Atomic Charges and Chemical Bonding in Y-Ga Compounds


Abstract
1. Introduction
2. Methods
3. Results and Discussion
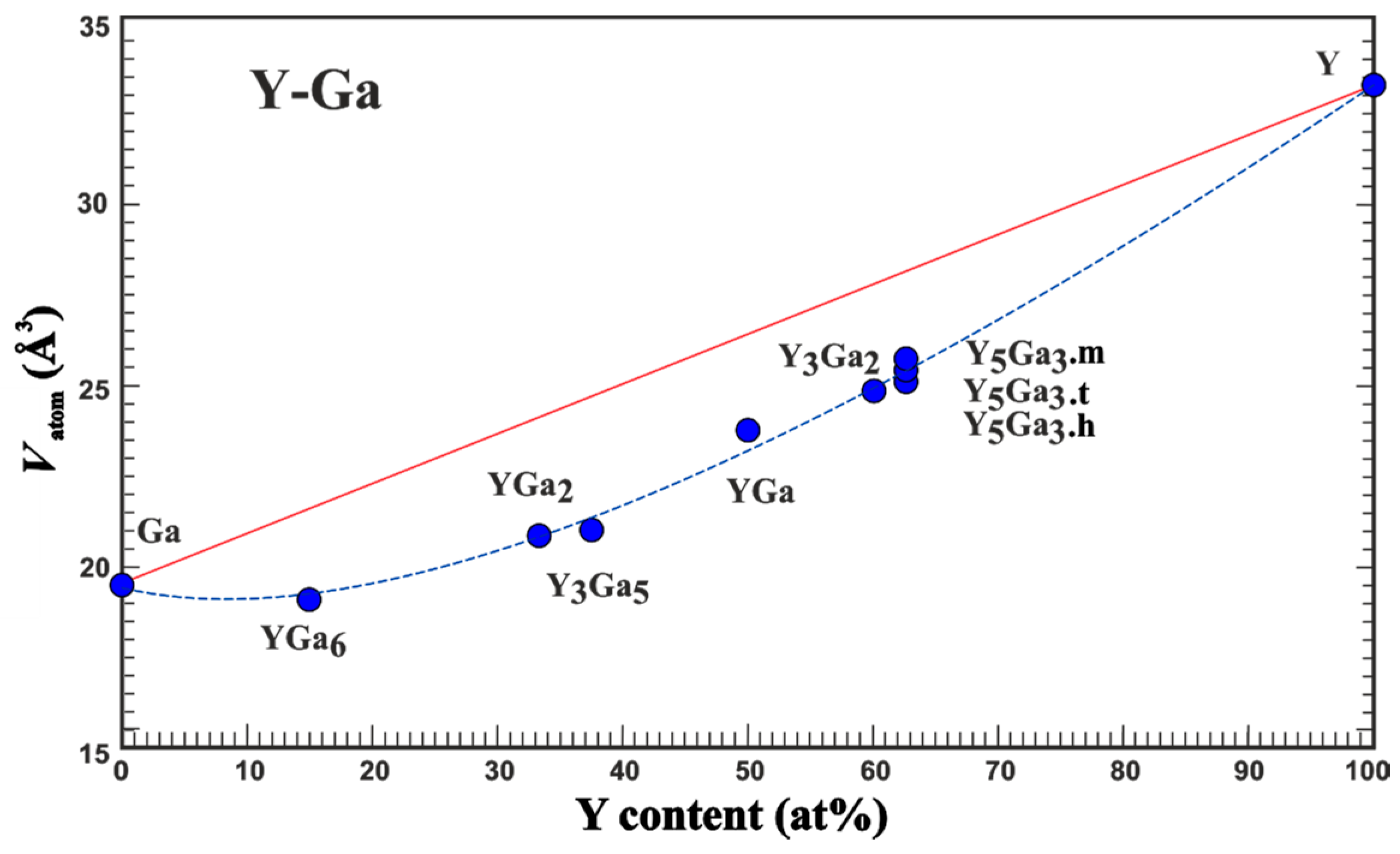
3.1. Average Atomic Volume
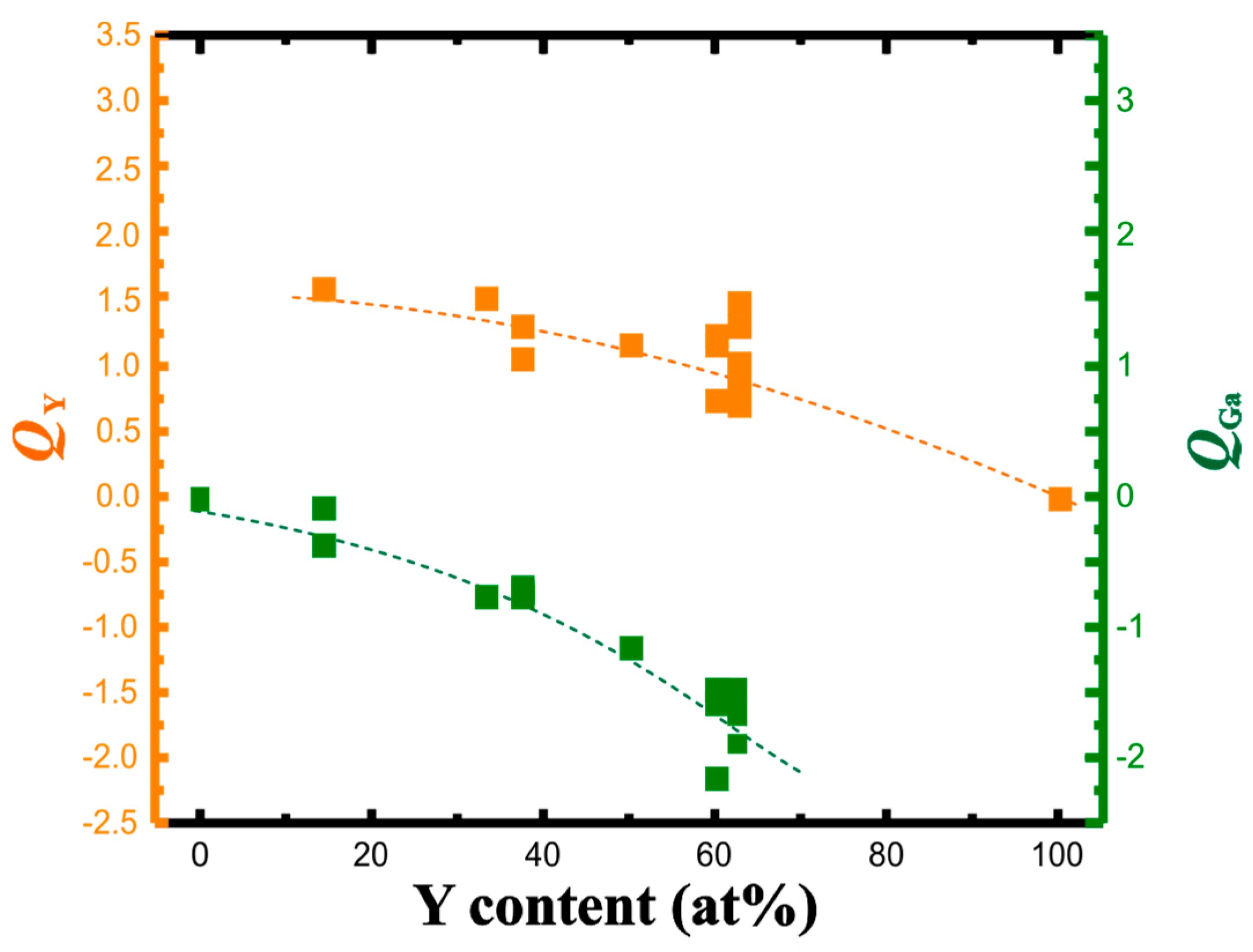
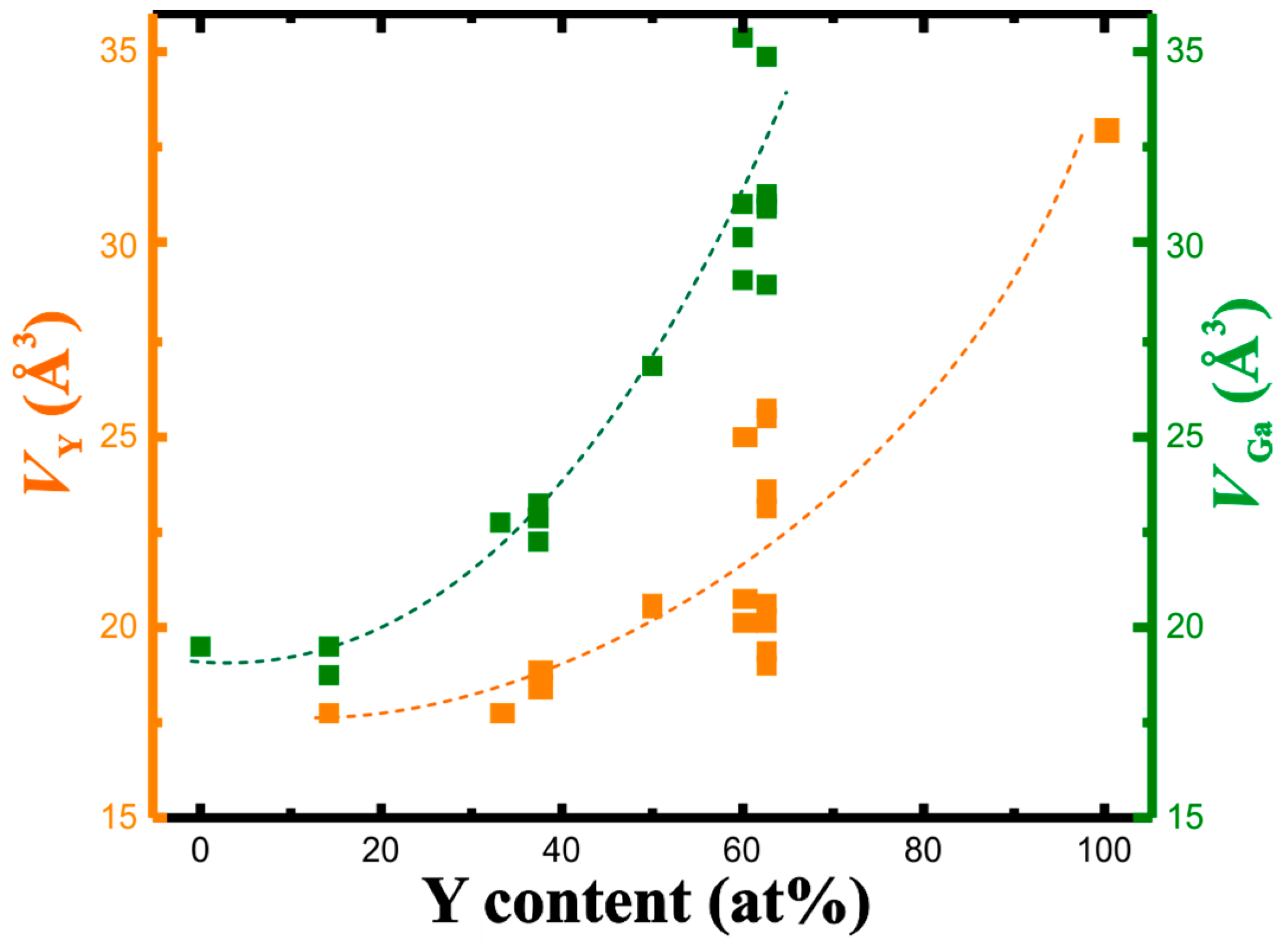
3.2. QTAIM Charges and Atomic Volumes
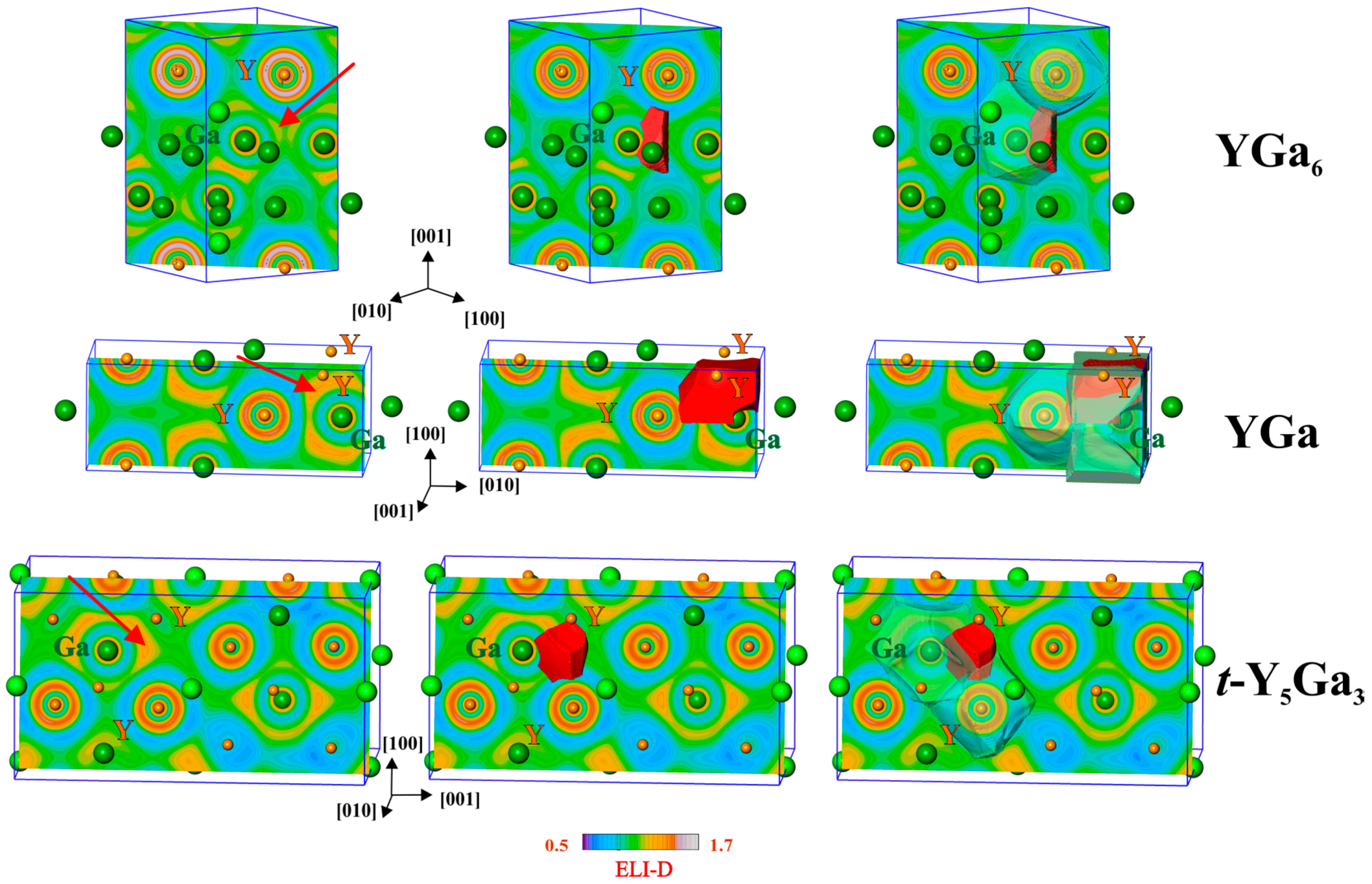
3.3. Yttrium–Gallium Interactions from Electron-Localizabilty Approach
4. Conclusions
Author Contributions
Conflicts of Interest
References and Note
- Barrett, C.S.; Massalski, T.B. Structure od Metals, 3rd ed.; Pergamon Press: Oxford, UK; New York, USA; Seoul, Korea; Tokyo, Japan, 1992; p. 306. [Google Scholar]
- Bader, R.F. Atoms in Molecules—A Quantum Theory; Clarendon Press: Oxford, UK, 1995. [Google Scholar]
- Kohout, M. Bonding indicators from electron pair density functionals. Faraday Discuss. 2007, 135, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.R.; Bezugly, V.; Kohout, M.; Grin, Y. Charge Decomposition Analysis of the Electron Localizability Indicator: A Bridge between the Orbital and Direct Space Representation of the Chemical Bond. Chem. Eur. J. 2007, 13, 5724–5741. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Savin, A.; Becke, A.D.; Flad, J.; Nesper, R.; Preuss, H.; von Schnering, H.G. A new look at electron localization. Angew. Chem. Int. Ed. Engl. 1991, 30, 409–412. [Google Scholar] [CrossRef]
- Grin, Y.; Savin, A.; Silvi, B. The ELF perspective of chemical bonding. In The Chemical Bond: Chemical Bonding Across the Periodic Table; Wiley-VCH: Weinheim, Germany, 2014; Volume 1, p. 345. [Google Scholar]
- Grin, Y.; Wagner, F.R.; Armbrüster, M.; Kohout, M.; Leithe-Jasper, A.; Schwarz, U.; Wedig, U.; von Schnering, H.G. CuAl2 revisited: Composition, crystal structure, chemical bonding, compressibility and Raman spectroscopy. J. Solid State Chem. 2006, 179, 1707–1719. [Google Scholar] [CrossRef]
- Baranov, A.; Kohout, M.; Wagner, F.R.; Grin, Y.; Bronger, W. Spatial chemistry of the aluminum-platinum compounds: A quantum chemical approach. Z. Kristallogr. 2007, 222, 527–531. [Google Scholar] [CrossRef]
- Amon, A.; Ormeci, A.; Bobnar, M.; Akselrud, L.G.; Avdeev, M.; Gumeniuk, R.; Burkhardt, U.; Prots, Y.; Hennig, C.; Leithe-Jasper, A.; Grin, Y. Cluster Formation in the Superconducting Complex Intermetallic Compound Be21Pt5. Acc. Chem. