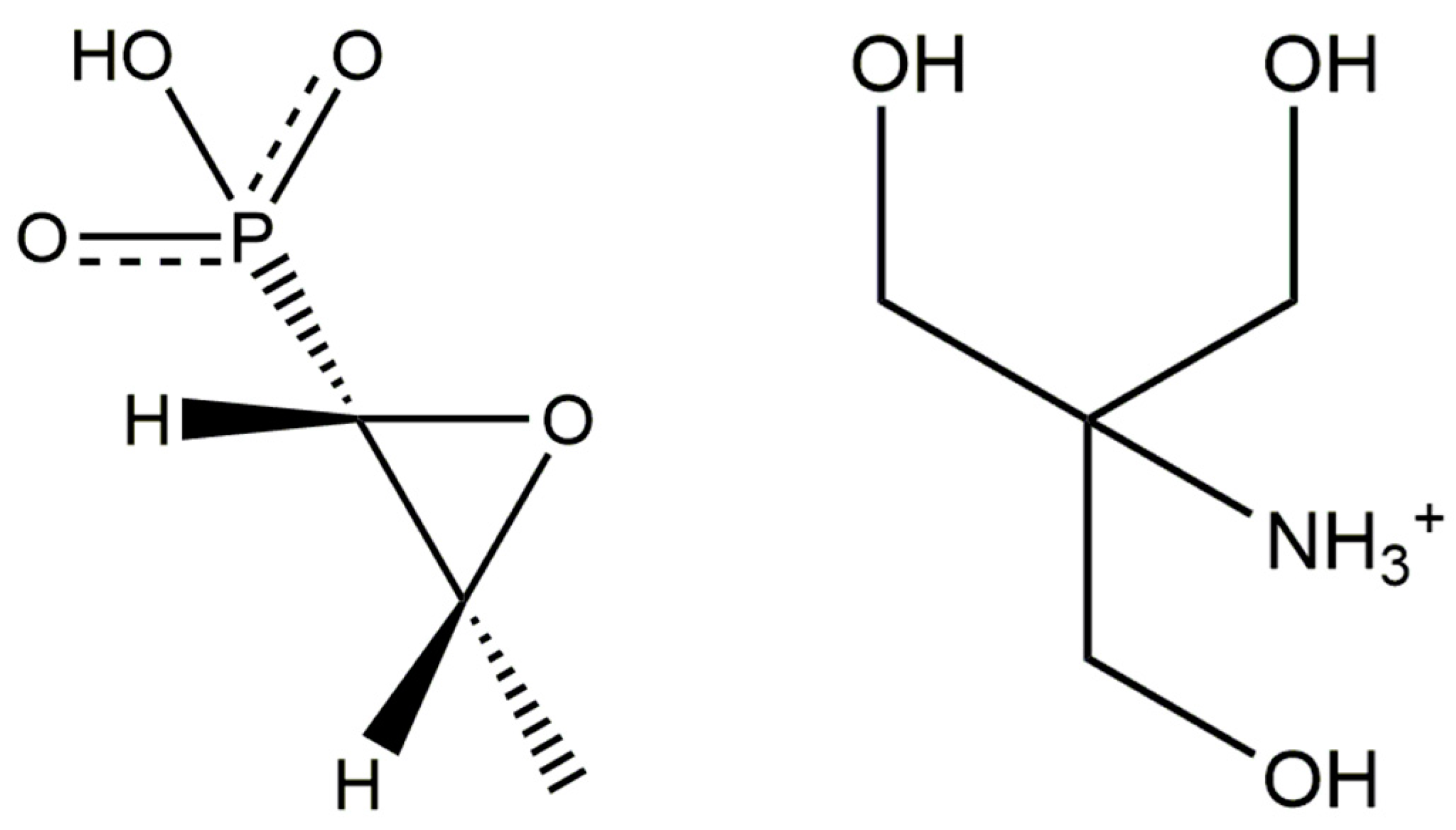
Crystal Structure of Fosfomycin Tromethamine, (C4H12NO3)(C3H6O4P), from Synchrotron Powder Diffraction Data and Density Functional Theory


Abstract
:1. Introduction
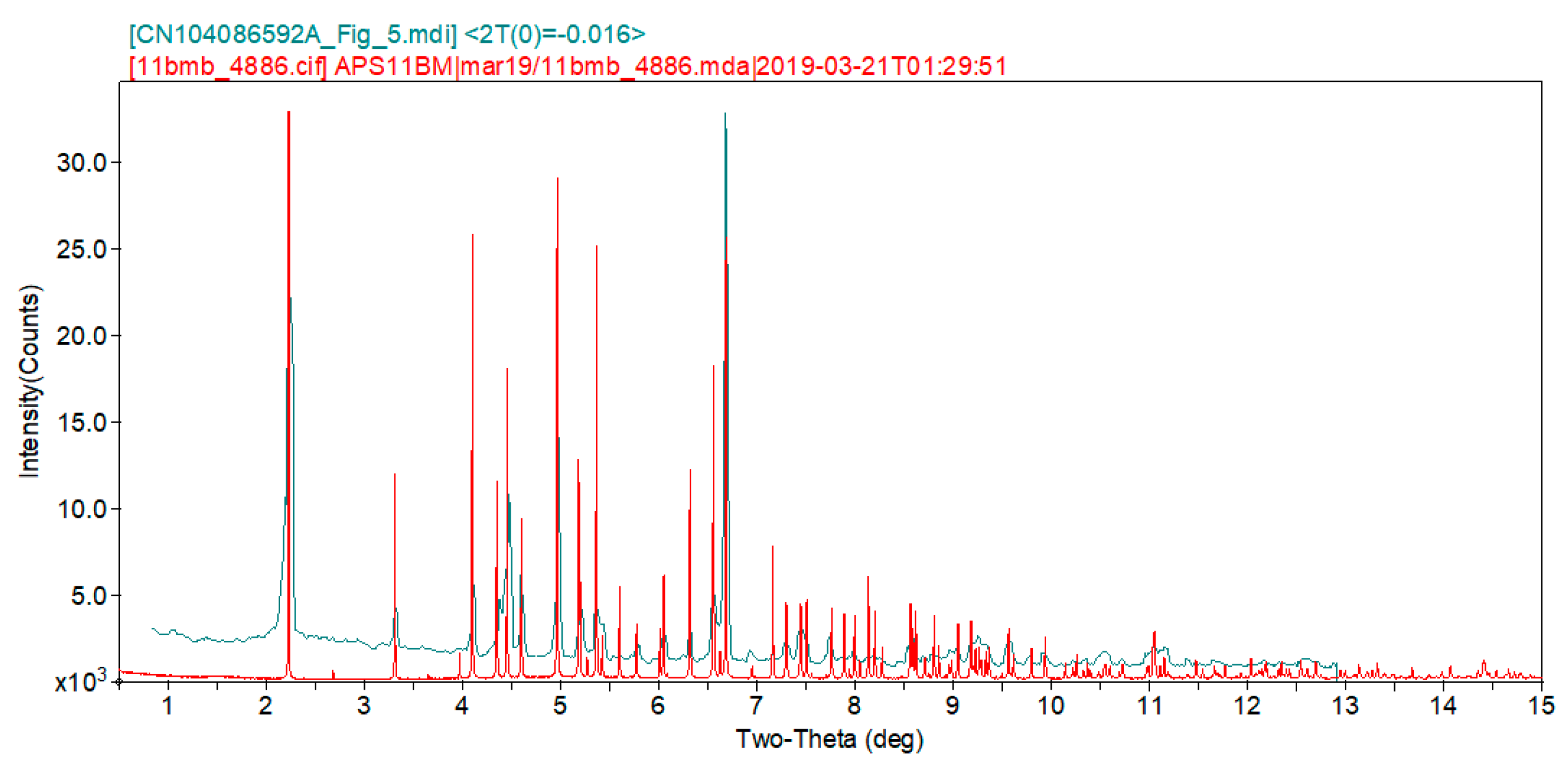
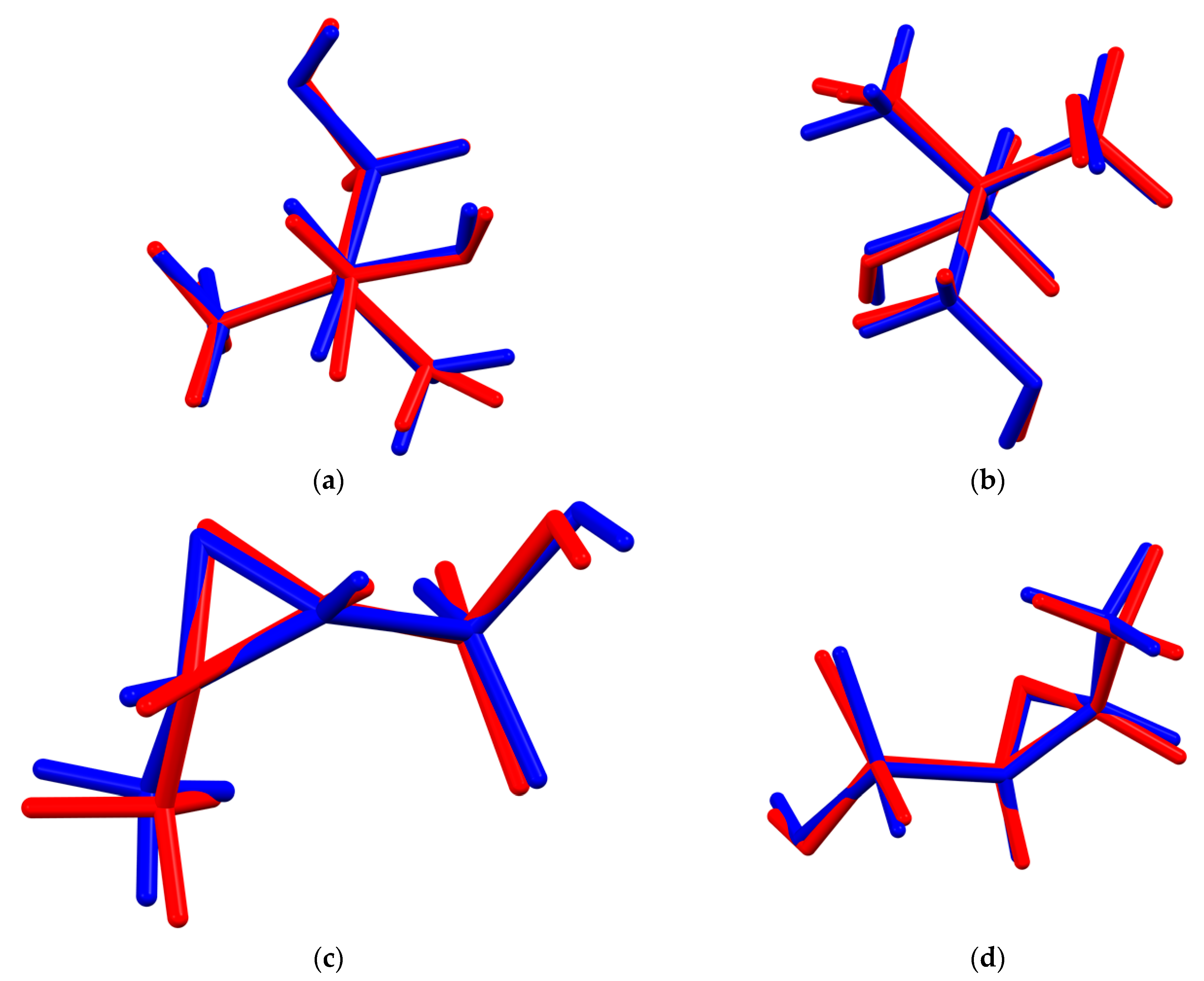
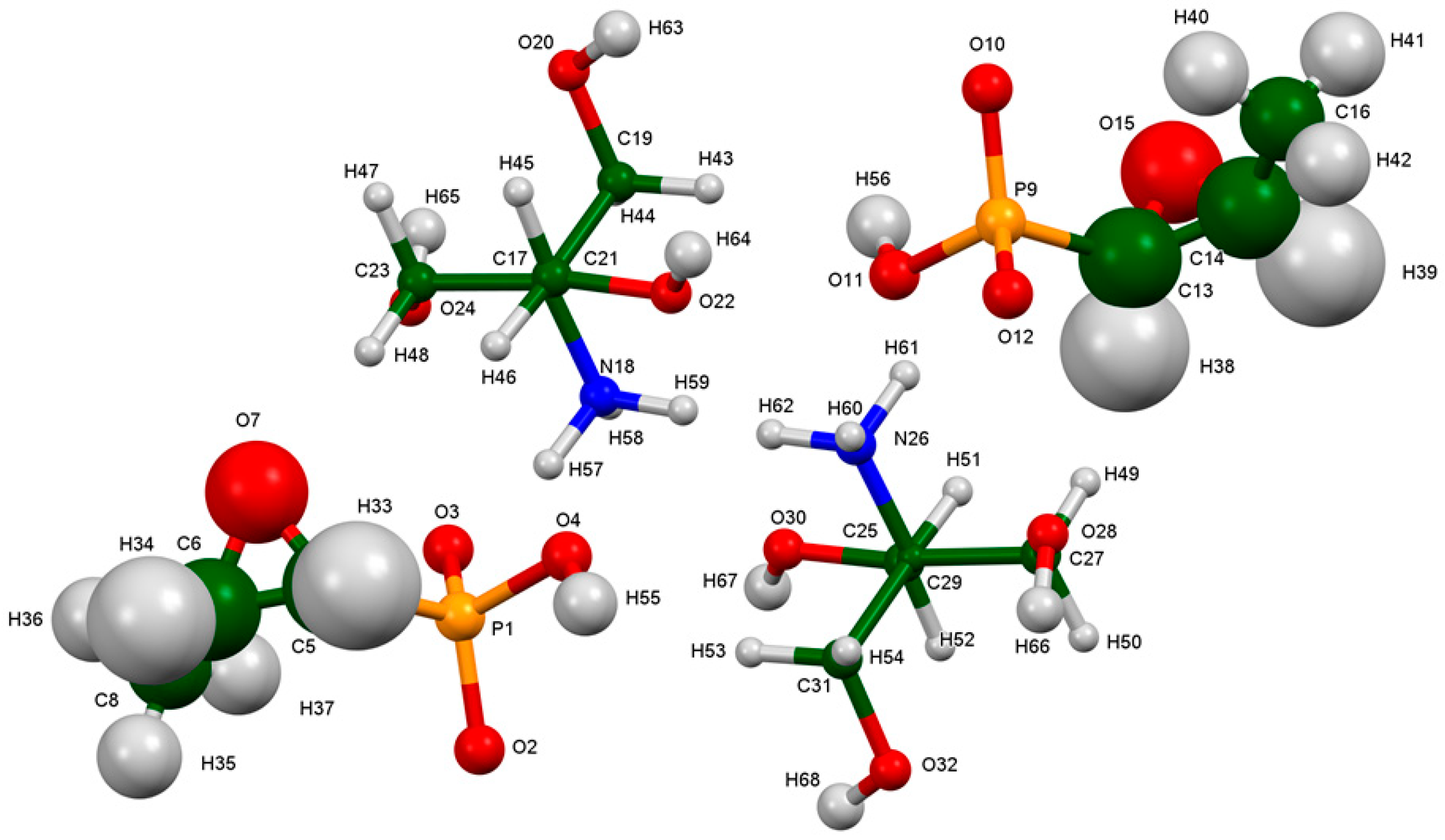
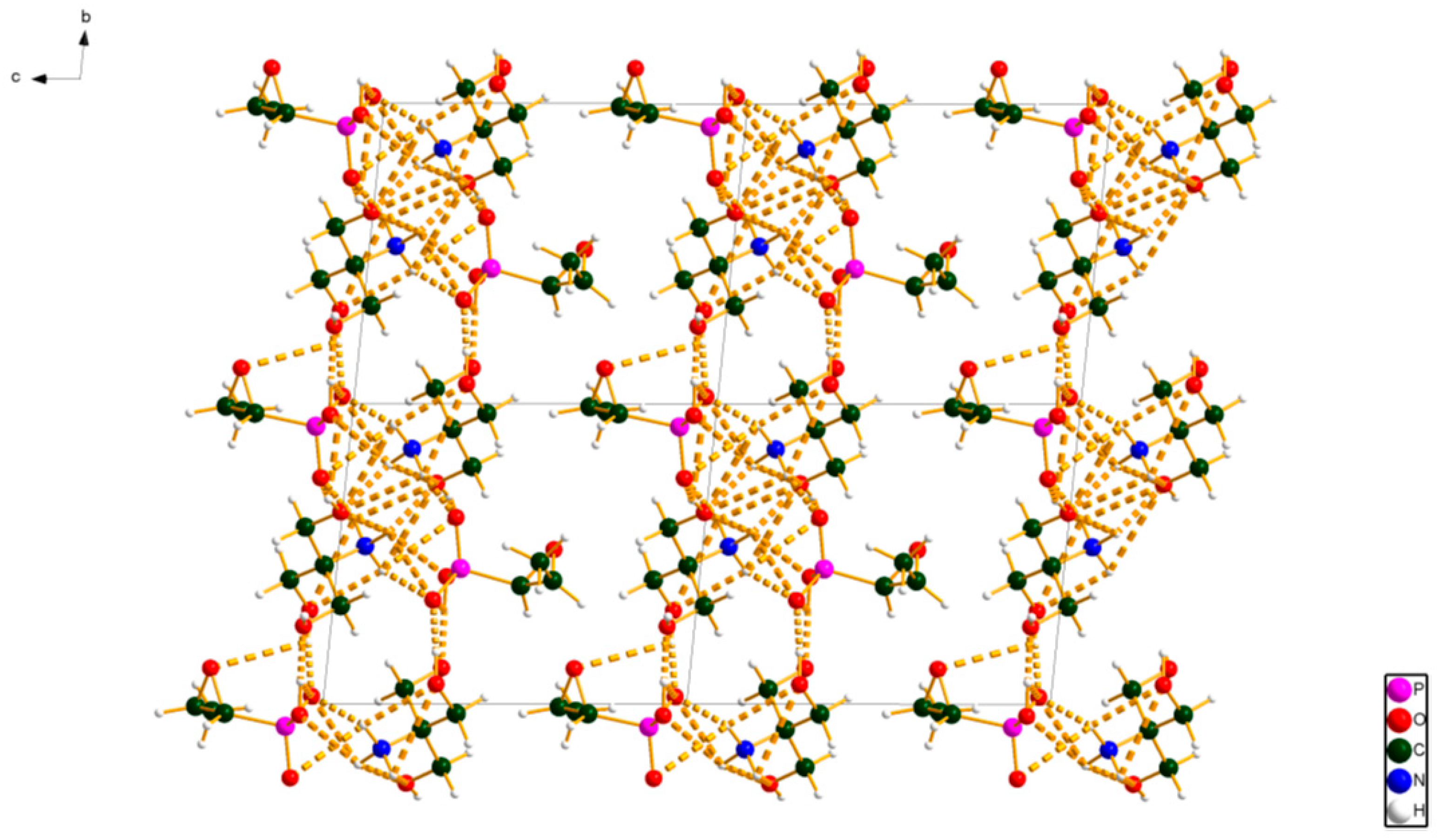
