Dislocation Structure and Mobility in Hcp Rare-Gas Solids: Quantum versus Classical



Abstract
1. Introduction
2. Methods Outline
2.1. Classical Simulations
2.2. Analysis Methods
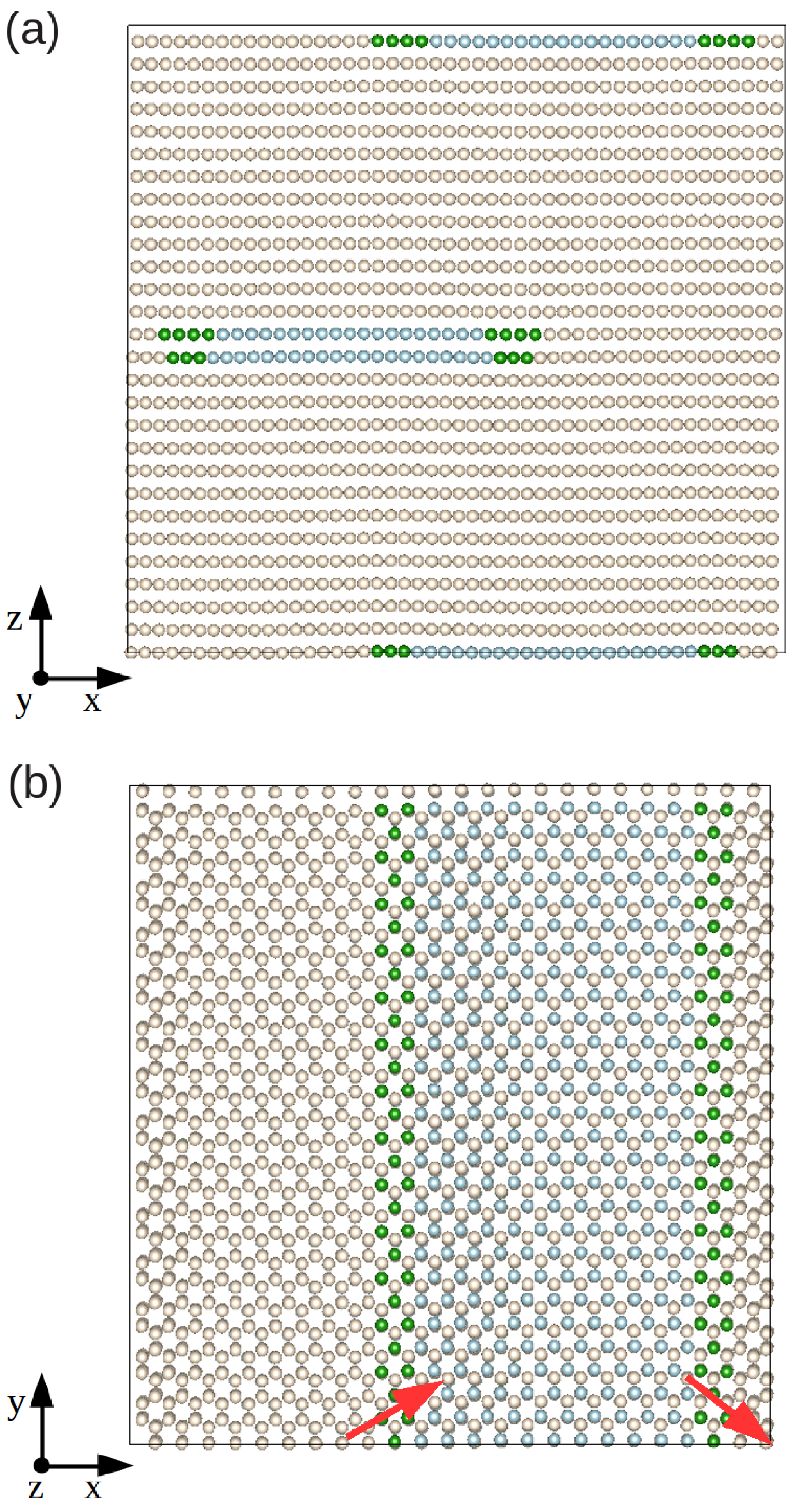
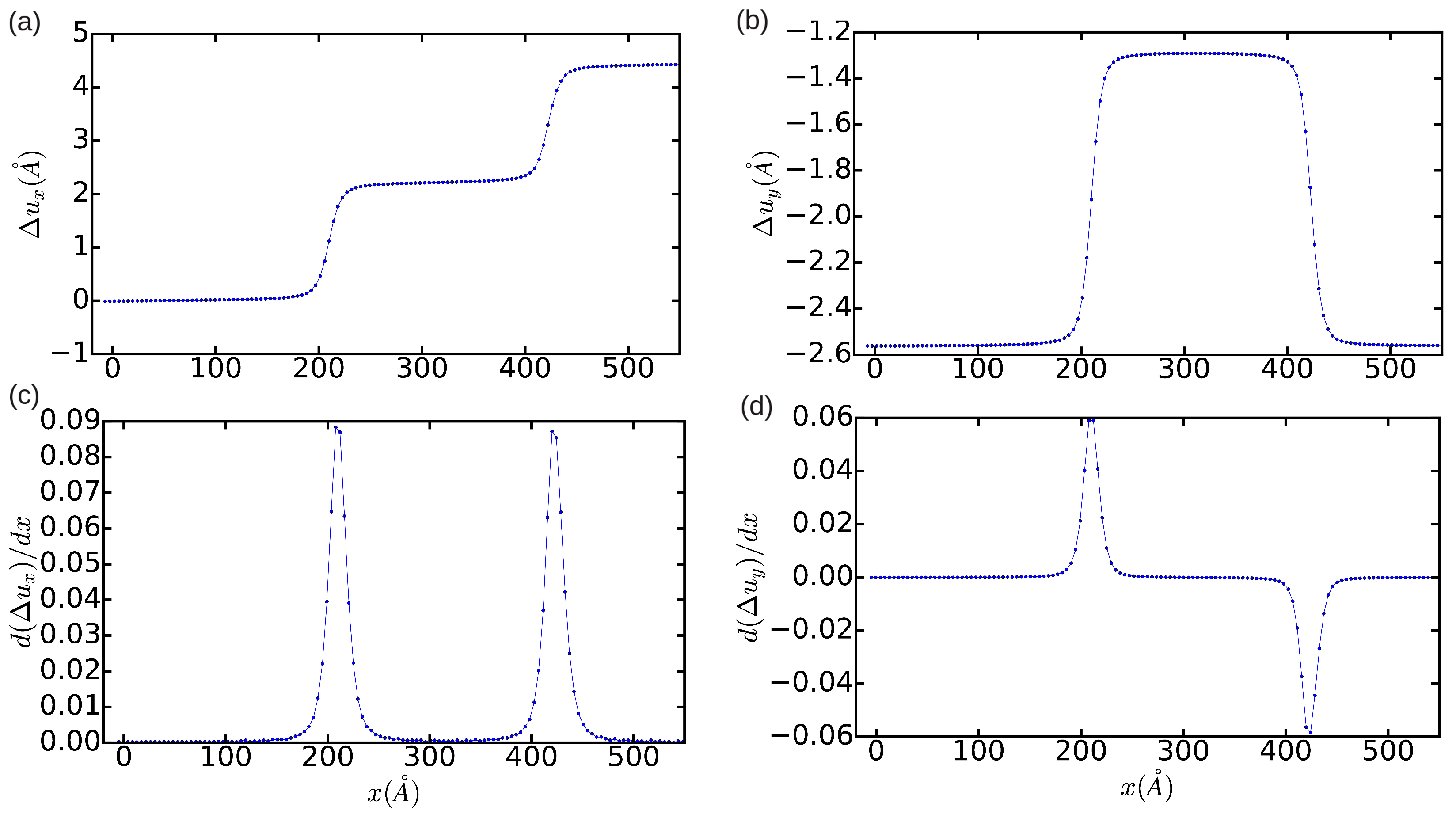
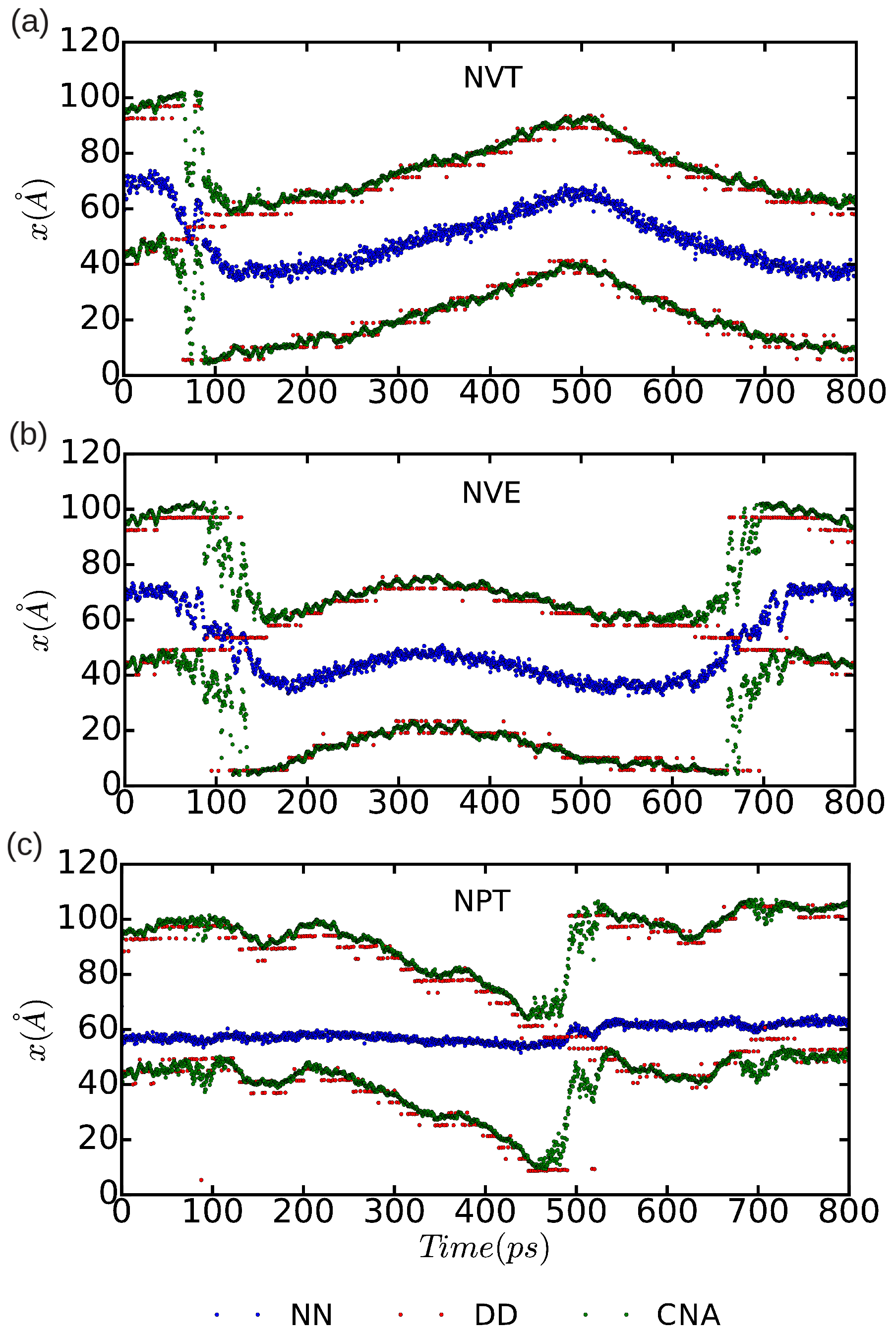
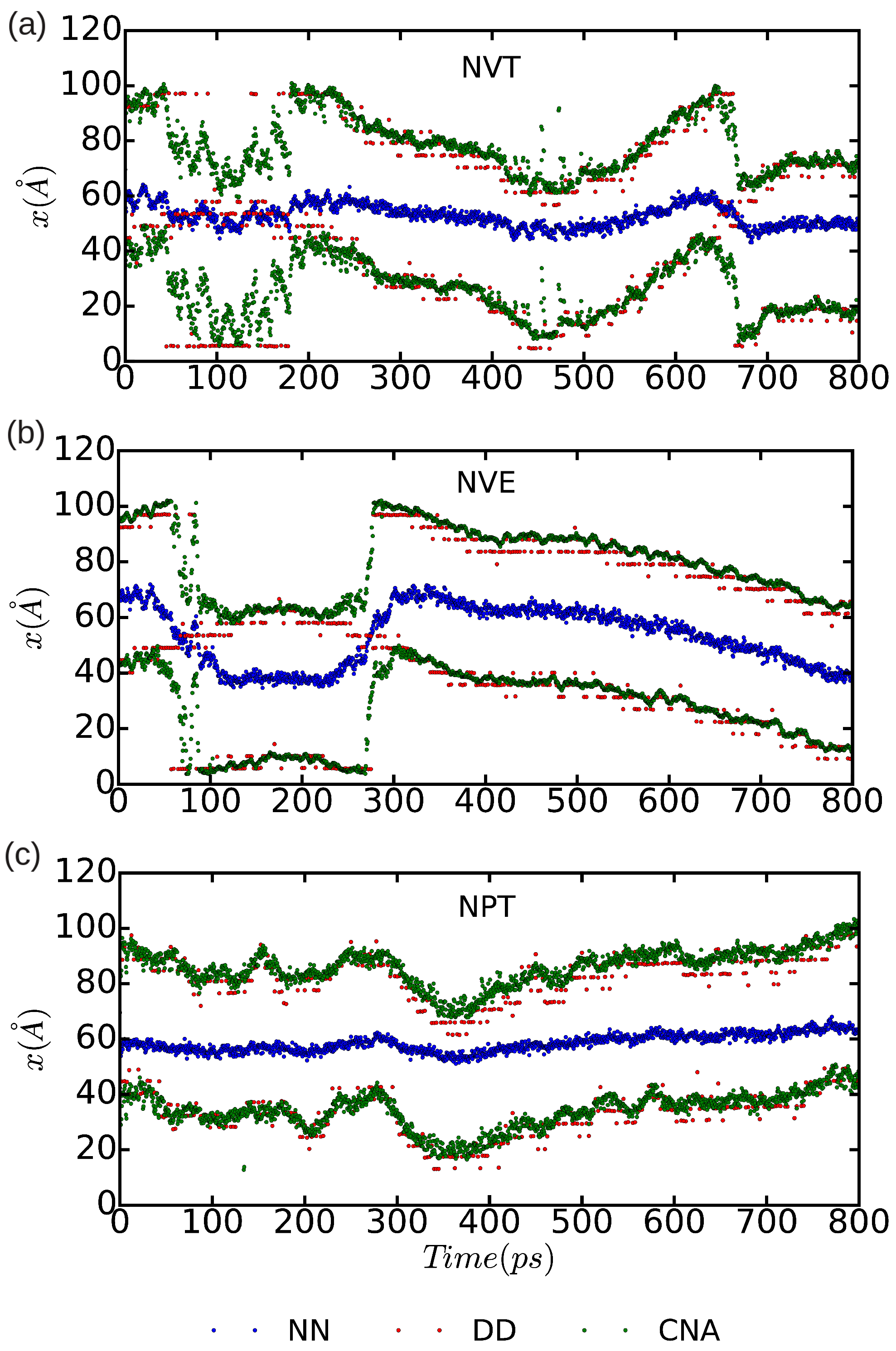
2.2.1. Differential Displacement Analysis (DD)
2.2.2. Nearest Neighbor Analysis (NN)
2.2.3. Common Neighbor Analysis (CNA)
3. Results and Discussion
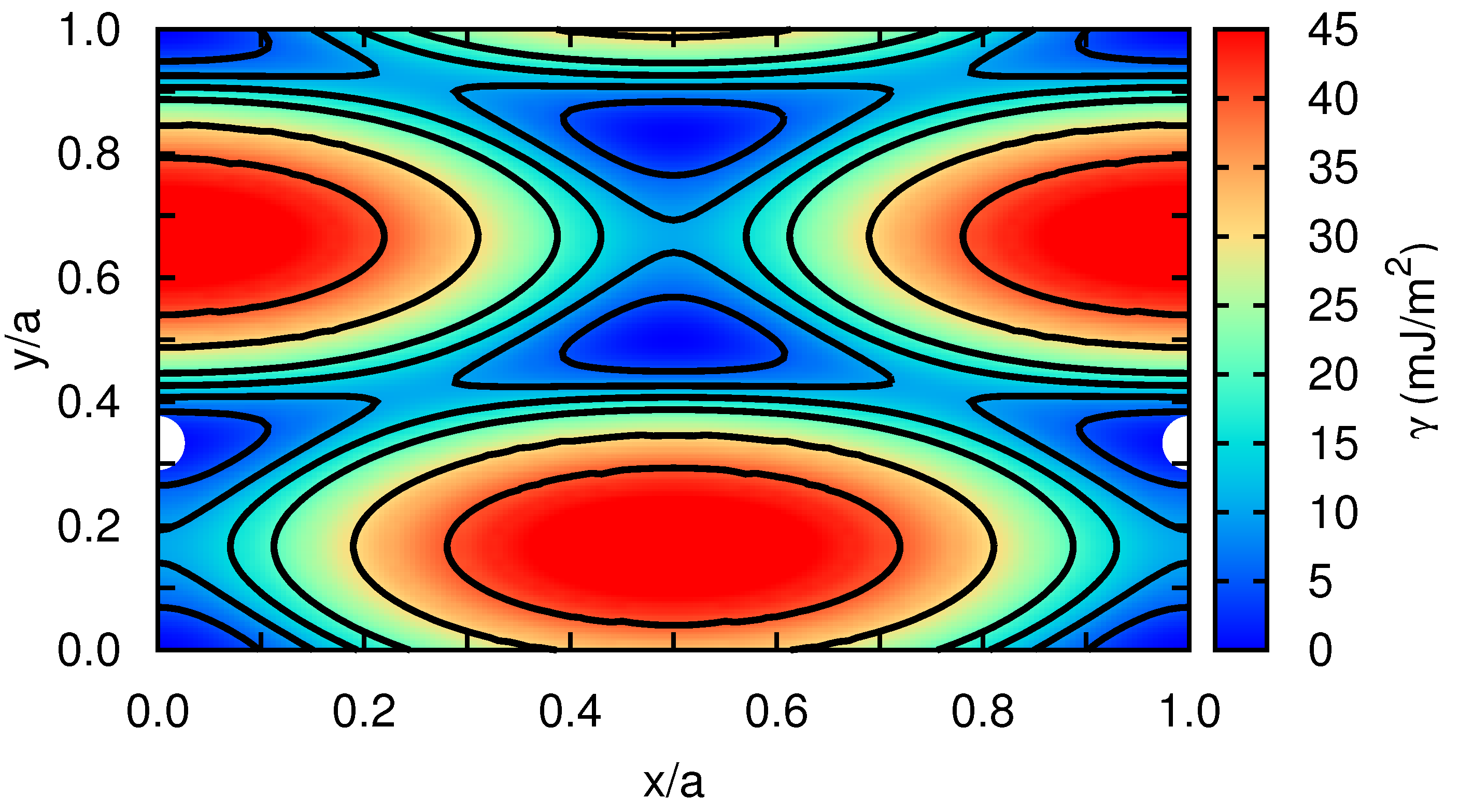
3.1. Edge Dislocation Structure
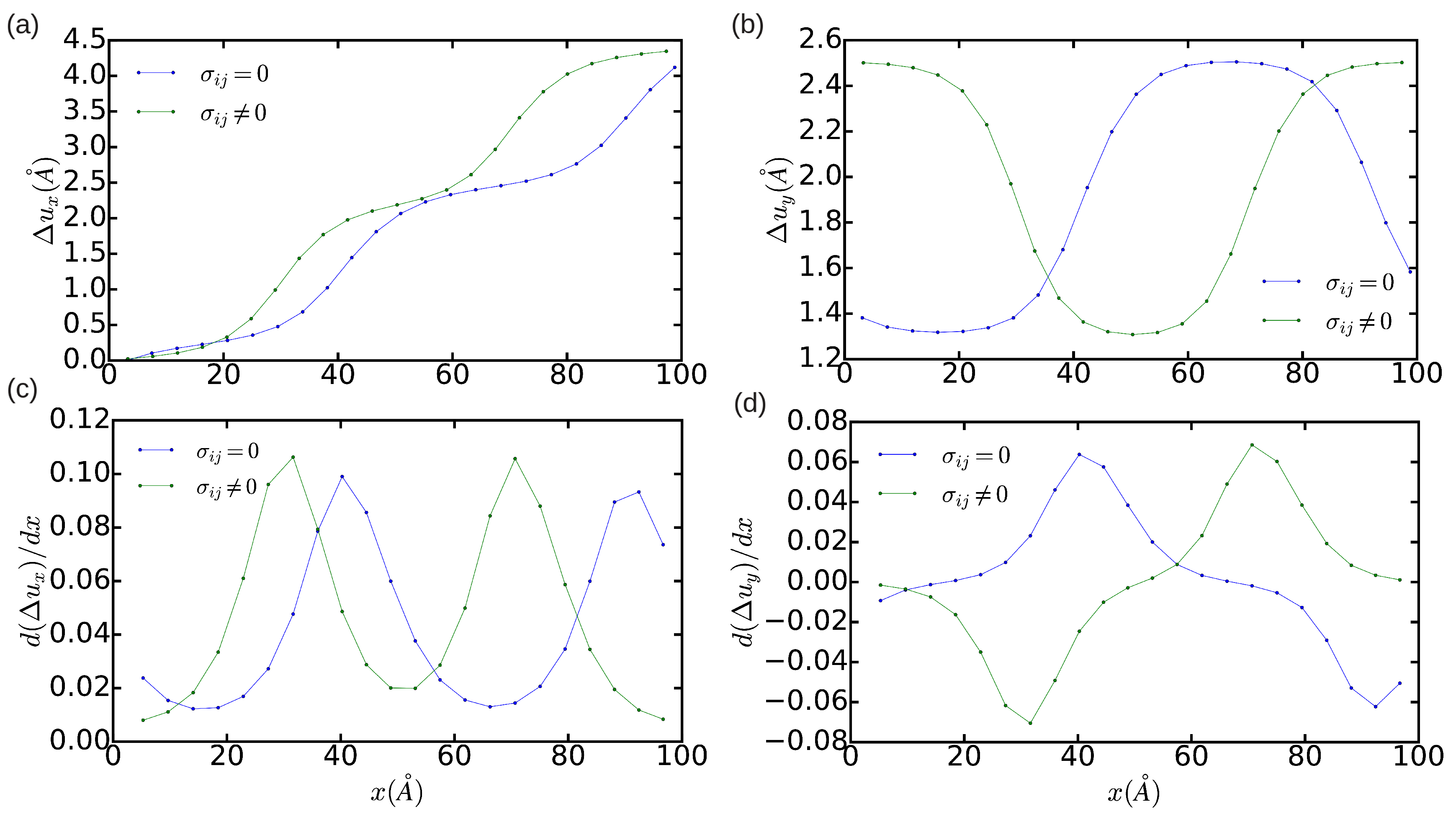
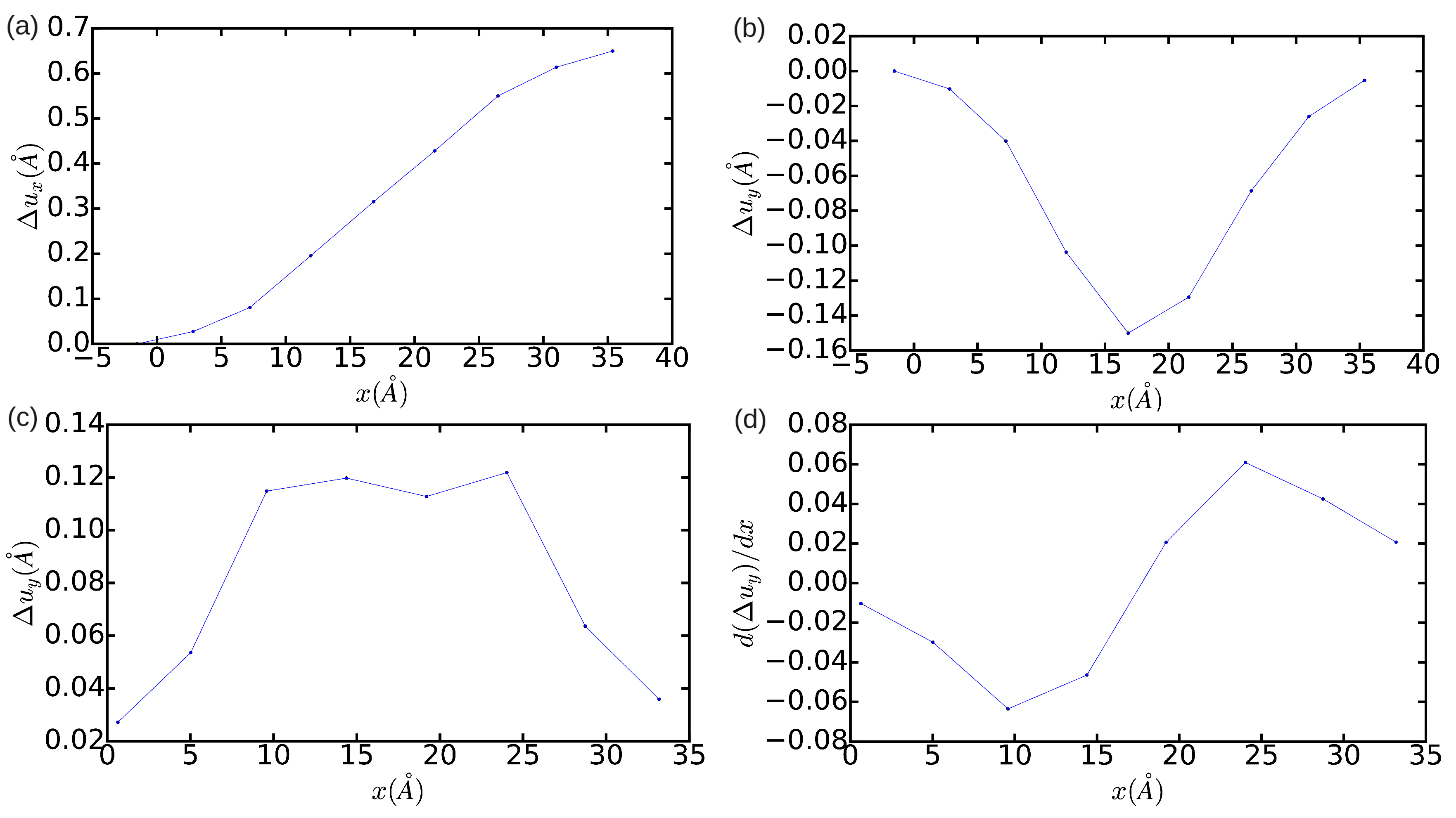
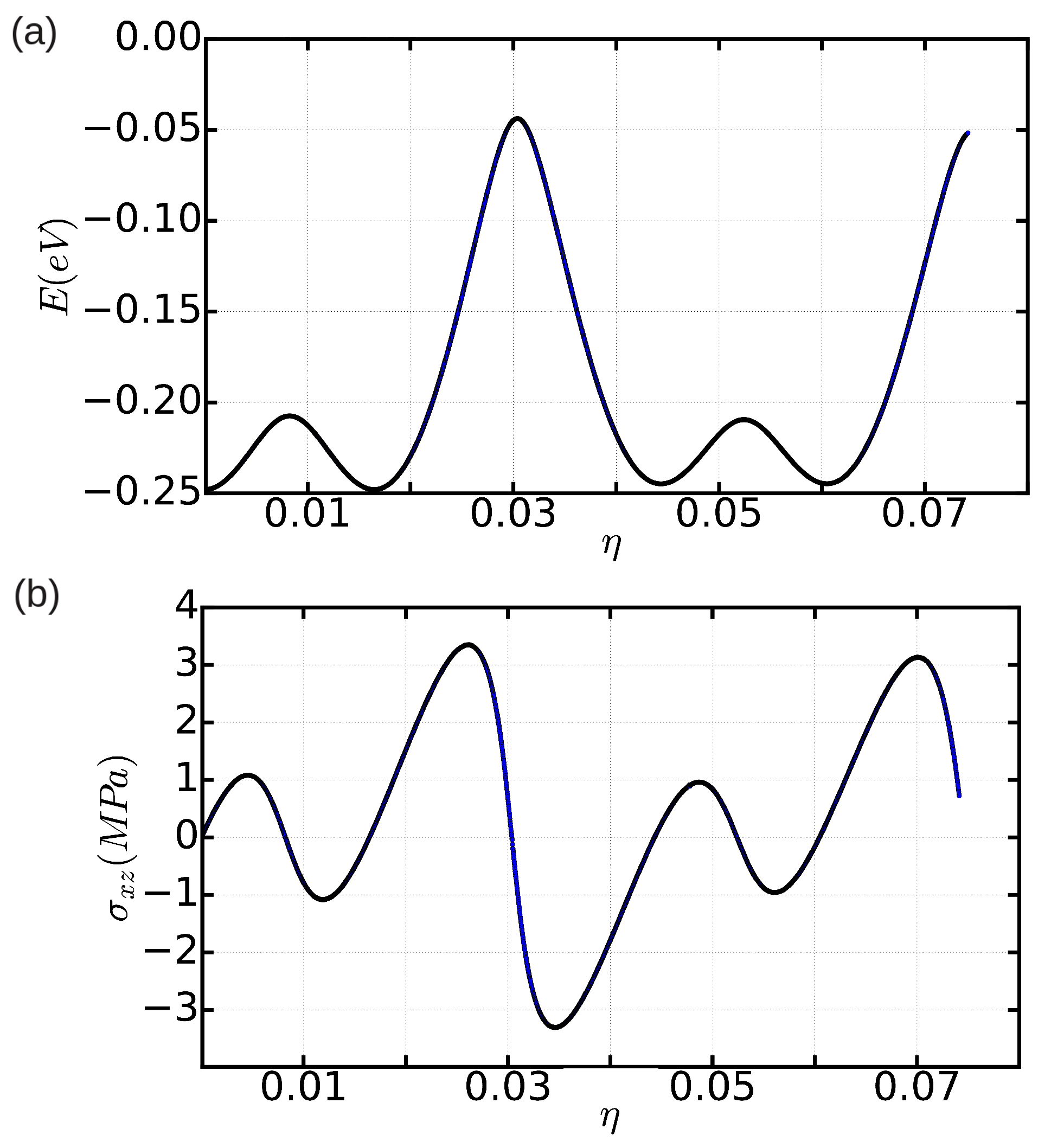
3.2. The Peierls Stress
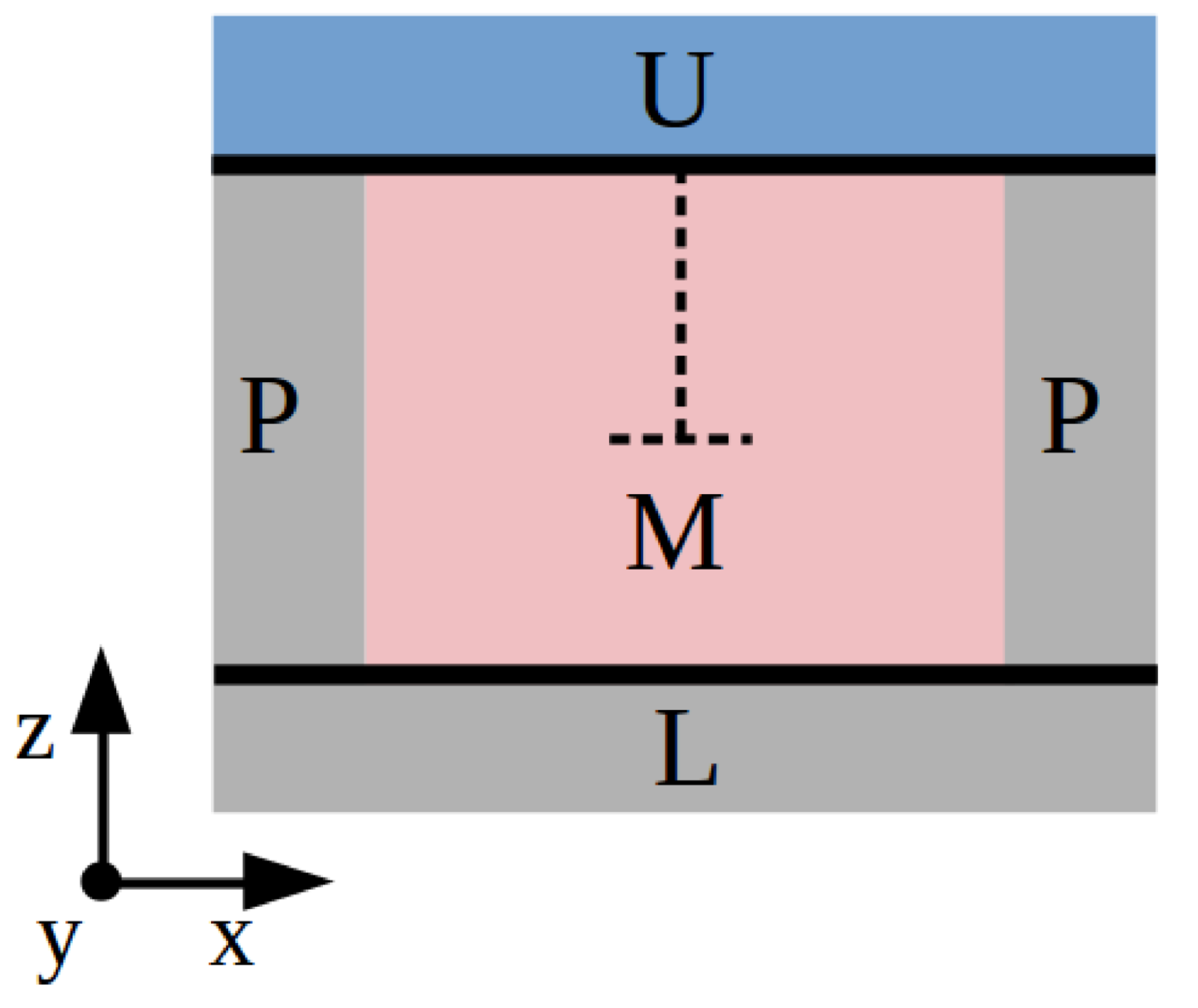
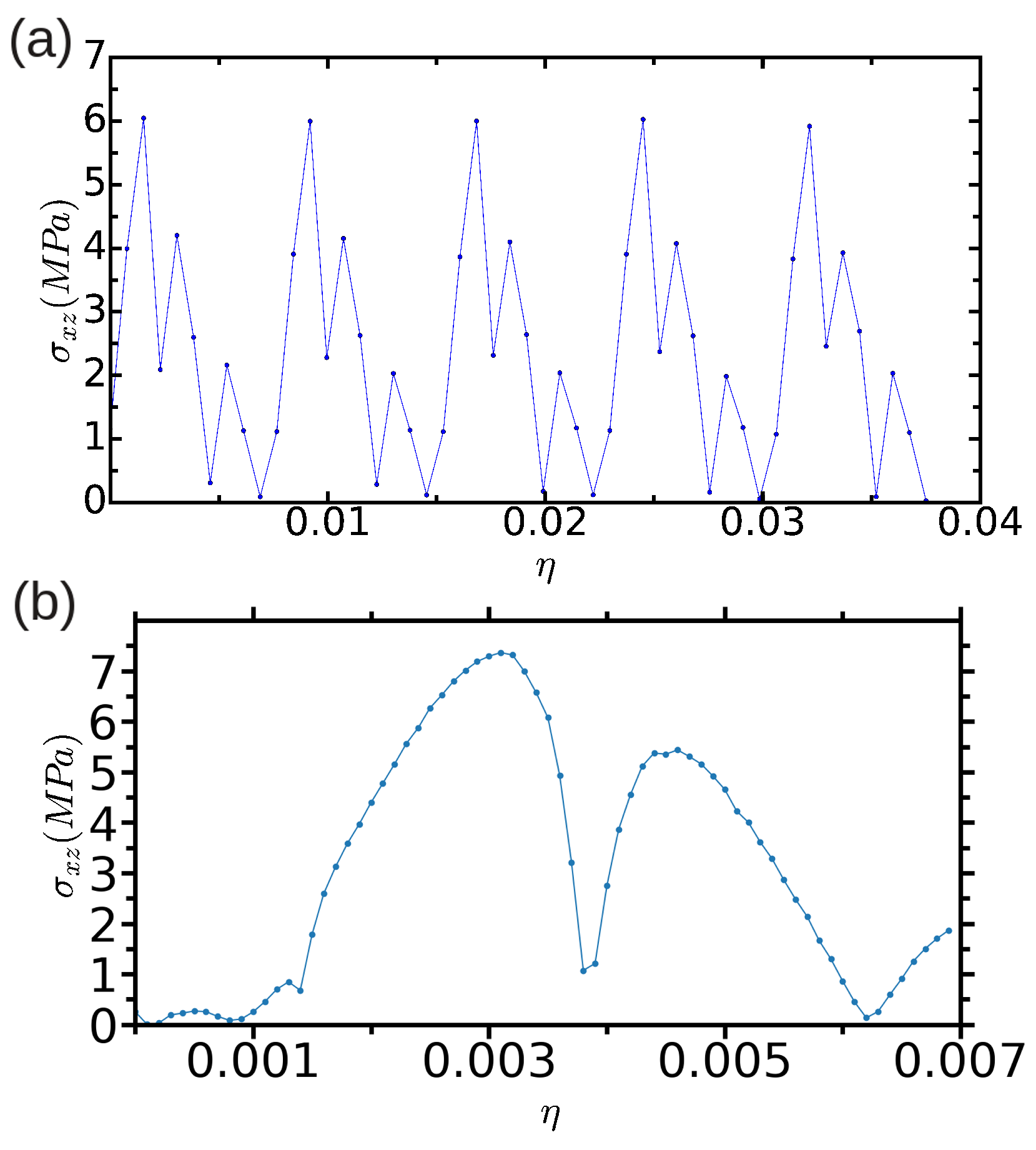
3.2.1. Method A: Fixed Boundary Conditions
3.2.2. Method B: Periodic Boundary Conditions
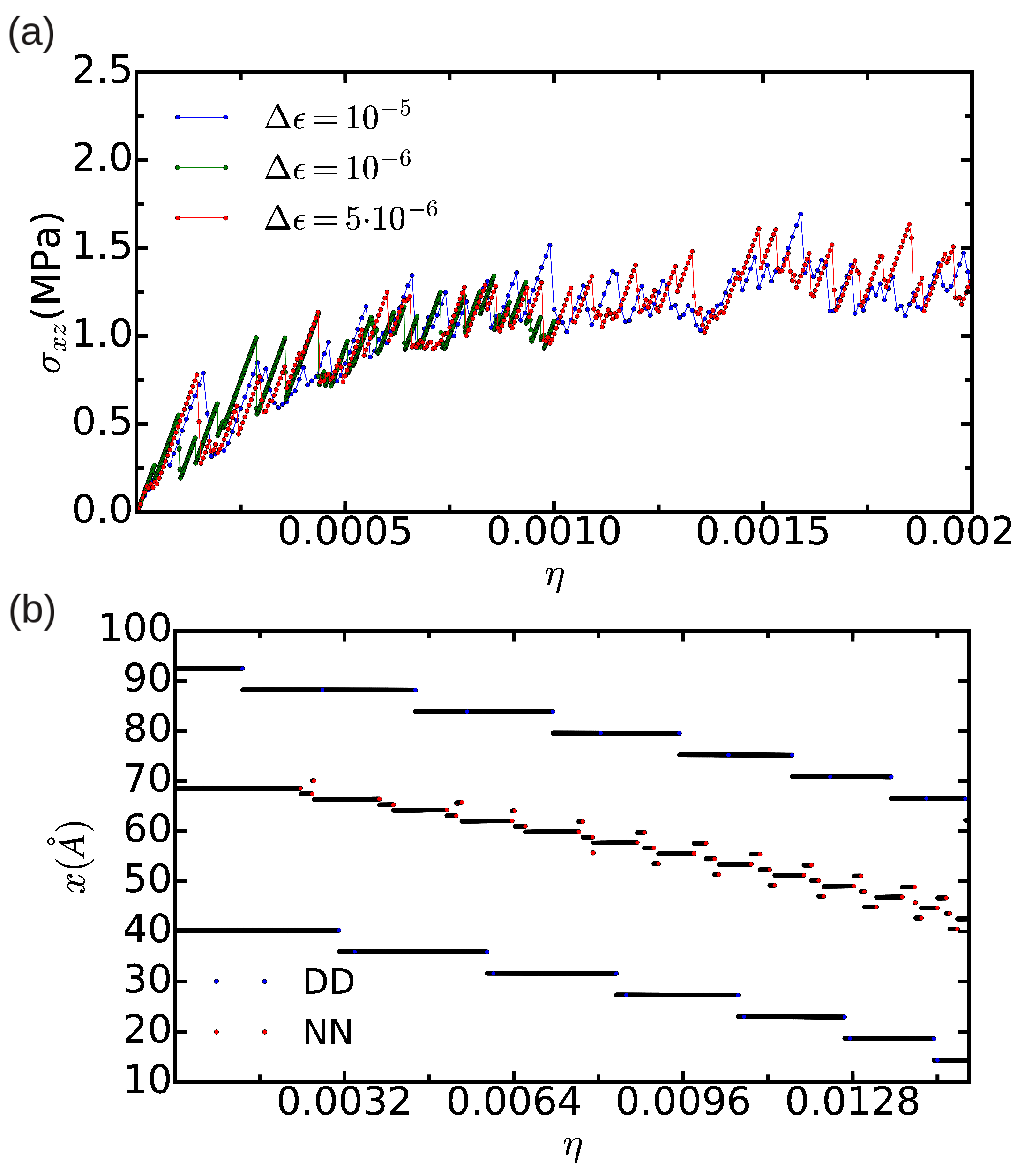
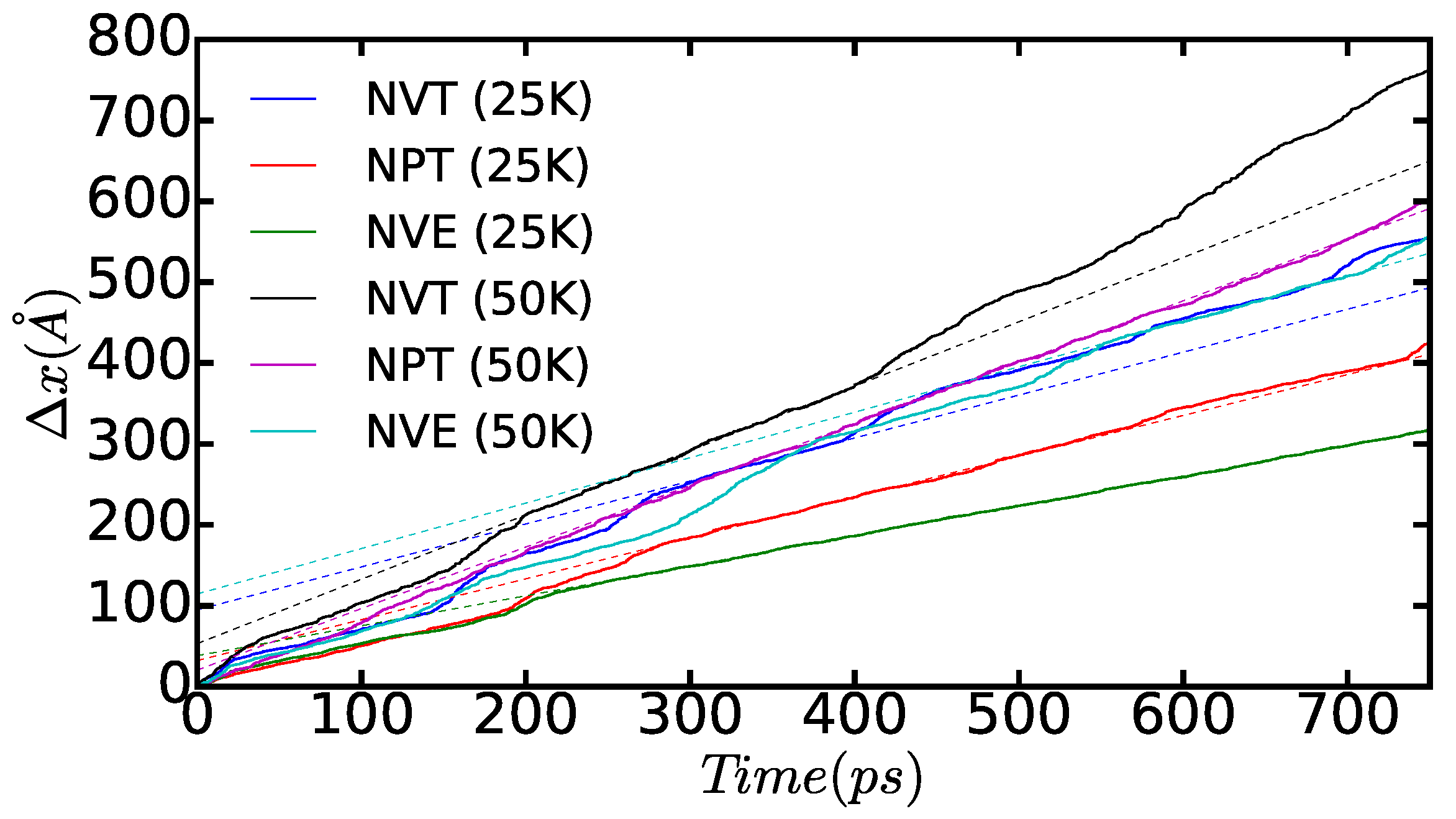
3.3. Dislocation Mobility: Finite-T Simulations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hirth, J.P.; Lothe, J. Theory of Dislocations, 2nd ed.; John Wiley: New York, NY, USA, 1982. [Google Scholar]