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Akselrud, L.; Bobnar, M.; Burkhardt, U.; Schmidt, M.; Zhao, J.-T.; Schwarz, U.; Grin, Y. Weak Interactions under Pressure: hp-CuBi and its Analogues. Angew. Chem. Int. Ed. 2017, 56, 5620–5624. [Google Scholar] [CrossRef] [PubMed]
- Ormeci, A.; Simon, A.; Grin, Y. Structural Topology and Chemical Bonding in Laves Phases. Angew. Chem. Int. Ed. 2010, 49, 8997–9001. [Google Scholar] [CrossRef] [PubMed]
- Kaltsoyannis, K.; Kerridge, A. Chemical Bonding in Lanthanides and Actinides. In The Chemical Bond Chemical Bonding Across the Periodic Table; Wiley-VCH: Weinheim, Germany, 2014; Volume 2, p. 337. [Google Scholar]
- Börrnert, C.; Grin, Y.; Wagner, F.R.W. Position-Space Bonding Indicators for Hexaborides of Alkali, Alkaline Earth, and Rare Earth Metals in Comparison to the Molecular Crystal K2[B6H6]. Z. Anorg. Allg. Chem. 2013, 639, 2013–2024. [Google Scholar] [CrossRef]
- Prots, Y.; Deppe, M.; Cardoso-Gil, R.; Cervellino, A.; Ormeci, A.; Geibel, C.; Grin, Y. Yb2Al15Pt6—Tshe most ordered variety of the Sc1.2Fe4Si9.8 aristotype. Chem. Met. Alloys 2014, 7, 85–99. [Google Scholar]