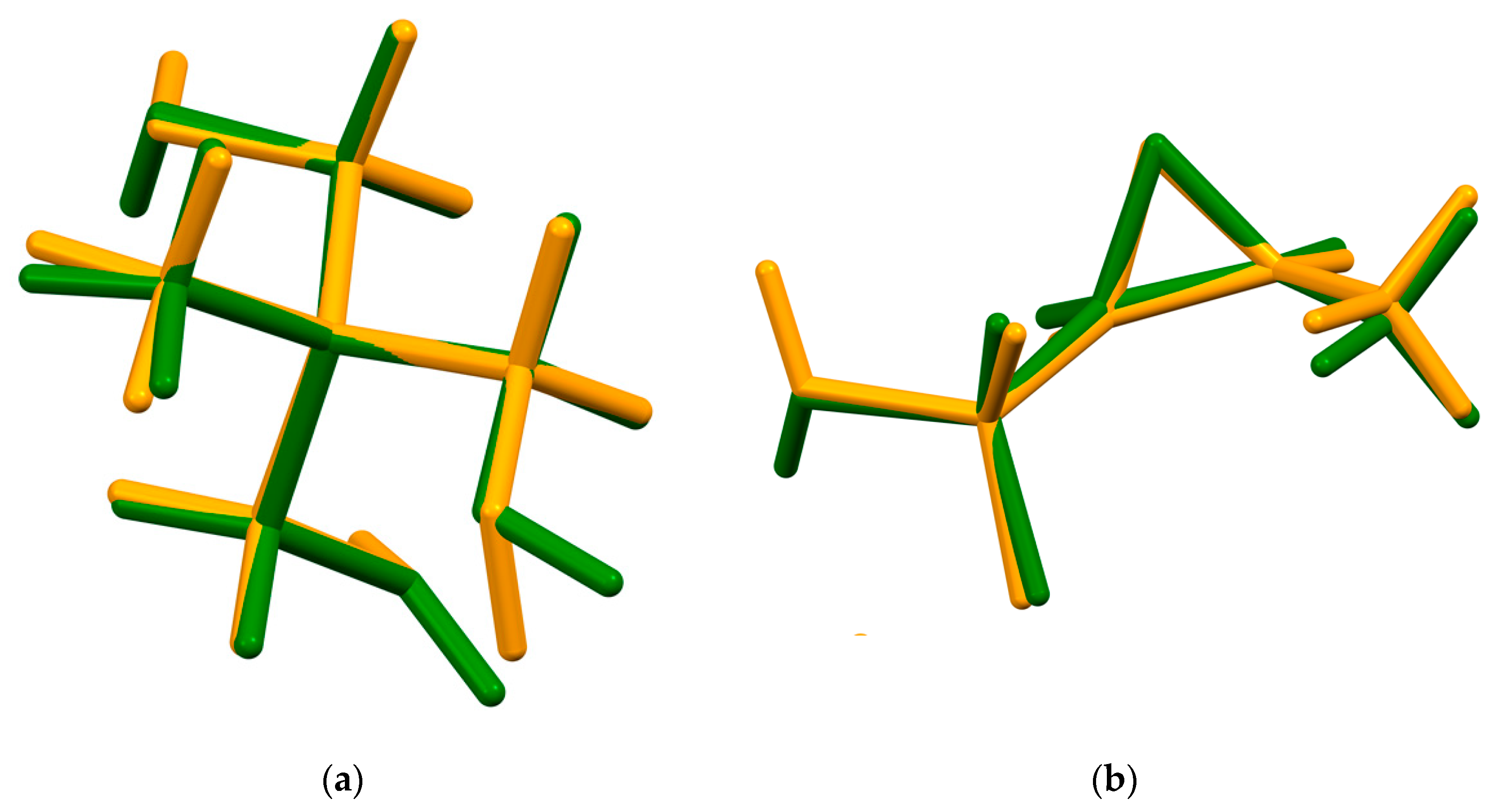
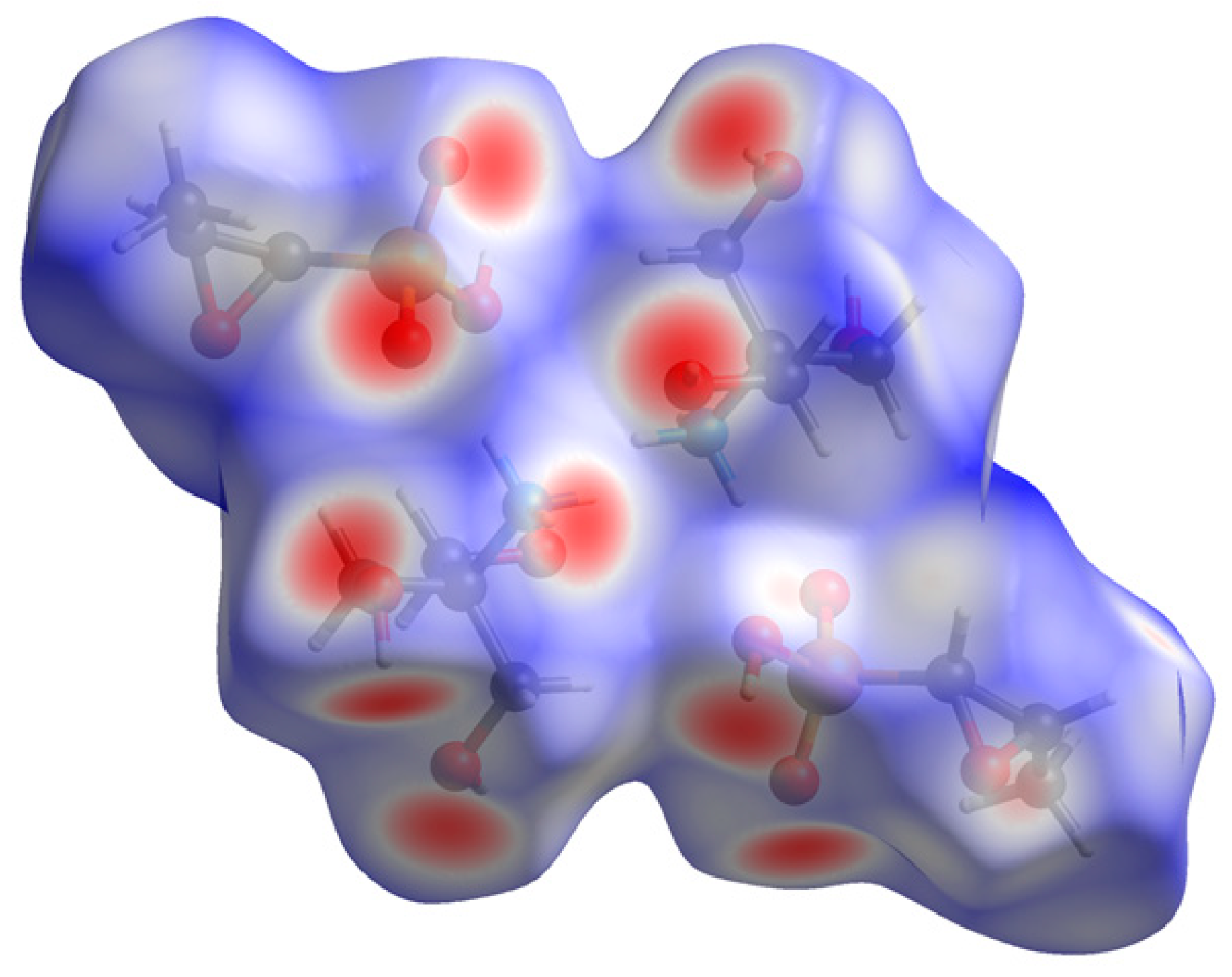
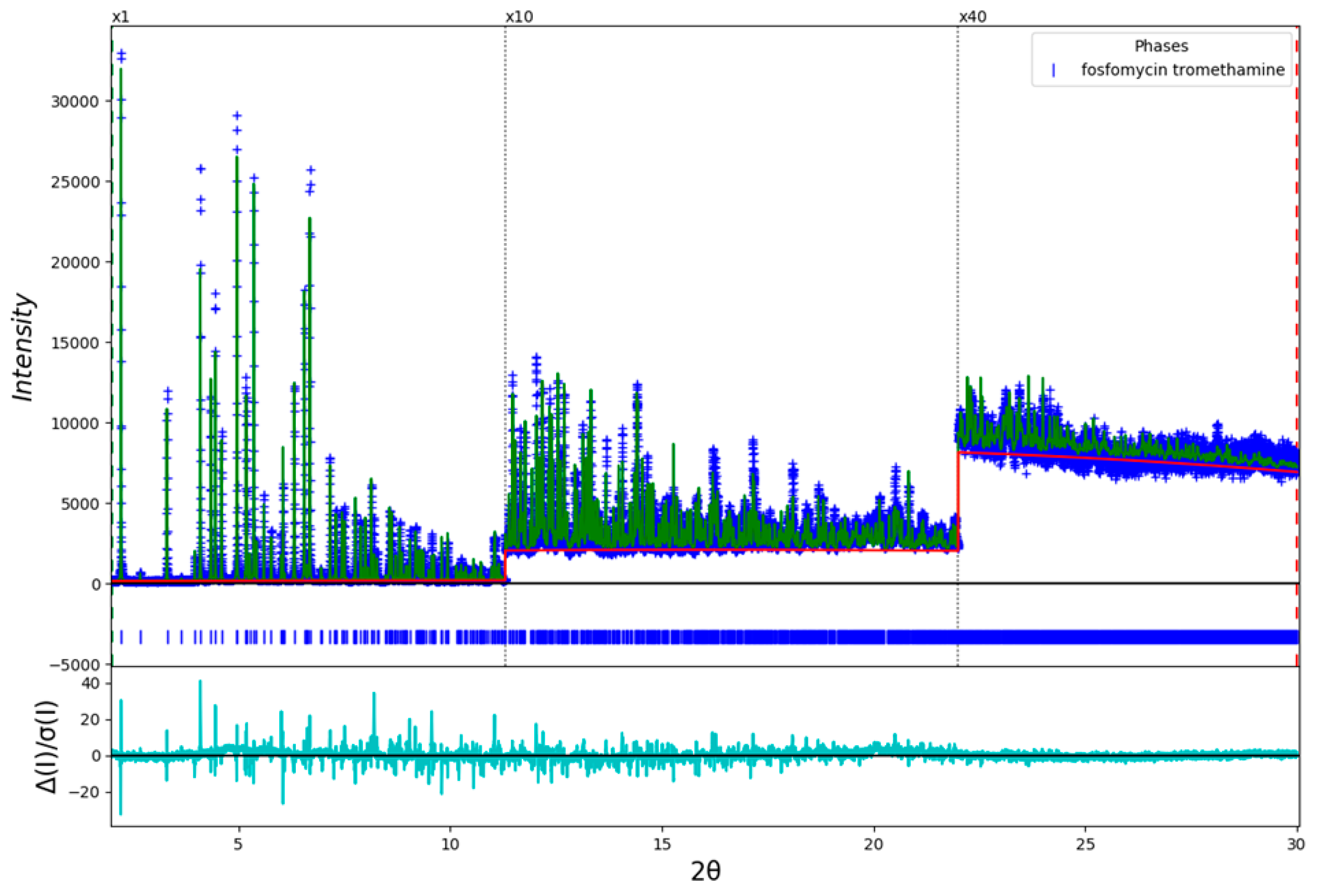
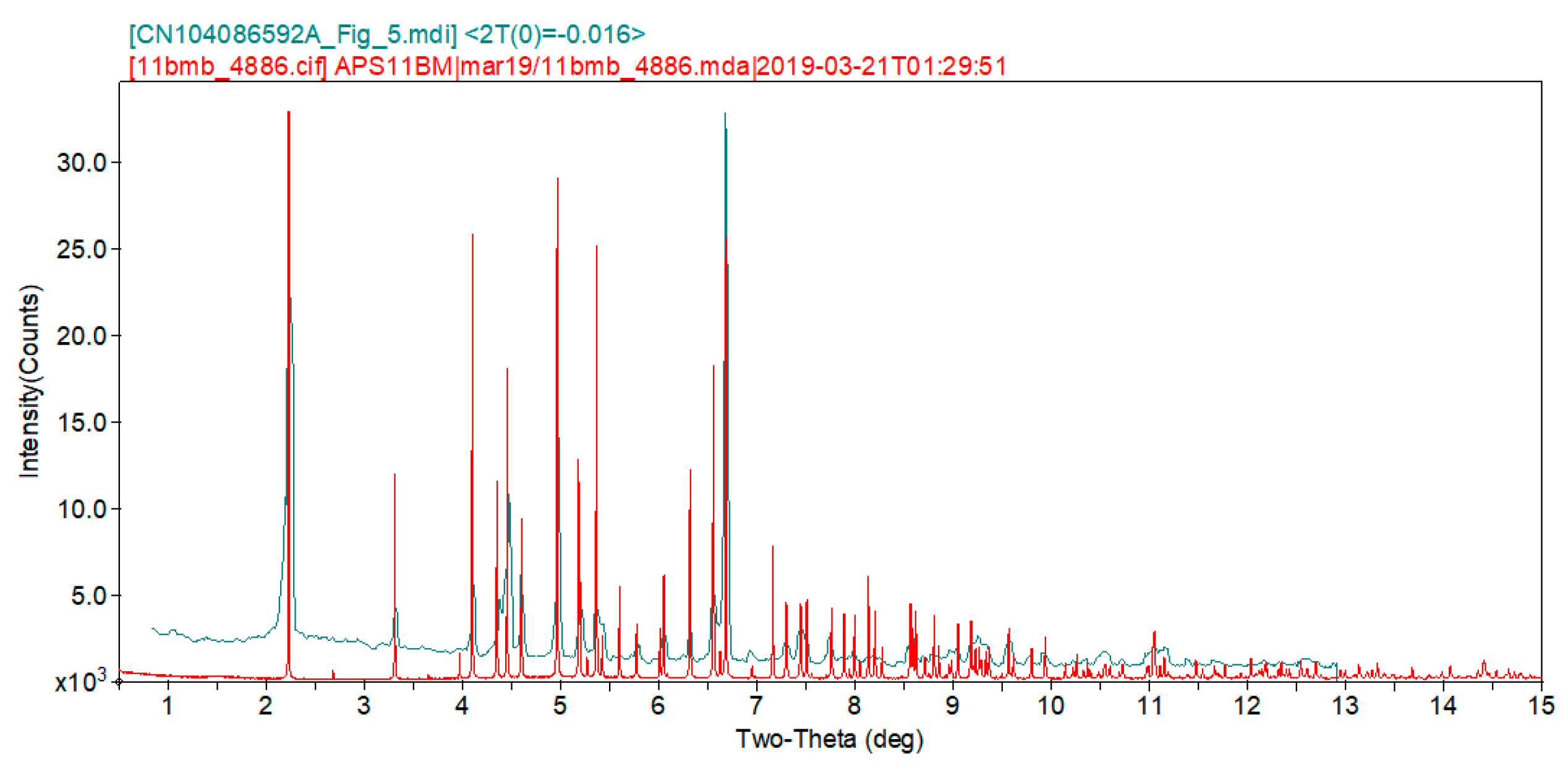
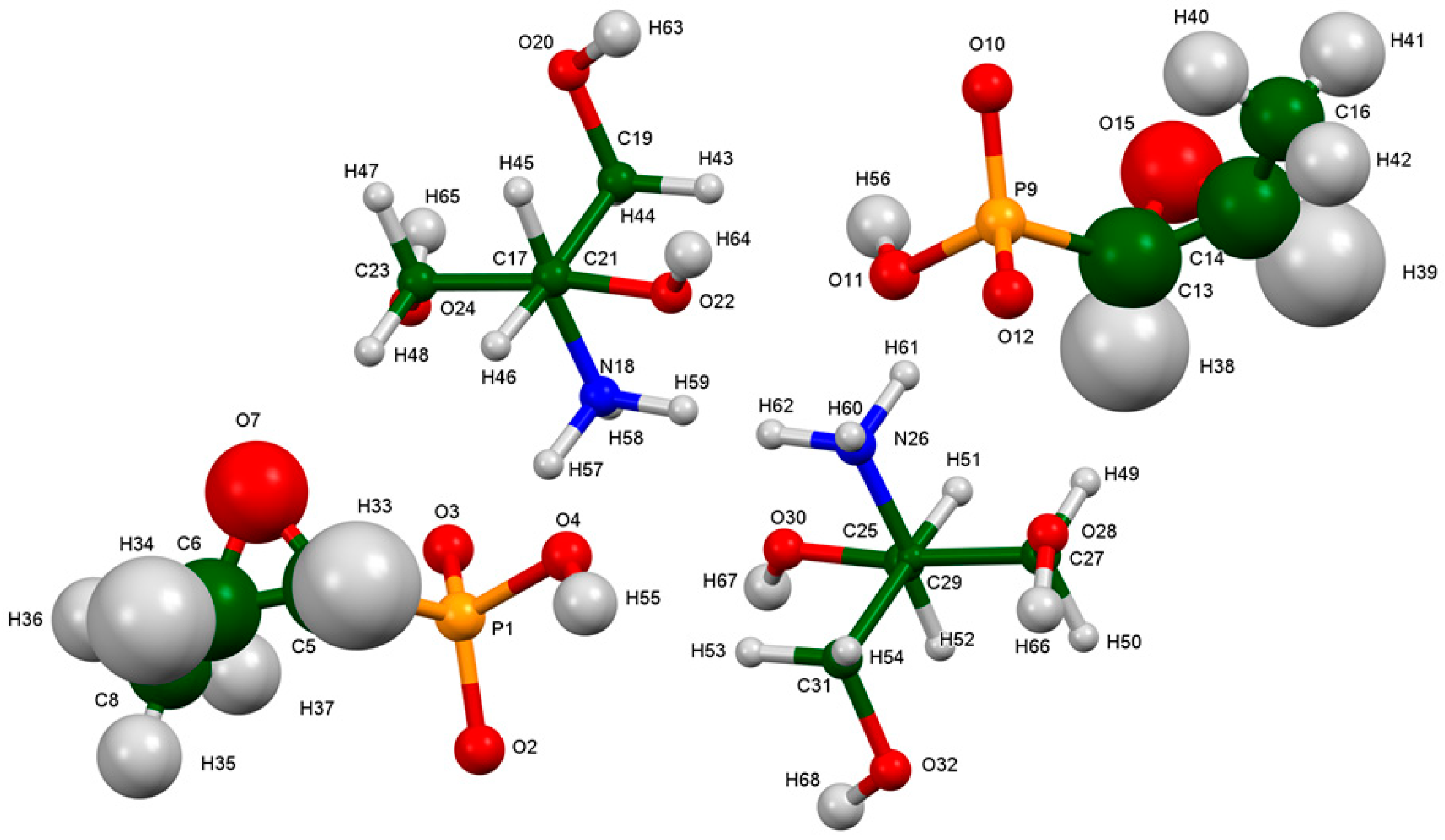
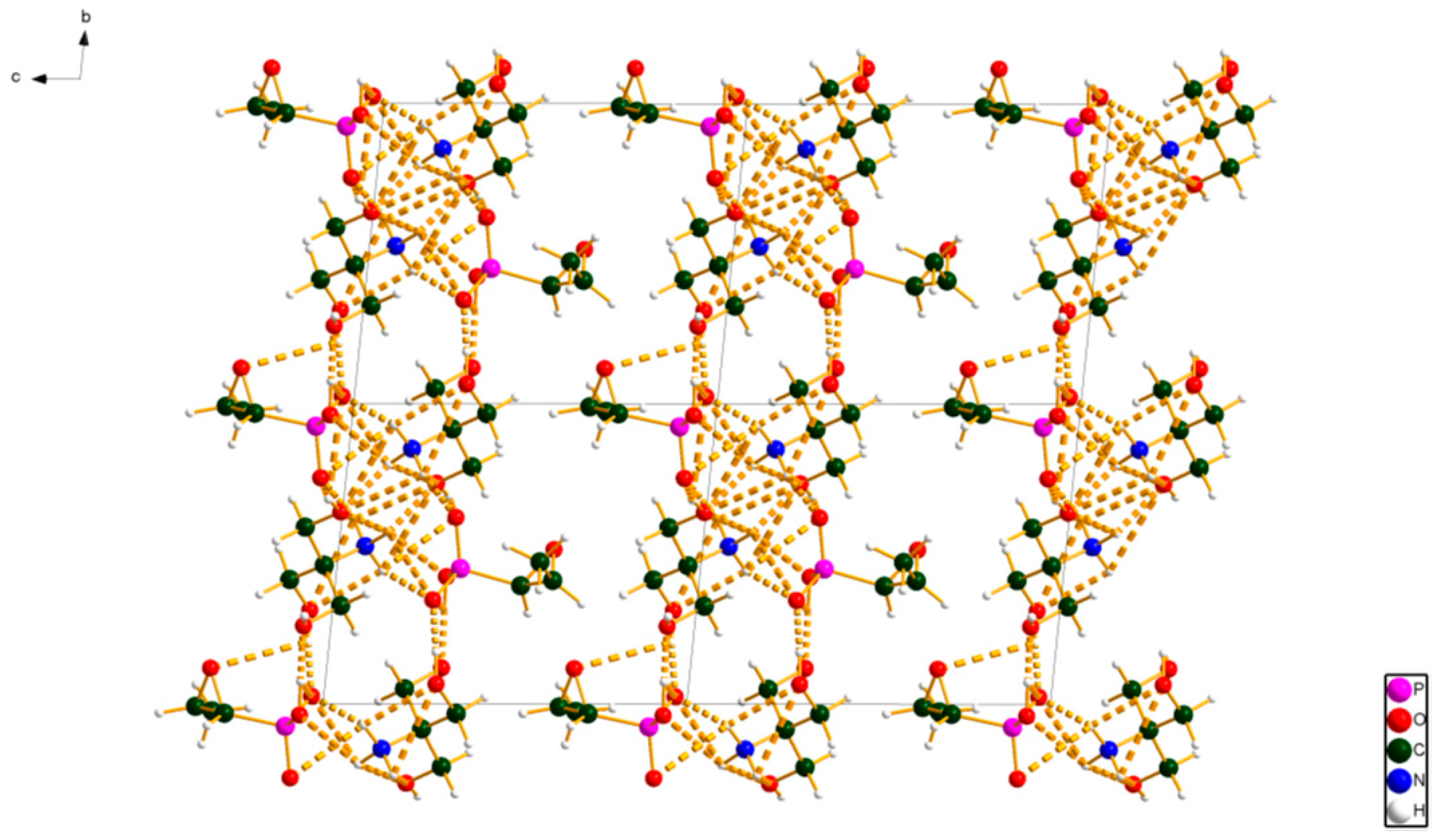
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Huang, S.; Chen, Y.; Meng, J.; Tang, S. Preparation Method of Fosfomycin Tromethamine. Patent CN104086592A, 8 October 2014. [Google Scholar]
