Trinodal Self-Penetrating Nets from Reactions of 1,4-Bis(alkoxy)-2,5-bis(3,2’:6’,3’’-terpyridin-4’-yl)benzene Ligands with Cobalt(II) Thiocyanate

 , , ,
, , ,  and
and 
Abstract

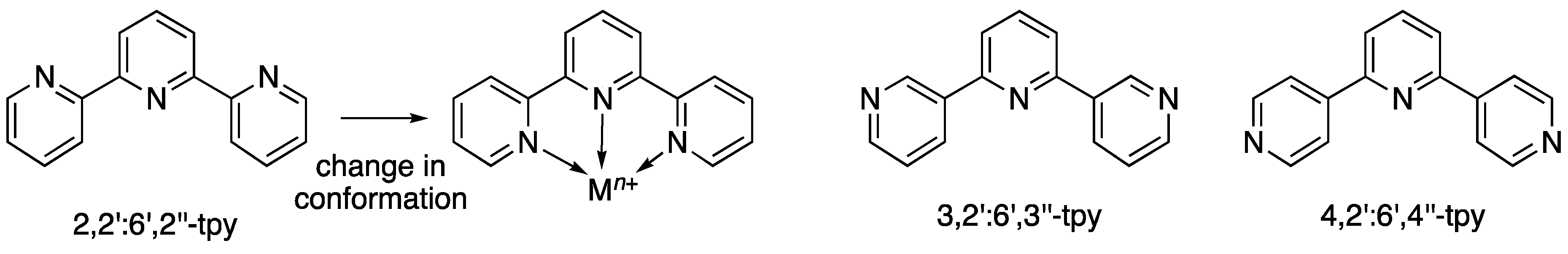
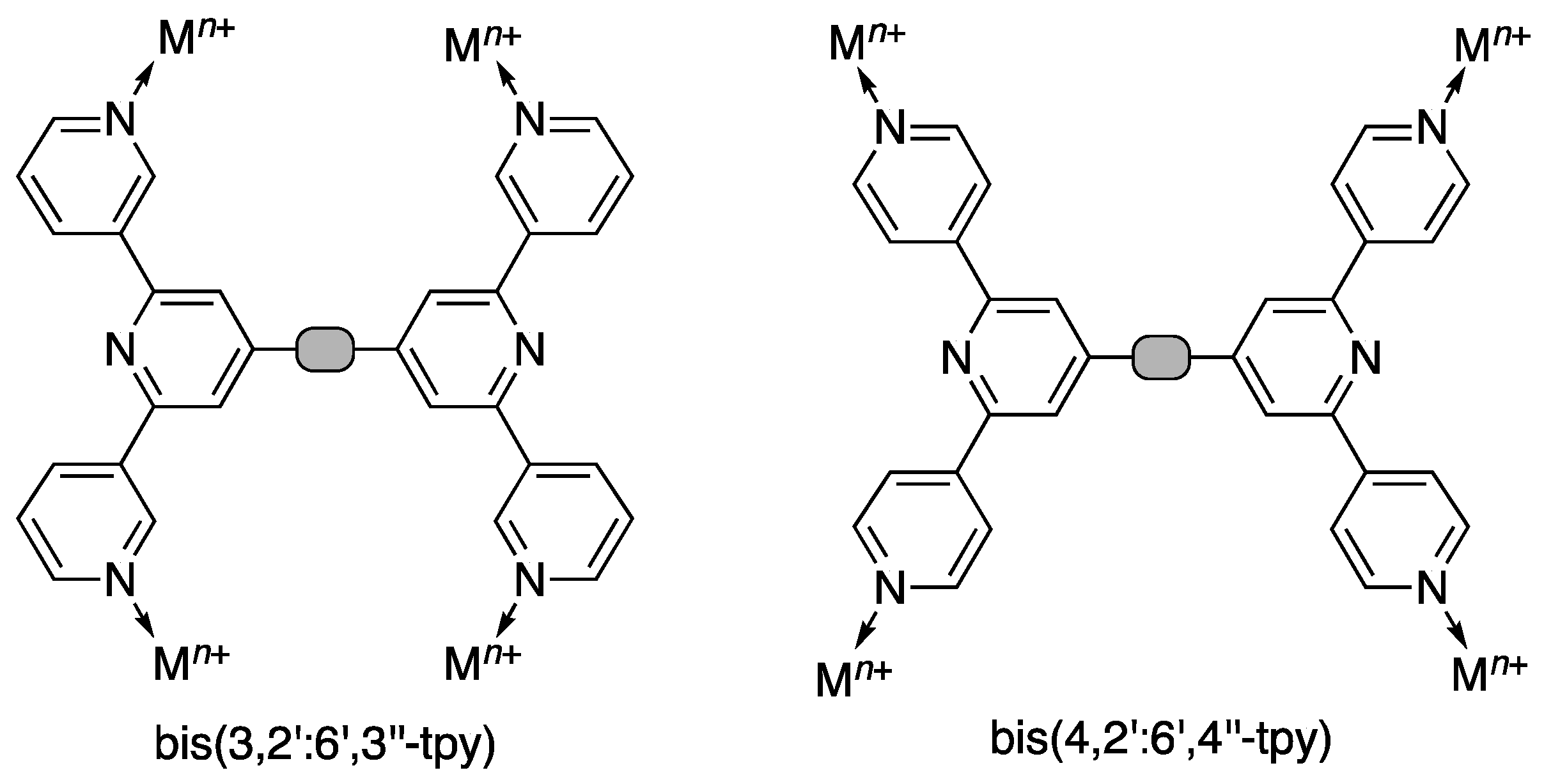
1. Introduction
2. Materials and Methods
2.1. General
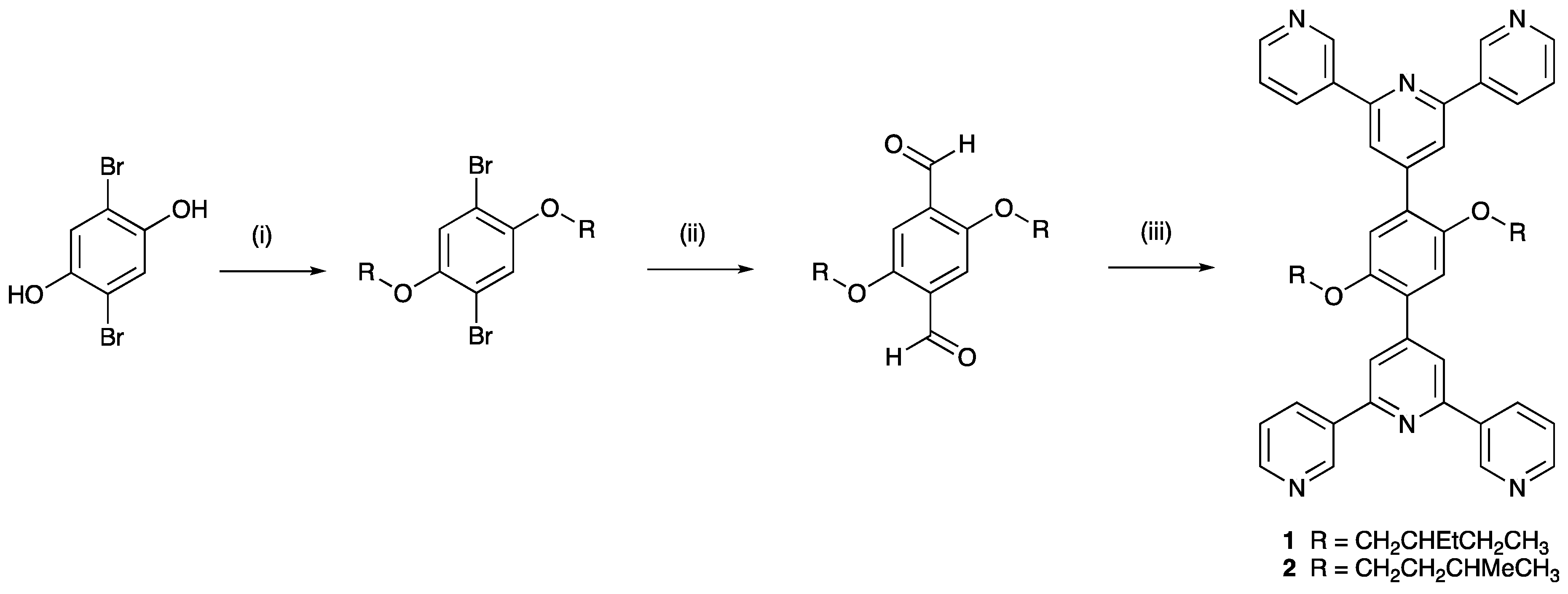
2.2. 1,4-Dibromo-2,5-bis(2-ethylbutoxy)benzene
2.3. 2,5-Bis(2-ethylbutoxy)benzene-1,4-dicarbaldehyde
2.4. 2,5-Bis(3-methylbutoxy)benzene-1,4-dicarbaldehyde
2.5. Synthesis of 1,4-Bis(2-ethylbutoxy)-2,5-bis(3,2’:6’,3’’-terpyridin-4’-yl)benzene (1)
2.6. Synthesis of 1,4-Bis(3-methylbutoxy)-2,5-bis(3,2’:6’,3’’-terpyridin-4’-yl)benzene (2)
2.7. Synthesis of [{Co(1)(NCS)2}·MeOH·3CHCl3]n
2.8. Synthesis of [{Co(2)(NCS)2}·0.8MeOH·1.8CHCl3]n
2.9. Crystallography
2.10. [{Co(1)(NCS)2}·MeOH.3CHCl3]n
2.11. [{Co(2)(NCS)2}·0.8MeOH·1.8CHCl3]n
3. Results and Discussion
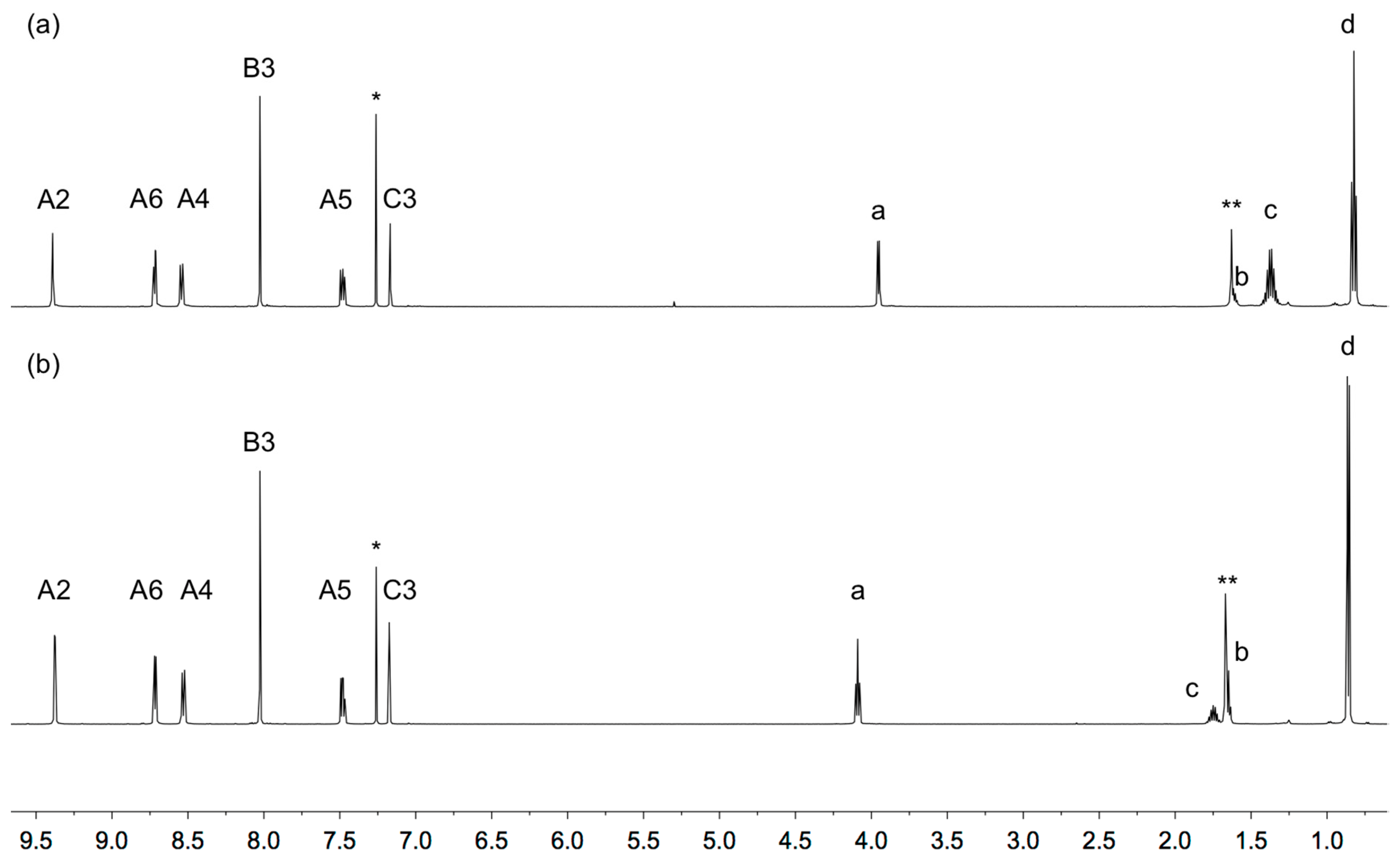
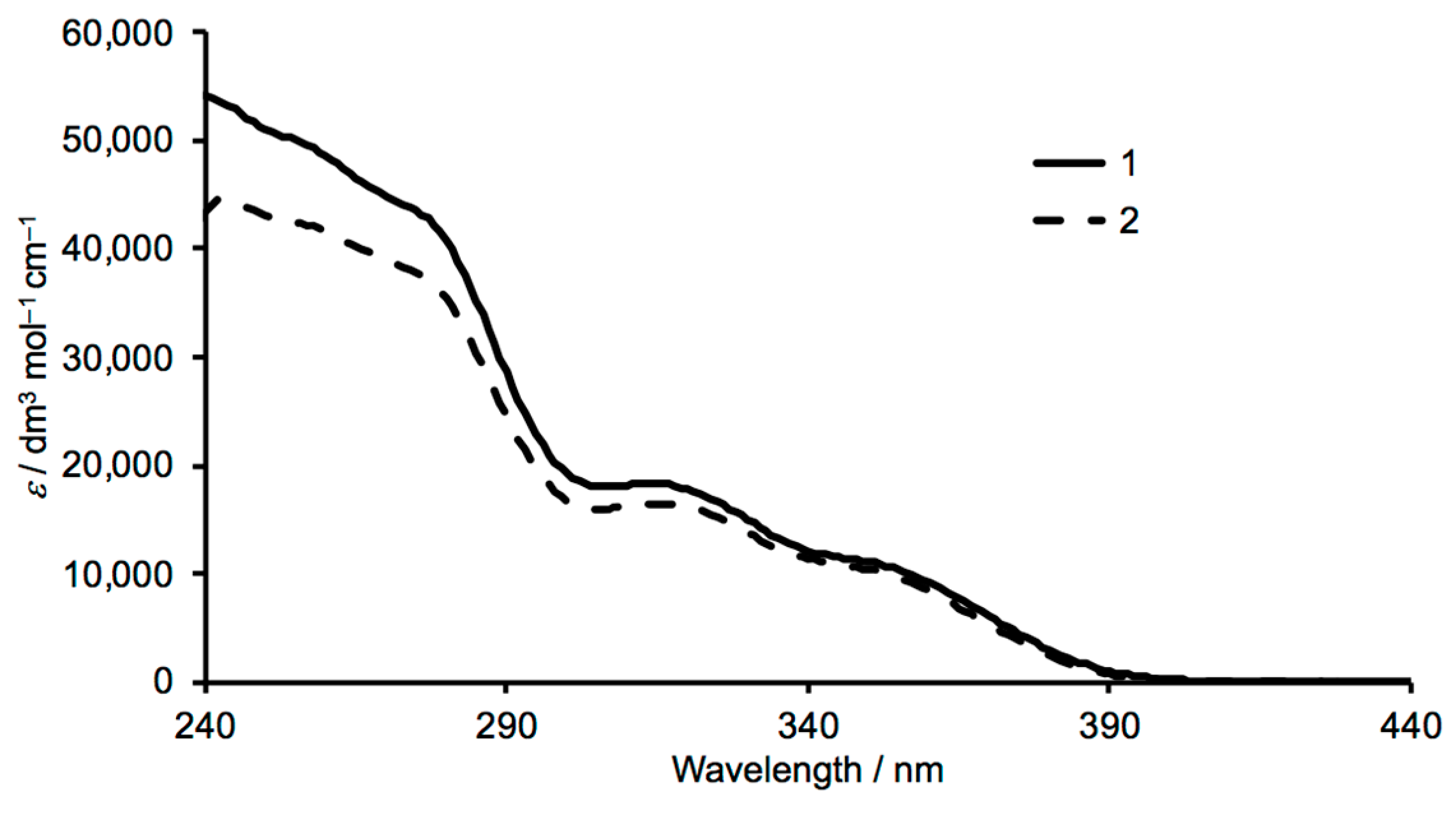