- Pöttgen, R.; Borrmann, H.; Grin, Y. Multi-Center vs. Two-Center Bonding within the Hetero-Polyanion in Eu2GaPt2 and its Prototype Ca2SiIr2. Z. Anorg. Allg. Chem. 2017, 643, 1338–1343. [Google Scholar] [CrossRef]
- Wirth, S.; Prots, Y.; Wedel, M.; Ernst, S.; Kirchner, S.; Fisk, Z.; Thompson, J.D.; Steglich, F.; Grin, Y. Structural Investigations of CeIrIn5 and CeCoIn5 on Macroscopic and Atomic Length Scales. J. Phys. Soc. Jpn. 2014, 83, 061009. [Google Scholar] [CrossRef]
- Yatsenko, S.P.; Semyannikov, A.A.; Semenov, G.B.; Chuntonov, K.A. Phase diagrams of rare earth metals with gallium. J. Less Common Met. 1979, 64, 185–199. [Google Scholar] [CrossRef]
- Sharma, B.D.; Donohue, J.A. A refinement of the crystal structure of gallium. Z. Kristallogr. 1962, 117, 293–300. [Google Scholar] [CrossRef]
- Gryniv, O.I.; Gryniv, I.A.; Grin, Y.; Yarmolyuk, Y.P. Synthesis and crystalline structure of the YGa6 compound. Dopov. Akad. Nauk Ukr. RSR Ser. B 1985, 5, 42–44. [Google Scholar]
- Bruzzone, G.; Ruggiero, A.F.; Bonino, G.B. Struttura di alcuni composti intermetallici dell‘ittrio. II. Composti con Al, Ga, In, Tl, Pb, Sn. Atti Accad. Naz. Lincei Cl. Sci. Fis. Mat. Nat. Rend. 1962, 33, 465–471. [Google Scholar]