- Kaduk, J.A.; Crowder, C.E.; Zhong, K.; Fawcett, T.G.; Suchomel, M.R. Crystal structure of atomoxetine hydrochloride (Strattera), C17H22NOCl. Powder Diffr. 2015, 29, 269–273. [Google Scholar] [CrossRef]
- Fawcett, T.G.; Kabekkodu, S.N.; Blanton, J.R.; Blanton, T.N. Chemical analysis by diffraction: The Powder Diffraction File™. Powder Diffr. 2017, 32, 63–71. [Google Scholar] [CrossRef]
- Orem, U.T. Silk Scientific, UN-SCAN-IT 7.0; Silk Scientific Corporation: Orem, UT, USA, 2013. [Google Scholar]
- MDI. Jade 9.8; Materials Data Inc.: Livermore, CA, USA, 2018. [Google Scholar]
- Van de Streek, J.; Neumann, M.A. Validation of molecular crystal structures from powder diffraction data with dispersion-corrected density functional theory (DFT-D). Acta Cryst. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Wavefunction, Inc. Spartan ’18 Version 1.3.0; Wavefunction Inc.: Irvine, CA, USA, 2018. [Google Scholar]
- Systèmes, D. Materials Studio 2019R1; BIOVIA: San Diego, CA, USA, 2018. [Google Scholar]
- Wheatley, A.M.; Kaduk, J.A. Crystal structures of ammonium citrates. Powder Diffr. 2019, 34, 35–43. [Google Scholar] [CrossRef]
- Rammohan, A.; Kaduk, J.A. Crystal structures of alkali metal (Group 1) citrate salts. Acta Cryst. Sect. B Cryst. Eng. Mater. 2018, 74, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chem. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Crawley, Australia, 2017; Available online: http://hirshfeldsurface.net (accessed on 30 May 2019).