- Hull, D.; Bacon, D.J. Introduction to Dislocations, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Bulatov, V.V.; Cai, W. Computer Simulations of Dislocations; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Nabarro, F.; Kubin, L.; Hirth, J. Dislocations in Solids; Elsevier: Amsterdam, The Netherlands, 2009; Volumes 1–13, pp. 14–16. [Google Scholar]
- Cazorla, C.; Boronat, J. Simulation and understanding of atomic and molecular quantum crystals. Rev. Mod. Phys. 2017, 89, 035003. [Google Scholar] [CrossRef]
- Ceperley, D.M. Path integrals in the theory of condensed helium. Rev. Mod. Phys. 1995, 67, 279. [Google Scholar] [CrossRef]
- McMahon, J.M.; Morales, M.A.; Pierleoni, C.; Ceperley, D.M. The properties of hydrogen and helium under extreme conditions. Rev. Mod. Phys. 2012, 84, 1607. [Google Scholar] [CrossRef]
- Herrero, C.P.; Ramírez, R. Path-integral simulation of solids. J. Phys. Condens. Matter 2014, 26, 233201. [Google Scholar] [CrossRef] [PubMed]
- Cazorla, C.; Boronat, J. First-principles modeling of quantum nuclear effects and atomic interactions in solid 4He at high pressure. Phys. Rev. B 2015, 91, 024103. [Google Scholar] [CrossRef]