- Yatsenko, S.P.; Gladyshevskii, E.I.; Tschuntonow, K.A.; Yarmolyuk, Y.P.; Grin, Y. Kristallstruktur von Tm3Ga5 und analoger Verbindungen. J. Less-Common Met. 1983, 91, 21–32. [Google Scholar] [CrossRef]
- Schob, O.; Parthé, E. AB Compounds with Sc, Y and Rare Earth Metals. I. Scandium and Yttrium Compounds with CrB and CsCl Structure. Acta Crystallogr. 1965, 19, 214–224. [Google Scholar] [CrossRef]
- Yatsenko, S.P.; Gladyshevskii, R.E.; Sitschewitsch, O.M.; Belskii, V.K.; Semyannikov, A.A.; Grin, Y.; Yarmolyuk, Y.P. Kristallstruktur von Gd3Ga2 und isotypen Verbindungen. J. Less-Common Met. 1986, 115, 17–22. [Google Scholar] [CrossRef]
- Zhao, J.-T.; Corbett, J.D. R5Ga3 compounds of selected rare earth metals R: Structures and properties. J. Alloys Compds. 1994, 210, 1–7. [Google Scholar] [CrossRef]
- Markiv, V.Y.; Belyavina, N.N. Crystal structure of new compounds of the composition R5Ga3 (R = Y, Tm, Lu). Dopov. Akad. Nauk Ukr. 1992, 10, 142–416. [Google Scholar]
- Schob, O.; Parthé, E. Sc5Ga3 and Y5Ga3 with D88 structure. Acta Crystallogr. 1964, 17, 1335–1336. [Google Scholar] [CrossRef]