3.1. Synthesis and Characterization of Ligands 1 and 2
3.2. Crystal Growth and Powder X-ray Diffraction
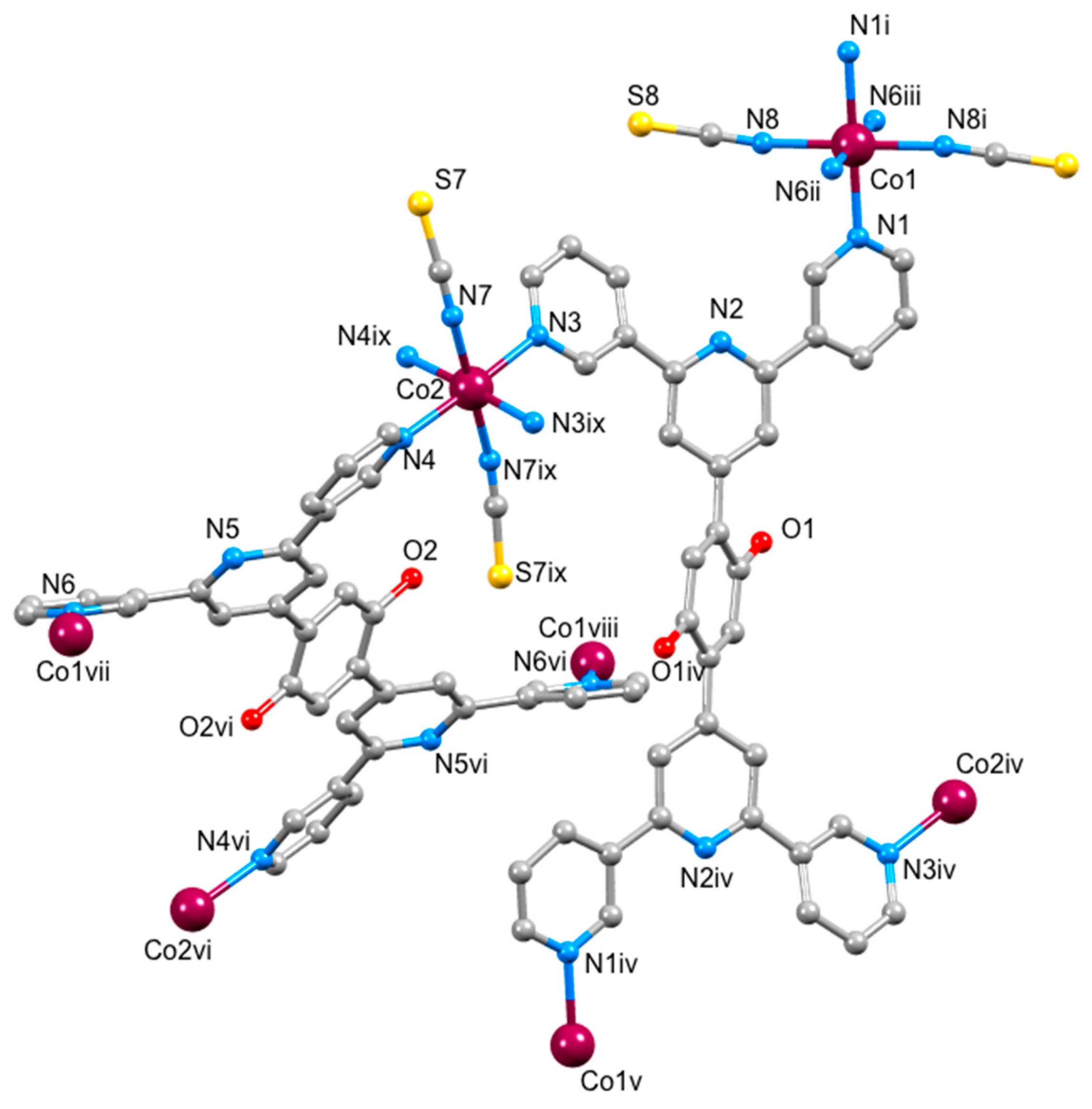
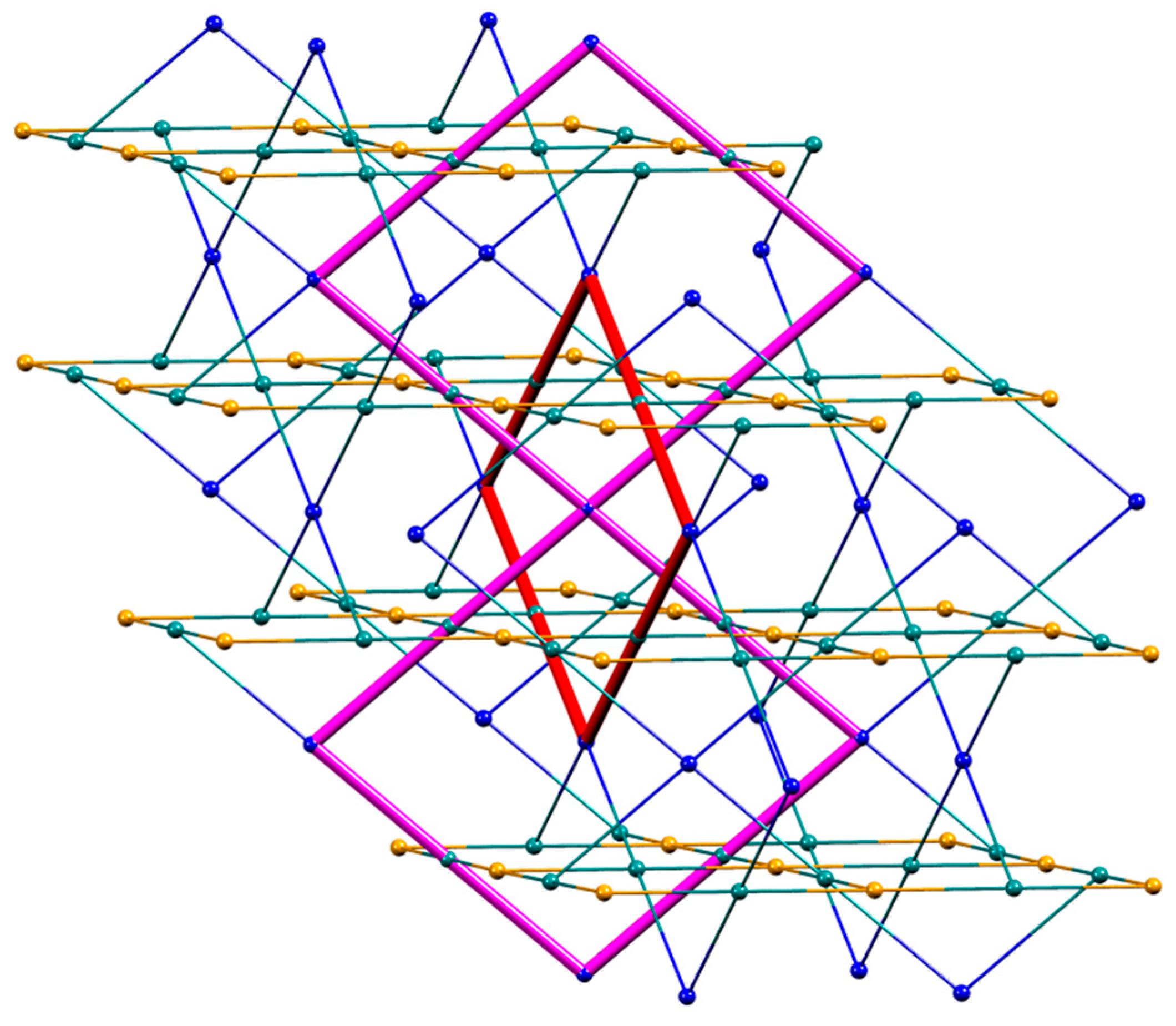
3.3. Single Crystal Structures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Constable, E.C. The Coordination Chemistry of 2,2′:6′,2″-Terpyridine and Higher Oligopyridines. Adv. Inorg. Chem. Radiochem. 1986, 30, 69–121. [Google Scholar] [CrossRef]
- Constable, E.C. 2,2′:6′,2″-Terpyridines: From chemical obscurity to common supramolecular motifs. Chem. Soc. Rev. 2007, 36, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Constable, E.C.; Housecroft, C.E. More hydra than Janus – Non-classical coordination modes in complexes of oligopyridine liigands. Coord. Chem. Rev. 2017, 359, 84–104. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E. Tetratopic bis(4,2’:6’,4’’-terpyridine) and bis(3,2’:6’,3’’-terpyridine) ligands as 4-connecting nodes in 2D-coordination networks and 3D-frameworks. J. Inorg. Organomet. Polym. Mater. 2018, 28, 414–427. [Google Scholar] [CrossRef]