- Haziot, A.; Rojas, X.; Fefferman, A.D.; Beamish, J.R.; Balibar, S. Giant plasticity of a quantum crystal. Phys. Rev. Lett. 2013, 110, 035301. [Google Scholar] [CrossRef] [PubMed]
- Landinez-Borda, E.J.; Cai, W.; de Koning, M. Dislocation Structure and Mobility in hcp 4He. Phys. Rev. Lett. 2016, 117, 045301. [Google Scholar] [CrossRef] [PubMed]
- Khater, H.A.; Bacon, D.J. Dislocation core structure and dynamics in two atomic models of α-zirconium. Acta Mater. 2010, 58, 2978–2987. [Google Scholar] [CrossRef]
- Kamimura, Y.; Edagawa, J.; Takeuchi, S. Experimental evaluation of the Peierls stresses in a variety of crystals and their relation to the crystal structure. Acta Mater. 2013, 61, 294–309. [Google Scholar] [CrossRef]
- Fefferman, A.D.; Souris, F.; Haziot, A.; Beamish, J.R.; Balibar, S. Dislocation networks in 4He crystals. Phys. Rev. B 2014, 89, 014105. [Google Scholar] [CrossRef]
- Balibar, S.; Beamish, J.; Fefferman, A.D.; Haziot, A.; Rojas, X.; Souris, F.C.R. Dislocations in a quantum crystal: Solid helium: A model and an exception. Physique 2016, 17, 264–275. [Google Scholar] [CrossRef]