- Spedding, F.H.; Sanden, B.; Beaudry, B.J. The Er-Y, Tb-Ho, Tb-Er, Dy-Ho, Dy-Er and Ho-Er phase systems. J. Less-Common Met. 1973, 31, 1–13. [Google Scholar] [CrossRef]
- Zhao, J.-T.; Seo, D.-K.; Corbett, J.D. Synthesis, structure and properties of CaGa, YGa and Y(Ga,Z) phases: A model for the transformation of a CrB to MoB-type structure in doped YGa phases. J. Alloys Compds. 2002, 334, 110–117. [Google Scholar] [CrossRef]
- Kohout, M.; Wagner, F.R. Modified version lmto47e of the Stuttgart TB-LMTO-ASA Code; Max-Planck-Institut für Chemische Physik fester Stoffe: Dresden, Germany, 2016. [Google Scholar]
- Jepsen, O.; Burkhardt, A.; Andersen, O.K. The Program TB-LMTO-ASA; Version 4.7; Max-Planck-Institut für Festkörperforschung: Stuttgart, Germany, 1999. [Google Scholar]
- Barth, U.; Hedin, L. A local exchange-correlation potential for the spin polarized case I. J. Phys. C 1972, 5, 1629–1642. [Google Scholar] [CrossRef]
- Andersen, O.K. Linear methods in band theory. Phys. Rev. B 1975, 12, 3060–3083. [Google Scholar] [CrossRef]
- Kohout, M. DGrid, version 4.6. Available online: http://www2.cpfs.mpg.de/~kohout/dgrid.html (accessed on 25 January 2018).