- Wild, A.; Winter, A.; Schluetter, F.; Schubert, U.S. Advances in the Field of π-Conjugated 2,2′:6′,2″-Terpyridines. Chem. Soc. Rev. 2011, 40, 1459–1511. [Google Scholar] [CrossRef]
- Yan, Y.; Huang, J. Hierarchical Assemblies of Coordination Supramolecules. Coord. Chem. Rev. 2010, 254, 1072–1080. [Google Scholar] [CrossRef]
- Constable, E.C. A Journey from Supramolecular Chemistry to Nanoscale Networks. Chimia 2013, 67, 388–392. [Google Scholar] [CrossRef][Green Version]
- Constable, E.C.; Ward, M.D. Synthesis and co-ordination behaviour of 6′,6″-bis(2-pyridyl)- 2,2′:4,4″:2″,2‴-quaterpyridine; ‘back-to-back’ 2,2′:6′,2″-terpyridine. J. Chem. Soc. Dalton Trans. 1990, 1405–1409. [Google Scholar] [CrossRef]
- Newkome, G.R.; Cardullo, F.; Constable, E.C.; Moorefield, C.N.; Cargill Thompson, A.M.W. Metallomicellanols: Incorporation of ruthenium(II) - 2,2’:6’,2"-terpyridine triads into cascade polymers. J. Chem. Soc. Chem. Commun. 1993, 925–927. [Google Scholar] [CrossRef]