- Fenichel, H.; Serin, B. Low-temperature specific heats of solid neon and solid xenon. Phys. Rev. 1966, 142, 490. [Google Scholar] [CrossRef]
- Plimpton, S. LAMMPS Molecular Dynamics Simulator. J. Comput. Phys. 1995, 117, 1. Available online: http://lammps.sandia.gov (accessed on 13 December 2017). [CrossRef]
- Packard, J.R.; Swenson, C.A. An experimental equation of state for solid xenon. J. Phys. Chem. Solids 1963, 24, 1405–1418. [Google Scholar] [CrossRef]
- Pollack, G.L. The solid state of rare gases. Rev. Mod. Phys. 1964, 36, 748. [Google Scholar] [CrossRef]
- Bernades, N. Quantum Mechanical Law of Corresponding States for Van der Waals Solids at 0° K. Phys. Rev. 1960, 120, 807. [Google Scholar] [CrossRef]
- Osetsky, Y.N.; Bacon, D.J. An atomic-level model for studying the dynamics of edge dislocations in metals. Model. Simul. Mater. Sci. Eng. 2003, 11, 427. [Google Scholar] [CrossRef]
- Vitek, V. Structure of dislocation cores in metallic materials and its impact on their plastic behaviour. Prog. Mater. Sci. 1992, 36, 1–27. [Google Scholar] [CrossRef]