- Bende, D.; Grin, Y.; Wagner, F.R. Chemical Bonding in MgAgAs-Type Compounds. In Heusler Alloys; Felser, C., Hirohata, A., Eds.; Springer: Cham, Switzerland; Heidelberg, Germany; New York, USA; Dodrecht, The Netherland; London, UK, 2016; p. 133ff. [Google Scholar]
- Dewhurst, K.; Sharma, S.; Nordström, L.; Granäs, O.; Gross, E.K.U. Program Elk, version 2.2.10. Available online: http://www.sourceforge.net (accessed on 25 January 2018).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Kohout, M.; Baranov, A.I. DGrid 4.7. Radebeul, Germany, 2012. [Google Scholar]
- Wagner, F.R. Program DISij, version 7.1.0; Max-Planck-Institut für Chemische Physik fester Stoffe: Dresden, Germany, 2015. [Google Scholar]
- Wang, S.; Yang, J.; Wu, L.; Wei, P.; Yang, J.; Zhang, W.; Grin, Y. Anisotropic multicenter bonding and high thermoelectric performance in electron-poor CdSb. Chem. Mater. 2015, 27, 1071–1081. [Google Scholar] [CrossRef]
- Kohout, M.; Wagner, F.R.; Grin, Y. Electron localization function for transition-metal compounds. Theor. Chem. Acc. 2002, 108, 150–156. [Google Scholar] [CrossRef]
- Baranov, A.I. Electron Localizability Indicators from Spinor Wavefunctions. J. Comp. Chem. 2014, 35, 565–585. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.R.; Bende, D.; Grin, Y. Heteropolar bonding and a position-space representation of the 8 − N rule. Dalton Trans. 2016, 45, 3236–3243. [Google Scholar] [CrossRef] [PubMed]
- Raub, S.; Jansen, G. A quantitative measure of bond polarity from the electron localization function and the theory of atoms in molecules. Theor. Chem. Acc. 2001, 106, 223–232. [Google Scholar] [CrossRef]
- Angyan, J.G.; Loos, M.; Meyer, I. Covalent Bond Orders and Atomic Valence Indices in the Topological Theory of Atoms in Molecules. J. Phys. Chem. 1994, 98, 5244–5248. [Google Scholar] [CrossRef]
- Wagner, F.R.; Baranov, A.I.; Grin, Y.; Kohout, M. A Position-Space View on Chemical Bonding in Metal Diborides with AlB2 Type of Crystal Structure. Z. Anorg. Allg. Chem. 2013, 639, 2025–2035. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure Type | Space Group | Unit Cell Dimensions | Atomic Coordinates | VAtom, Å3 | QAtom | Charge Transfer per Ga Atom, e | Reference for Crystallo-Graphic Data | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a, Å | b, Å | c, Å | β, deg | VUnit Cell, Å3 | Atom | x | y | z | |||||||
| Ga | Ga | Cmca | 4.523 | 7.661 | 4.524 | 156.76 | Ga | 0 | 0.1549 | 0.0810 | 19.60 | 0 | 0 | 19 | |
| YGa6 | PuGa6 | P4/nbm | 5.947 | 7.549 | 225.87 | Y | ¼ | ¾ | 0 | 17.84 | +1.60 | 0.27 | 20 | ||
| Ga1 | ¼ | ¼ | 0.1610 | 18.75 | −0.08 | ||||||||||
| Ga2 | 0.4359 | 1 − x | 03480 | 19.53 | −0.36 | ||||||||||
| YGa2 | AlB2 | P6/mmm | 4.211 | 4.112 | 63.15 | Y | 0 | 0 | 0 | 17.70 | +1.54 | 0.77 | 21 | ||