- Fu, J.-H.; Wang, S.-Y.; Chen, Y.-S.; Prusty, S.; Chan, Y.-T. One-Pot Self-Assembly of Stellated Metallo-Supramolecules from Multivalent and Complementary Terpyridine-Based Ligands. J. Am. Chem. Soc. 2019. [Google Scholar] [CrossRef]
- Lü, J.; Han, L.-W.; Alsmail, N.H.; Blake, A.J.; Lewis, W.; Cao, R.; Schröder, M. Control of Assembly of Dihydropyridyl and Pyridyl Molecules via Directed Hydrogen Bonding. Cryst. Growth Des. 2015, 15, 4219–4224. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.; Perez-Krap, C.; Suyetin, M.; Alsmail, N.H.; Yan, Y.; Yang, S.; Lewis, W.; Bicjoutskaia, E.; Tang, C.C.; Blake, A.J.; et al. Polycatenated 2D Hydrogen-Bonded Binary Supramolecular Organic Frameworks (SOFs) with Enhanced Gas Adsorption and Selectivity. Cryst. Growth Des. 2018, 18, 2555–2562. [Google Scholar] [CrossRef] [PubMed]
- Klein, Y.M.; Constable, E.C.; Housecroft, C.E.; Prescimone, A. A 3-dimensional {42.84} lvt net built from a ditopic bis(3,2’:6’,3’’-terpyridine) tecton bearing long alkyl tails. CrystEngComm 2015, 17, 2070–2073. [Google Scholar] [CrossRef]
- Klein, Y.M.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. A double-stranded 1D-coordination polymer assembled using the tetravergent ligand 1,1’-bis(4,2’:6’,4’’-terpyridin-4’-yl)ferrocene. Inorg. Chem. Comm. 2016, 70, 118–120. [Google Scholar] [CrossRef]
- Klein, Y.M.; Prescimone, A.; Karpacheva, M.; Constable, E.C.; Housecroft, C.E. Sometimes the same, sometimes different: Understanding self-assembly algorithms in coordination networks. Polymers 2018, 10, 1369. [Google Scholar] [CrossRef] [PubMed]
- Constable, E.C.; Housecroft, C.E.; Vujovic, S.; Zampese, J.A. 2D→2D Parallel interpenetration of (4,4) sheets constructed from a ditopic bis(4,2’:6’,4’’-terpyridine). CrystEngComm 2014, 16, 3494–3497. [Google Scholar] [CrossRef]