- Bravais, A. Etudes Cristallographiques; Gauthier Villars: Paris, France, 1866. [Google Scholar]
- Friedel, G. Etudes sur la loi de Bravais. Bull. Soc. Fr. Mineral. 1907, 30, 326–455. [Google Scholar]
- Donnay, J.D.H.; Harker, D. A new law of crystal morphology extending the law of Bravais. Am. Mineral. 1937, 22, 446–447. [Google Scholar]
- Lee, P.L.; Shu, D.; Ramanathan, M.; Preissner, C.; Wang, J.; Beno, M.A.; Von Dreele, R.B.; Ribaud, L.; Kurtz, C.; Antao, S.M.; et al. A twelve-analyzer detector system for high-resolution powder diffraction. J. Synch. Rad. 2008, 15, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Toby, B.H.; Lee, P.L.; Ribaud, L.; Antao, S.M.; Kurtz, C.; Ramanathan, M.; Von Dreele, R.B.; Beno, M.A. A dedicated powder diffraction beamline at the Advanced Photon Source: Commissioning and early operational results. Rev. Sci. Inst. 2008, 79, 085105. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Toby, B.H.; Von Dreele, R.B. GSAS II: The genesis of a modern open source all purpose crystallography software package. J. Appl. Crystallogr. 2013, 46, 544–549. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Erba, A.; Zicovich-Wilson, C.M.; Civalleri, B.; Casassa, S.; Maschio, L.; Ferrabone, M.; De La Pierre, M.; D-Arco, P.; et al. CRYSTAL14: A program for the ab initio investigation of crystalline solids. Int. J. Quantum Chem. 2014, 114, 1287–1317. [Google Scholar] [CrossRef]
- Gatti, C.; Saunders, V.R.; Roetti, C. Crystal-field effects on the topological properties of the electron-density in molecular crystals—The case of urea. J. Chem. Phys. 1994, 101, 10686–10696. [Google Scholar] [CrossRef]
- Peintinger, M.F.; Vilela Oliveira, D.; Bredow, T. Consistent Gaussian Basis Sets of Triple-Zeta Valence with Polarization quality for Solid-State Calculations. J. Comput. Chem. 2013, 34, 451–459. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
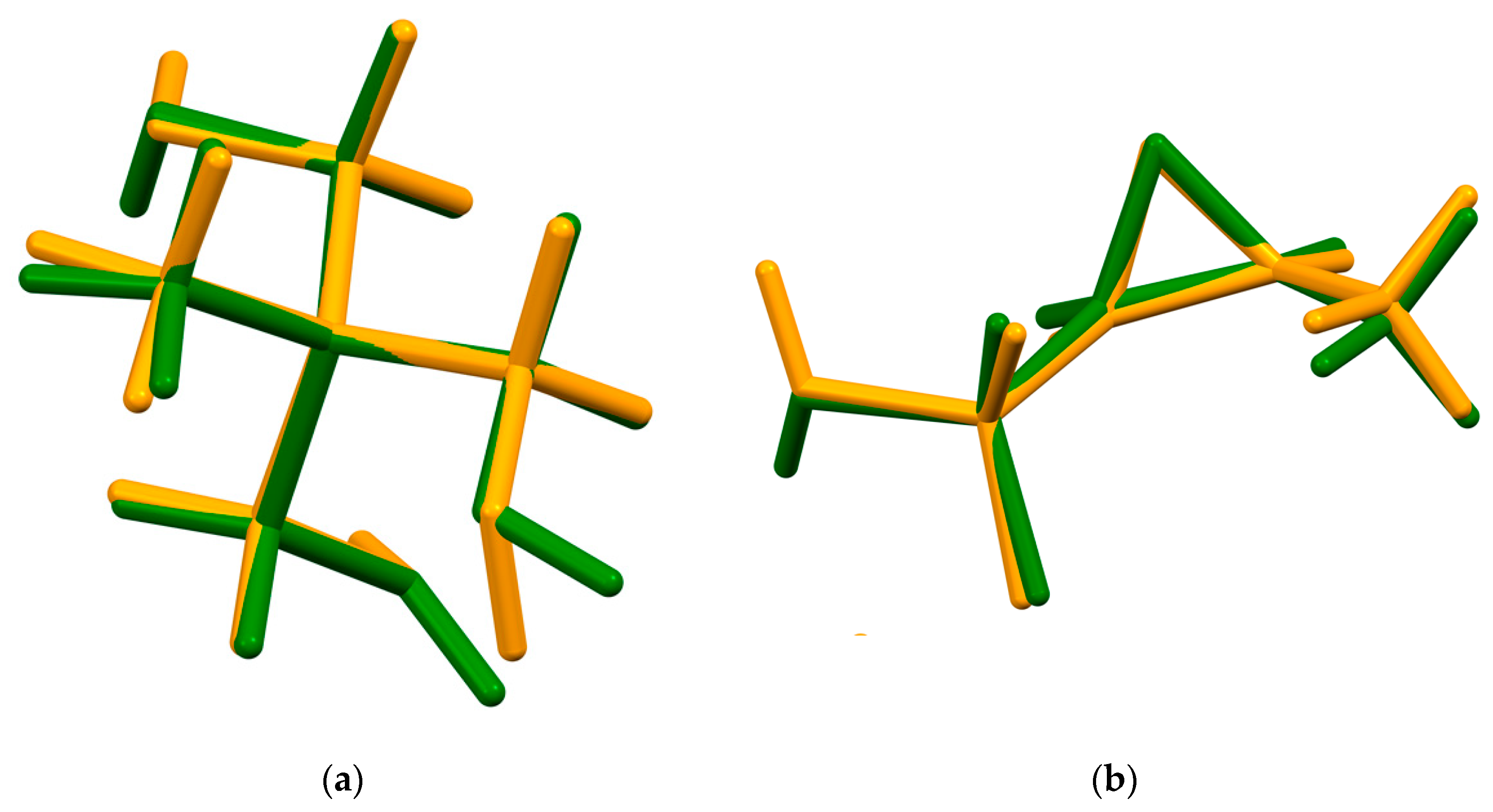{kind=link}
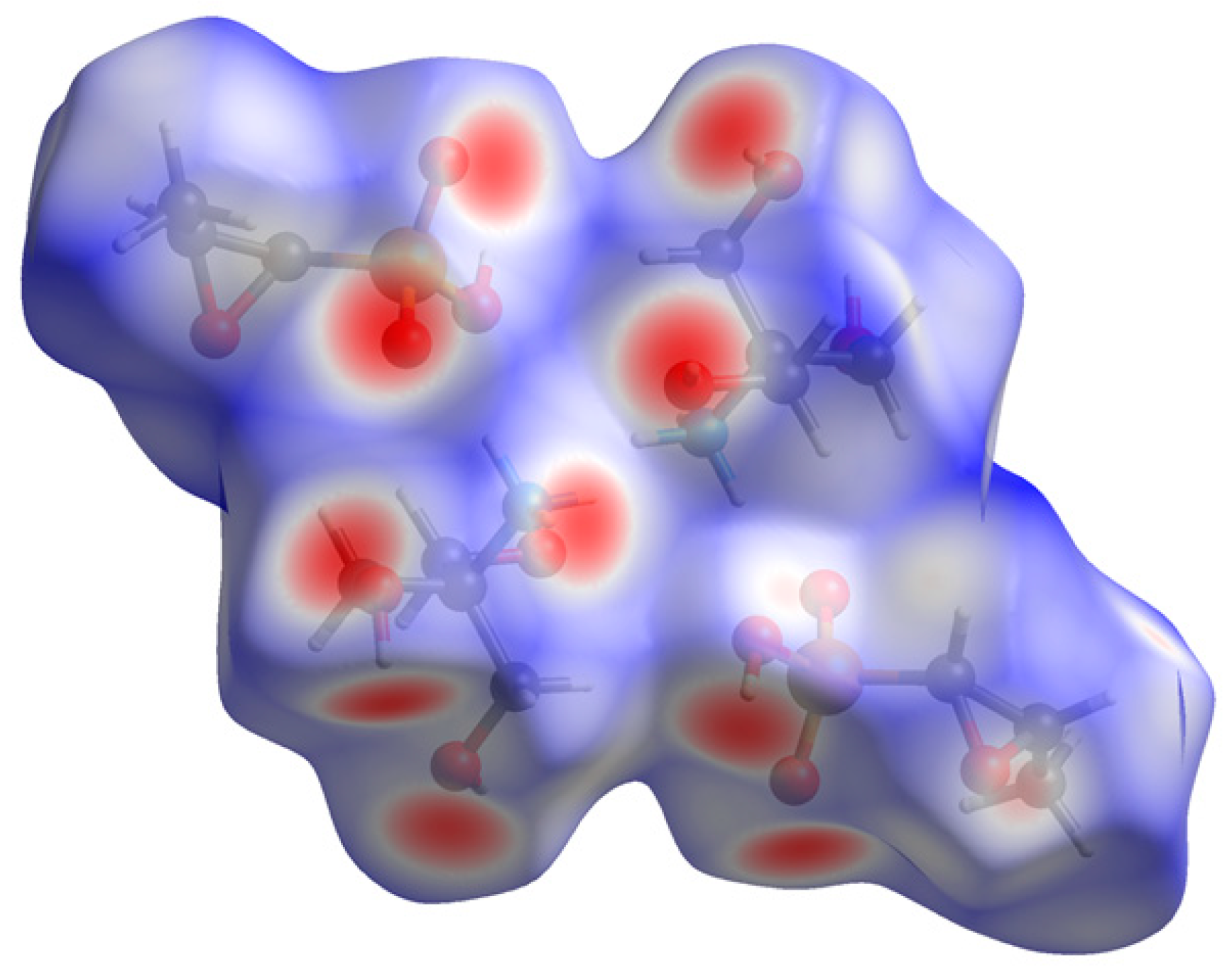{kind=link}
{kind=link}
| Fragment | Rms Δ, Å | Max. Δ, Å |
|---|---|---|
| cation 1 | 0.065 | 0.100 |
| cation 2 | 0.086 | 0.151 |
| anion 1 | 0.223 | 0.327 |
| anion 2 | 0.172 | 0.258 |
| H-Bond | D-H, Å | H⋅⋅⋅A, Å | D⋅⋅⋅A, Å | D-H⋯A, ̊ | Overlap, e | E, kcal/mol |
|---|---|---|---|---|---|---|
| N26-H61⋅⋅⋅O12 | 1.045 | 1.748 | 2.765 | 163.0 | 0.075 | 6.3 |
| N26-H60⋅⋅⋅O2 | 1.041 | 1.859 | 2.843 | 156.2 | 0.064 | 5.8 |
| N18-H58⋅⋅⋅O10 | 1.041 | 1.843 | 2.820 | 155.0 | 0.065 | 5.9 |
| N18-H57⋅⋅⋅O3 | 1.047 | 1.748 | 2.774 | 165.4 | 0.075 | 6.3 |
| N26-H62⋅⋅⋅O22 | 1.030 | 2.021 | 2.833 | 133.8 | 0.043 | 4.8 |
| N26-H62⋅⋅⋅O30 | 1.030 | 2.369 | 2.742 | 100.0 | 0.013 | 2.6 |
