1. Introduction
Crystallization is widespread in nature, in everyday live, meteorology (ice and snow), and even in biology, e.g., biomineralization of bone, teeth, and shells. Crystals are present in healthy (insulin) as well as in diseased human organisms (e.g., kidney and gallbladder stones, uric acid crystals in gout). Protein crystals also bear significant scientific, medical, and industrial relevance. Crystalline drug formulations are most appropriate to maintain protein stability during storage, transport, and upon administration.
Reports on protein crystallization date back some 180 years. Friedrich Ludwig Hünefeld observed crystallization of the hemoglobin from earthworm blood by accident [
1]. Before the introduction of X-ray crystallography in biology 1934 [
2], biochemists and physiologists used protein crystallization for purification and characterization purposes only. To date, relatively large and well-diffracting crystals are needed for X-ray (and neutron) diffraction studies, which are the most powerful methods for structure-function studies of biomolecules. The knowledge of 3D protein molecule structures and protein-substrate interactions is vital when it comes to understanding the mechanisms of life and the human genome, and developing novel, protein-based pharmaceuticals for structure-guided drug design and controlled drug delivery. Not surprisingly, already in 1962, the Nobel Prize in Chemistry was awarded jointly to Max Perutz and John Kendrew “for their studies of the structures of globular proteins”, (hemoglobin and myoglobin). Other Nobel Prizes for X-ray structure determinations of bio-molecules and complexes have followed.
Unfortunately, growing crystals suitable for X-ray crystallography remains, even nowadays, the major stumbling block in the whole study. Despite intensive endeavors, there is no recipe for growing crystals of newly-expressed proteins. Instead, a tedious trial-and-error approach is applied. The numerous state-of-the-art crystallization tools employed, such as robots, automation and miniaturization of crystallization trials, Dynamic Light Scattering, crystallization screening kits, etc., do not exclude the need for researchers’ creativity and acumen. Different approaches to evoke protein crystallization have been attempted with and without success.
Because of the lack of seed crystals, spontaneous crystallization is used with newly-expressed proteins. Inevitably, it starts with the formation of the smallest sized stable crystalline particles possible under the conditions present, coined as ‘nuclei’. Therefore, the step that determines the difference between success and failure of the crystallization effort is to compel the protein molecules scattered homogeneously in the solution to form stable crystal nuclei; once nucleated, the crystals continue their growth spontaneously. So, a detailed understanding of crystal nucleation in general, and of the protein crystal nucleation, are obligatory. Such knowledge is needed because, being the first crystallization stage, nucleation predetermines important features of the subsequent crystal growth, such as polymorph selection, number of nucleated crystals, crystal size distribution, and frequently, crystal quality.
Protein crystal polymorphism is the ability of a same chemical composition substance to exist in more than one crystal structure. Whilst having identical chemical properties, polymorphs can differ markedly in their dissolvability and bioavailability (the fraction of an administered dose of unchanged drug that reaches the systemic circulation). Thus, depending on the crystal polymorphic form the same molecule may have, or may not have a therapeutic effect [
3]. Furthermore, in the worst case, a change of the polymorphic form may render a drug toxic. Hence, of crucial importance is to identify all relevant polymorphs which are decisive for the therapeutic function of drug formulation.
The molecular-kinetic mechanism of protein crystal nucleation is extremely complex. This process involves a subtle interplay between physical and biochemical factors which enables a highly-precise self-assembly of biological macromolecules into stable clusters. For instance, a physical requirement for successful protein crystallization is that the protein-protein attraction strength should be moderate—the attraction should be large enough to promote crystallization while not being so large as to provoke amorphous precipitation. In other words, the pair-wise protein attraction must be carefully fine-tuned, which is achieved by selecting proper crystallization conditions. However, although some physical laws established previously for the crystallization of small inorganic molecules rule protein crystal nucleation as well, it is the large size of the protein molecules and their highly inhomogeneous and patchy surfaces that make protein crystal nucleation so peculiar. Physical and biochemical aspects of protein crystal nucleation can be distinguished in an appropriately-designed experimental setting, e.g., see [
4].