- Vujovic, S.; Constable, E.C.; Housecroft, C.E.; Morris, C.D.; Neuburger, M.; Prescimone, A. Engineering 2D→2D parallel interpenetration using long alkoxy-chain substituents. Polyhedron 2015, 92, 77–83. [Google Scholar] [CrossRef]
- Klein, Y.M.; Prescimone, A.; Neuburger, M.; Constable, E.C.; Housecroft, C.E. What a difference a tail makes: 2D→2D parallel interpenetration of sheets to interpenetrated nbo networks using ditopic-4,2’:6’,4’’-terpyridine ligands. CrystEngComm 2017, 19, 2894–2902. [Google Scholar] [CrossRef]
- Cave, G.W.V.; Raston, C.L. Efficient synthesis of pyridine via a sequential solventless aldol condensation and Michael addition. J. Chem. Soc. Perkin Trans. 1 2001, 3258–3264. [Google Scholar] [CrossRef]
- Beddoe, S.V.F.; Fitzpatrick, A.J.; Price, J.R.; Mallo, N.; Beves, J.E.; Morgan, G.G.; Kitchen, J.A.; Keene, T.D. A Bridge Too Far: Testing the Limits of Polypyridyl Ligands in Bridging Soluble Subunits of a Coordination Polymer. Cryst. Growth Des. 2017, 17, 6603–6612. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Constable, E.C. Ditopic and tetratopic 4,2’:6’,4"-Terpyridines as Structural Motifs in 2D- and 3D-Coordination Assemblies. Chimia 2019, 73, 462–467. [Google Scholar] [CrossRef]
- Yoshida, J.; Nishikiori, S.; Hidetaka, Y. Bis(3-cyano-pentane-2,4-dioato) Co(II) as a Linear Building Block for Coordination Polymers: Combinations with Two Polypyridines. J. Coord. Chem. 2013, 66, 2191–2200. [Google Scholar] [CrossRef]
- Shao, P.; Li, Z.; Luo, J.; Wang, H.; Qin, J. A Convenient Synthetic Route to 2,5-Dialkoxyterephthalaldehyde. Synth. Commun. 2005, 35, 49–53. [Google Scholar] [CrossRef]
- Jin, J.-Y.; Jin, Z.-Z.; Xia, Y.; Zhou, Z.-Y.; Wu, X.; Zhu, D.-X.; Su, Z.-M. Design and synthesis of 1,4-bis[4-(1,1-dicyanovinyl)styryl]-2,5-bis(alkoxy)benzenes as red organic electroluminescent PPV analogs. Polymer 2007, 48, 4028–4033. [Google Scholar] [CrossRef]
- Software for the Integration of CCD Detector System Bruker Analytical X-ray Systems, version 2; Bruker AXS: Madison, WI, USA, 2013.