| N26-H60⋅⋅⋅O28 | 1.041 | 2.541* | 2.807 | 93.7 | 0.017 | 3.0 |
| N18-H59⋅⋅⋅O30 | 1.031 | 1.996 | 2.832 | 136.3 | 0.048 | 5.1 |
| N18-H59⋅⋅⋅O22 | 1.031 | 2.357 | 2.731 | 99.9 | 0.015 | 2.8 |
| N18-H58⋅⋅⋅O24 | 1.041 | 2.561* | 2.809 | 92.6 | 0.016 | 2.9 |
| O11-H56⋅⋅⋅O32 | 1.002 | 1.651 | 2.652 | 176.6 | 0.076 | 15.1 |
| O4-H55⋅⋅⋅O20 | 0.995 | 1.713 | 2.701 | 171.8 | 0.071 | 14.6 |
| O30-H67⋅⋅⋅O12 | 0.989 | 1.677 | 2.648 | 166.5 | 0.068 | 14.3 |
| O24-H65⋅⋅⋅O2 | 1.001 | 1.602 | 2.600 | 175.0 | 0.075 | 15.0 |
| O22-H64⋅⋅⋅O3 | 0.991 | 1.659 | 2.634 | 166.8 | 0.067 | 14.1 |
| O32-H68⋅⋅⋅O28 | 0.991 | 1.697 | 2.678 | 169.3 | 0.071 | 14.6 |
| O28-H66⋅⋅⋅O10 | 0.995 | 1.637 | 2.623 | 170.2 | 0.067 | 14.1 |
| O20-H63⋅⋅⋅O24 | 0.989 | 1.706 | 2.683 | 168.5 | 0.070 | 14.5 |
| C31-H53⋅⋅⋅O4 | 1.091 | 2.402 | 3.346 | 143.9 | 0.019 | |
| C31-H53⋅⋅⋅O30 | 1.091 | 2.532* | 2.894 | 98.0 | 0.011 | |
| C29-H51⋅⋅⋅O12 | 1.095 | 2.569 | 3.345 | 127.0 | 0.011 | |
| C27-H50⋅⋅⋅O15 | 1.092 | 2.613 | 3.508 | 138.6 | 0.010 | |
| C27-H49⋅⋅⋅O12 | 1.092 | 2.468 | 3.282 | 130.3 | 0.015 | |
| C23-H48⋅⋅⋅O3 | 1.094 | 2.569 | 3.353 | 127.8 | 0.012 | |
| C21-H46⋅⋅⋅O3 | 1.096 | 2.618 | 3.388 | 126.6 | 0.010 | |
| C19-H43⋅⋅⋅O11 | 1.091 | 2.381 | 3.327 | 144.1 | 0.019 | |
| C19-H43⋅⋅⋅O22 | 1.091 | 2.550 | 2.909 | 98.0 | 0.011 | |
| C16-H40⋅⋅⋅O10 | 1.089 | 2.530 | 3.373 | 133.4 | 0.017 | |
| C6-H34⋅⋅⋅O32 | 1.091 | 2.662 | 3.668 | 153.1 | 0.014 | |
| C5-H33⋅⋅⋅O15 | 1.092 | 2.384 | 3.407 | 155.4 | 0.021 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butler, Z.R.; Kaduk, J.A.; Gindhart, A.M.; Blanton, T.N. Crystal Structure of Fosfomycin Tromethamine, (C4H12NO3)(C3H6O4P), from Synchrotron Powder Diffraction Data and Density Functional Theory. Crystals 2019, 9, 384. https://doi.org/10.3390/cryst9080384
Butler ZR, Kaduk JA, Gindhart AM, Blanton TN. Crystal Structure of Fosfomycin Tromethamine, (C4H12NO3)(C3H6O4P), from Synchrotron Powder Diffraction Data and Density Functional Theory. Crystals. 2019; 9(8):384. https://doi.org/10.3390/cryst9080384
Chicago/Turabian StyleButler, Zachary R., James A. Kaduk, Amy M. Gindhart, and Thomas N. Blanton. 2019. "Crystal Structure of Fosfomycin Tromethamine, (C4H12NO3)(C3H6O4P), from Synchrotron Powder Diffraction Data and Density Functional Theory" Crystals 9, no. 8: 384. https://doi.org/10.3390/cryst9080384
APA StyleButler, Z. R., Kaduk, J. A., Gindhart, A. M., & Blanton, T. N. (2019). Crystal Structure of Fosfomycin Tromethamine, (C4H12NO3)(C3H6O4P), from Synchrotron Powder Diffraction Data and Density Functional Theory. Crystals, 9(8), 384. https://doi.org/10.3390/cryst9080384