- Stukowski, A. Structure identification methods for atomistic simulations of crystalline materials. Model. Simul. Mater. Sci. Eng. 2012, 20, 045021. [Google Scholar] [CrossRef]
- Honeycutt, J.D.; Andersen, H.C. Molecular dynamics study of melting and freezing of small Lennard-Jones clusters. J. Phys. Chem. 1987, 91, 4950–4963. [Google Scholar] [CrossRef]
- Bacon, D.J.; Vitek, V. Atomic-scale modeling of dislocations and related properties in the hexagonal-close-packed metals. Metall. Mater. Trans. A 2002, 33, 721–733. [Google Scholar] [CrossRef]
- Boninsegni, M.; Kuklov, A.B.; Pollet, L.; Prokofev, N.V.; Svistunov, B.V.; Troyer, M. Luttinger liquid in the core of a screw dislocation in helium-4. Phys. Rev. Lett. 2007, 99, 035301. [Google Scholar] [CrossRef] [PubMed]
- Ackland, G.J.; Wooding, S.J.; Bacon, D.J. Defect, surface and displacement-threshold properties of α-zirconium simulated with a many-body potential. Philos. Mag. A 1995, 71, 553–565. [Google Scholar] [CrossRef]
- Mendelev, M.I.; Ackland, G.J. Development of an interatomic potential for the simulation of phase transformations in zirconium. Philos. Mag. Lett. 2007, 87, 349–359. [Google Scholar] [CrossRef]