- Sheldrick, G.M. ShelXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. Superflip - A Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Palatinus, L.; Prathapa, S.J.; van Smaalen, S. EDMA: A Computer Program for Topological Analysis of Discrete Electron Densities. J. Appl. Cryst. 2012, 45, 575–580. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0 - New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Spek, A.L. Platon Squeeze: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- LeBail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mat. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Pawley, G.S. Unit-cell refinement from powder diffraction scans. J. Appl. Cryst. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent Advances in Magnetic Structure Determination by Neutron Powder Diffraction. Phys. B 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Roisnel, T.; Rodríguez-Carvajal, J. WinPLOTR: A Windows tool for powder diffraction patterns analysis. Mater. Sci. Forum 2001, 118–123. [Google Scholar] [CrossRef]
- Prasad, T.K.; Suh, M.P. Metal-organic frameworks incorporating various alkoxy pendant groups: Hollow tubular morphologies, X-ray single-crystal structures, and selective carbon dioxide adsorption properties. Chem. Asian J. 2015, 10, 2257–2263. [Google Scholar] [CrossRef]
- Delgado-Friedrichs, O.; O’Keeffe, M. Identification of and symmetry computation for crystal nets. Acta Crystallogr. Sect. A 2003, 59, 351–360. [Google Scholar] [CrossRef]
- Batten, S.R.; Neville, S.M.; Turner, D.R. Coordination Polymers: Design, Analysis and Application; RSC Publishing: Cambridge, UK, 2009; ISBN 978-0-85404-837-3. [Google Scholar]
- O’Keeffe, M.; Peskov, M.A.; Ramsden, S.J.; Yaghi, O.M. The Reticular Chemistry Structure Resource (RCSR) database of, and symbols for, crystal nets. Acc. Chem. Res. 2008, 41, 1782–1789. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond a | [{Co(1)(NCS)2}.MeOH.3CHCl3]n | Corresponding Bond Lengths in [{Co(2)(NCS)2}.0.8MeOH.1.8CHCl3]n |
|---|---|---|
| Bond Length/Å | Bond Length/Å | |
| Co1–N1 | 2.171(6) | 2.175(6) |
| Co1–N6 ii | 2.229(6) | 2.244(6) |
| Co1–N8 | 2.065(6) | 2.091(5) |
| Co2–N3 | 2.136(6) | 2.138(6) |
| Co2–N4 | 2.145(6) | 2.134(6) |
| Co2–N7 | 2.074(6) | 2.107(7) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manfroni, G.; Prescimone, A.; Batten, S.R.; Klein, Y.M.; Gawryluk, D.J.; Constable, E.C.; Housecroft, C.E. Trinodal Self-Penetrating Nets from Reactions of 1,4-Bis(alkoxy)-2,5-bis(3,2’:6’,3’’-terpyridin-4’-yl)benzene Ligands with Cobalt(II) Thiocyanate. Crystals 2019, 9, 529. https://doi.org/10.3390/cryst9100529
Manfroni G, Prescimone A, Batten SR, Klein YM, Gawryluk DJ, Constable EC, Housecroft CE. Trinodal Self-Penetrating Nets from Reactions of 1,4-Bis(alkoxy)-2,5-bis(3,2’:6’,3’’-terpyridin-4’-yl)benzene Ligands with Cobalt(II) Thiocyanate. Crystals. 2019; 9(10):529. https://doi.org/10.3390/cryst9100529
Chicago/Turabian StyleManfroni, Giacomo, Alessandro Prescimone, Stuart R. Batten, Y. Maximilian Klein, Dariusz J. Gawryluk, Edwin C. Constable, and Catherine E. Housecroft. 2019. "Trinodal Self-Penetrating Nets from Reactions of 1,4-Bis(alkoxy)-2,5-bis(3,2’:6’,3’’-terpyridin-4’-yl)benzene Ligands with Cobalt(II) Thiocyanate" Crystals 9, no. 10: 529. https://doi.org/10.3390/cryst9100529
APA StyleManfroni, G., Prescimone, A., Batten, S. R., Klein, Y. M., Gawryluk, D. J., Constable, E. C., & Housecroft, C. E. (2019). Trinodal Self-Penetrating Nets from Reactions of 1,4-Bis(alkoxy)-2,5-bis(3,2’:6’,3’’-terpyridin-4’-yl)benzene Ligands with Cobalt(II) Thiocyanate. Crystals, 9(10), 529. https://doi.org/10.3390/cryst9100529