Substantial difference, on both the molecular and macroscopic levels, is established by the crystallization of protein and small (inorganic) molecules. On a molecular-scale, independently of the spatial orientation of the meeting species, every hit between small molecules in supersaturated media has the potential to contribute for the formation of a crystal bond. The reason for this is that small molecules possess spherical interaction fields and a constant interaction potential. In contrast, the surface of protein molecules is highly patchy and heterogeneous, and only a limited number of discrete patches on it become attractive molecule portions under crystallization conditions. Due to the strict selection of the crystalline bonding patches, a successful collision between protein molecules, resulting in the formation of a crystalline connection, requires not only sufficiently close approach of the species, but also, their proper spatial orientation. Macroscopically, the difference between small molecules and proteins is manifested through the notorious reluctance of proteins to crystallize. Furthermore, although requiring unusually high supersaturation, protein crystal nucleation and growth occur much more slowly than that with small-molecule substances.
The so-called bond selection mechanism (BSM) was devised to explain the reduced rate of the protein crystal nucleation [
5,
6,
7,
8]. It accounts for the biochemical constraint associated with the strict selection of crystalline bonds, which also enforces specific bond orientations. Principally similar to BSM is the increasingly popular ‘sticky patch’ model, which was derived from colloid chemistry and soft matter physics, e.g., see [
9,
10]. The severe steric restriction to the protein crystal bond formation (arising due to the small size of the contacting patches) is mitigated to some degree by ‘sticky’ collisions, i.e., where two biomolecules in a water environment remain trapped close to each other after their first encounter. In contrast to small molecules, large biomolecules perform rotational diffusion, which involves multiple collisions (about nine collisions after their first encounter) [
11]. During rotational diffusion, the biomolecules get a good chance of reorienting toward proper spatial positioning of the crystallization patches on the two meeting protein molecules; successful encounters become much more probable.
Bond selection is also a factor in protein crystal growth [
12]; for a molecule to bond at the kink site, it must be in an adequate orientation. Typically, the protein crystals grow under unusually high supersaturations, i.e., around 100%, and even more. Despite this high supersaturation, their growth proceeds more slowly than that observed with small molecule crystals; a typical value of the step kinetic coefficient for inorganic crystals grown from solution is 2 to 3 orders of magnitude higher than that of protein crystals [
13,
14]. This fact can be explained because of a low probability of proper spatial orientation of an incoming protein molecule for its incorporation into the kink site. Perhaps, the higher protein concentration that is used at crystallization conditions is needed to mitigate this decelerating impact (through a higher attachment attempt frequency).
The aim of this review is to consider some biochemically-conditioned peculiarities of the protein crystal nucleation (and growth). Recently-discovered molecular scale pathways for protein crystal nucleation are discussed first. Novel proof for the BSM are presented. The so-called EBDE method (equilibration between crystal bond and destructive energies) is re-substantiated, and protein crystal nucleation in pores is revisited on this basis. Another aim of the present paper is to report a novel consideration of the crystallization in solution flow.
2. Classical or Two-Step Crystal Nucleation Mechanisms: What Is Currently Known?
Despite nucleation significance and nearly a century-long period of intensive study, crystallization onset is still debated. Quite often, classical nucleation theory (CNT) is employed to explain protein crystal nucleation (e.g., [
15]). However, while providing an adequate explanation of the fluctuation-based nucleation mechanism and the origin of the nucleation barrier, there is an inadequacy between some measurements of crystal nucleation rates and CNT predictions. (It is worth noting that nucleation rates calculations according CNT suffer from uncertainty in determining the energy of the interface arising between the new phase and the mother phase. While such energies are not measurable for those nanoparticles, interface energy variation of only 10% can alter the nucleation rate by many orders of magnitude. The reason is that the nucleation rate depends exponentially on the nucleation energy barrier, which in turn is determined by the interface free energy in power three.) CNT also failed to account for observations in biomineralization processes that are responsible for the formation of invertebrate mineralized skeletal elements, e.g., the mollusk shell nacre layer (aragonite polymorph) and the sea urchin spicule (calcite polymorph) [
16]. To explain this inadequacy, the so-called two-step nucleation mechanism (TSNM) has been proposed [
17].