- Keyse, R.J.; Venables, J.A. Stacking fault energy and crystal stability of solid krypton and xenon. J. Phys. C Sol. Stat. Phys. 1985, 18, 4435. [Google Scholar] [CrossRef]
- Cazorla, C.; Boronat, J. Atomic kinetic energy, momentum distribution, and structure of solid neon at zero temperature. Phys. Rev. B 2008, 77, 024310. [Google Scholar] [CrossRef]
- Boronat, J.; Cazorla, C.; Colognesi, D.; Zoppi, M. Quantum hydrogen vibrational dynamics in LiH: Neutron-scattering measurements and variational Monte Carlo simulations. Phys. Rev. B 2004, 69, 174302. [Google Scholar] [CrossRef]
- Shin, I.; Carter, E.A. Possible origin of the discrepancy in Peierls stresses of fcc metals: First-principles simulations of dislocation mobility in aluminum. Phys. Rev. B 2013, 88, 064106. [Google Scholar] [CrossRef]
- Cazorla, C. The role of density functional theory methods in the prediction of nanostructured gas-adsorbent materials. Coord. Chem. Rev. 2015, 300, 142–163. [Google Scholar] [CrossRef]
- Wang, G.; Strachan, A.; Cagin, T.; Goodard, W.A., III. Calculating the Peierls energy and Peierls stress from atomistic simulations of screw dislocation dynamics: application to bcc tantalum. Model. Simul. Mater. Sci. Eng. 2004, 12, S371. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: New York, NY, USA, 1954. [Google Scholar]
- Cazorla, C.; Lutsyshyn, Y.; Boronat, J. Elastic constants of solid 4He under pressure: Diffusion Monte Carlo study. Phys. Rev. B 2012, 85, 024101. [Google Scholar] [CrossRef]
- Cazorla, C.; Lutsyshyn, Y.; Boronat, J. Elastic constants of incommensurate solid 4He from diffusion Monte Carlo simulations. Phys. Rev. B 2013, 87, 214522. [Google Scholar] [CrossRef]
- Olmsted, D.; Hardikar, K.Y.; Phillips, R. Lattice resistance and Peierls stress in finite size atomistic dislocation simulations. Model. Simul. Mater. Sci. Eng. 2001, 9, 215. [Google Scholar] [CrossRef]
- Yadav, S.K.; Ramprasad, R.; Misra, A.; Liu, X.-Y. Core structure and Peierls stress of edge and screw dislocations in TiN: A density functional theory study. Acta Mater. 2014, 74, 268–277. [Google Scholar] [CrossRef]