| Ga | ⅓ | ⅔ | ½ | 22.83 | −0.77 | ||||||||||
| Y3Ga5 | Tm3Ga5 | Pnma | 11.450 | 9.674 | 6.098 | 675.46 | Y1 | 0.3521 | 0.0092 | 0.1099 | 18.49 | +1.31 | 0.74 | 22 | |
| Y2 | 0.0736 | ¼ | 0.1524 | 18.93 | +1.06 | ||||||||||
| Ga1 | 0.0909 | 0.5527 | 0.1244 | 22.22 | −0.76 | ||||||||||
| Ga2 | 0.0052 | ¼ | 0.6280 | 23.23 | −0.74 | ||||||||||
| Ga3 | 0.2520 | ¼ | 0.8250 | 22.91 | −0.70 | ||||||||||
| Ga4 | 0.2980 | ¼ | 0.3970 | 22.94 | −0.72 | ||||||||||
| YGa | α-ITl | Cmcm | 4.302 | 10.860 | 4.073 | 190.29 | Y | 0 | 0.138 | ¼ | 20.61 | +1.15 | 1.15 | 23 | |
| Ga | 0 | 0.417 | ¼ | 26.91 | -1.15 | ||||||||||
| Y3Ga2 | Gd3Ga2 | I4/mcm | 11.620 | 14.860 | 2006.46 | Y1 | 0.0666 | 0.2029 | 0.1356 | 20.78 | +1.16 | 1.65 | 24 | ||
| Y2 | 0.1688 | x + ½ | 0 | 25.02 | +0.74 | ||||||||||
| Y3 | 0 | ½ | 0.1510 | 20.14 | +1.22 | ||||||||||
| Ga1 | 0.1777 | x + ½ | 0.2013 | 29.18 | −1.48 | ||||||||||
| Ga2 | 0.6160 | x − ½ | 0 | 31.06 | −2.13 | ||||||||||
| Ga3 | 0 | 0 | 0 | 35.46 | −1.47 | ||||||||||
| Ga4 | 0 | 0 | ¼ | 30.22 | −1.58 | ||||||||||
| t-Y5Ga3 | Ba5Si3 | P4/ncc | 7.6474 | 14.147 | 827.36 | Y1 | 0 | ½ | 0.2603 | 19.39 | +1.30 | 1.59 | 25 | ||
| Y2 | 0.1723 | 0.1621 | 0.1027 | 23.61 | +0.87 | ||||||||||
| Ga1 | 0 | 0.5 | 0.994 | 34.94 | −1.88 | ||||||||||
| Ga2 | 0.3768 | x | ¼ | 29.03 | −1.45 | ||||||||||
| m-Y5Ga3 | Y5Ga3 | C2/m | 8.635 | 14.894 | 6.510 | 91.19 | 837.07 | Y1 | 0.7300 | 0 | 0.226 | 25.77 | +0.79 | 1.60 | 26 |
| Y2 | 0 | 0.1800 | 0 | 19.03 | +1.48 | ||||||||||
| Y3 | 0 | 0.151 | ½ | 20.24 | +1.03 | ||||||||||
| Y4 | 0.378 | 0.123 | 0.264 | 25.49 | +0.71 | ||||||||||
| Ga1 | 0.110 | 0 | 0.198 | 31.16 | −1.51 | ||||||||||
| Ga2 | 0.212 | 0.301 | 0.293 | 31.03 | −1.65 | ||||||||||
| h-Y5Ga3 | Mn5Si3 | P63/mcm | 8.576 | 6.479 | 412.67 | Y1 | 0.236 | 0 | ¼ | 20.67 | +1.01 | 1.66 | 27 | ||
| Y2 | ⅓ | ⅔ | 0 | 23.19 | +0.98 | ||||||||||
| Ga | 0.5991 | 0 | ¼ | 31.41 | −1.66 | ||||||||||
| Y | Mg | P63/mmc | 3.6482 | 5.7318 | 66.08 | Y | ⅓ | ⅔ | ¼ | 33.04 | 0 | 28 | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grin, Y.; Fedorchuk, A.; Faria, R.J.; Wagner, F.R. Atomic Charges and Chemical Bonding in Y-Ga Compounds. Crystals 2018, 8, 99. https://doi.org/10.3390/cryst8020099
Grin Y, Fedorchuk A, Faria RJ, Wagner FR. Atomic Charges and Chemical Bonding in Y-Ga Compounds. Crystals. 2018; 8(2):99. https://doi.org/10.3390/cryst8020099
Chicago/Turabian StyleGrin, Yuri, Anatolij Fedorchuk, Rodolfo J. Faria, and Frank R. Wagner. 2018. "Atomic Charges and Chemical Bonding in Y-Ga Compounds" Crystals 8, no. 2: 99. https://doi.org/10.3390/cryst8020099
APA StyleGrin, Y., Fedorchuk, A., Faria, R. J., & Wagner, F. R. (2018). Atomic Charges and Chemical Bonding in Y-Ga Compounds. Crystals, 8(2), 99. https://doi.org/10.3390/cryst8020099