Though frequently contested, CNT has been confirmed by atomic force microscopy [
18]. Molecular-scale images of sub-critical and super-critical crystals landing on apoferritin crystals under supersaturated conditions reveal a classical nucleation pathway. Yau and Vekilov observe that pre-nucleation clusters of apoferritin are also crystalline, and have the same molecular arrangement as those in bulk crystals. So, the nucleation pathway complies with the classical crystal nucleation pathway. Sleutel et al. [
19] have also observed that glucose isomerase 2D crystal nucleation proceeds following the classical pattern, and proved the existence of a critical crystal size. This notwithstanding, a question arises of whether CNT can give a reliable physical rendition of protein crystal nucleation process?
TSNM denies the simultaneous densification and ordering during a single nucleation event. The theoretical basis for TSNM has been laid by ten Wolde and Frenkel [
20]. Performing numerical simulations, they predict that the thermodynamically-favored nucleation pathway for colloidal and protein-like substances entails the initial formation of a liquid cluster to be subsequently transformed into a crystalline nucleus. However, this prediction is valid only for conditions close to the critical point of a phase diagram (the intersection of the liquid-liquid binodal and spinodal). Nucleation pathways that follow the CNT would result in different phase diagram regions. In reality, due to protein aggregation or gelation, the critical point is hardly accessible in protein crystallization experiments. This means that most protein crystallization experiments do not occur under conditions prescribed by ten Wolde and Frenkel (and the TSNM of proteins has not been experimentally demonstrated with proteins crystallizing under such conditions).
TSNM does not contest the basic CNT concept of a fluctuation-based nucleation mechanism. In its initial formulation, TSNM assumes nucleation initiation via a high-density liquid phase appearing in the bulk solution, with crystal nuclei being formed inside this dense liquid phase during the second TSNM step [
21]. The intermediate phase preserves some similarity to the mother phase, since it is only densified. Therefore, the phase-transition energy barrier is lowered below the one needed for a direct crystal nucleus formation occurring via the CNT mechanism. The fact that TSNM breaks up a single large activation barrier into two smaller ones (the second barrier being for the ordering step) makes it intuitively more attractive [
22]. For whatever reason, the TSNM idea has gained increasing popularity over the years, and despite the limited extent of clear experimental proof for the case of protein crystal nucleation, the TSNM idea is broadly accepted as a scientific fact, superseding CNT.
It is mainly mesoscale observations that corroborate TSNM. Sauter et al. [
23] have reported a two-step pathway of protein nucleation for the protein/precipitant system β-lactoglobulin/CdCl
2. Vivares et al. [
24] have observed TSNM formation of glucose isomerase crystals in a concentrated liquid phase. Using Dynamic Light Scattering, Schubert et al. [
25] have observed the occurrence of liquid dense protein clusters with growing-over-time nanocrystals formed inside, as verified by transmission electron microscopy (TEM).
However, due to the molecular scale of the processes involved, insights into the earliest crystal nucleation stages have remained only very partially understood until recently. The remarkable advancement in instrumental techniques has enabled molecular-scale observation of protein crystal nucleation. For instance, no intermediate condensed liquid droplets, but only amorphous solid particles consisting of lysozyme molecules, are observed [
26], without the formation of crystalline phases inside such amorphous particles. This speaks about the challenges faced by TSNM initial formulation.