- Balasubramanian, N. The temperature dependence of the dislocation velocity-stress exponent. Scr. Metall. 1996, 3, 21–24. [Google Scholar] [CrossRef]
- Fitzgerald, S.P. Kink pair production and dislocation motion. Sci. Rep. 2016, 6, 39708. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T = 25 K | ||
| T = 50 K | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sempere, S.; Serra, A.; Boronat, J.; Cazorla, C. Dislocation Structure and Mobility in Hcp Rare-Gas Solids: Quantum versus Classical. Crystals 2018, 8, 64. https://doi.org/10.3390/cryst8020064
Sempere S, Serra A, Boronat J, Cazorla C. Dislocation Structure and Mobility in Hcp Rare-Gas Solids: Quantum versus Classical. Crystals. 2018; 8(2):64. https://doi.org/10.3390/cryst8020064
Chicago/Turabian StyleSempere, Santiago, Anna Serra, Jordi Boronat, and Claudio Cazorla. 2018. "Dislocation Structure and Mobility in Hcp Rare-Gas Solids: Quantum versus Classical" Crystals 8, no. 2: 64. https://doi.org/10.3390/cryst8020064
APA StyleSempere, S., Serra, A., Boronat, J., & Cazorla, C. (2018). Dislocation Structure and Mobility in Hcp Rare-Gas Solids: Quantum versus Classical. Crystals, 8(2), 64. https://doi.org/10.3390/cryst8020064