Liquid-cell TEM reveals diverse nucleation pathways. Van Driessche et al. [
27] used vitrified samples plunge frozen at various time intervals. In doing so, the authors imaged the nanoscale structure of pre-nucleation clusters along the way to the crystalline nuclei. Looking into the earliest stages of glucose isomerase crystal nucleation [
28], Van Driessche et al. have shown distinct nucleation pathways for two glucose isomerase polymorphic crystal forms. For the rhombic polymorph, the authors have not observed any amorphous precursors of crystal nuclei being formed prior the appearance of crystalline nanoparticles (the latter being as small as 100 nm). Therefore, Van Driessche et al. [
27] concluded homogeneous crystal nucleation from solution following the rules of the CNT. With the needle-like polymorphic form of glucose isomerase, they observed crystalline nanorods that measured 12 by 2 molecules, and appeared almost immediately after setting crystallization conditions. The smallest glucose isomerase rods captured at very early time points (just 20 s after adding a crystallizing precipitate) have the same crystalline order as the bulk crystals, which is in accordance with CNT. Afterwards, however, the rods undergo grouping and oriented attachment into larger structures, referred to as fibers. These fibers (having widths of 40 nm and lengths varying between 100 nm up to multiple microns) have a significant deviation from the crystallographic packing, as predicted by the two-step model. On the basis of these observations, Van Driessche et al. [
27] have hypothesized that the formation of a crystalline nucleus takes place by the lateral merging of fibers mediated by oriented attachment. In conclusion, the authors found indications that elements of both CNT and TSNM may act during the nucleation of glucose isomerase crystals. A two-step crystal nucleation has also been seen using time-resolved potentiometry and turbidimetry combined with Dynamic Light Scattering, small-angle X-ray scattering, and in situ imaging by cryo-TEM (on frozen-hydrated cryo-samples) [
29]. The authors have studied small molecule (Portland cement) crystallization.
Most recently, Sleutel and Van Driessche have introduced some balanced views on the subject [
30]. Assessing up-to-date results from cryo-TEM in situ imaging with its strengths and weaknesses, the authors offer new insights into the earliest stages of the protein crystal nucleation. For proteins, neither the suggested ubiquity of TSNM can be confirmed unambiguously, nor CNT rejected completely; the reality might be diverse. The experimental proof thus far can only affirm the absence of a universal nucleation mechanism in all crystallization cases. The authors’ review represents a substantial leap in our understanding of how protein molecules, scattered homogeneously in the solution, are constrained to form stable crystal nuclei [
30] (see also [
31]).
3. Bond Selection during Protein Crystallization
Interactions between bio-molecules and protein–substrate interactions play a central role in many complex cellular processes (such as signaling, transcription, inhibition, translation, and regulation), and are responsible for the stability and shelf-life of pharmaceutical formulations. Highly-selective and directional protein-protein interactions govern the protein crystal nucleation as well. The formation of protein crystal lattice contacts (i.e., regions on the surface of a protein that are in contact with regions on the surfaces of neighboring proteins in the crystal lattice) is a typical biochemically-rooted peculiarity of protein crystallization [
4].
The study of protein crystal bond formation benefits from the comparison with the physiological protein-protein interactions. Due to their enormous biological importance, the latter have been investigated much more thoroughly than the former. A fundamental postulate for any person working with/on proteins is that only the surface structure of a protein molecule dictates its ability to bind to partners; the protein intramolecular interactions in the bulk do not participate in protein crystal lattice binding, because those interactions are concealed under the amino-acid residues situated at the molecular surface. Although it is impossible to observe the elementary acts of protein crystal bond formation, knowledge about lattice contacts, which are the result of the protein crystallization, are available from Protein Data Bank data, e.g., [
32,
33,
34,
35]. This information shows the clear differences between physiological protein-protein interactions and biologically non-functional protein crystal bonds. It is well known that the former arise due to hydrophobic areas that occupy relatively large portions on the protein molecule surface. Besides, the physiological protein-protein bonds are extremely specific and strong. In contrast, protein crystal lattice contacts are hydrophilic, polar, and occupy relatively small fractions of the molecule surfaces [
32,
36]. The intra-crystalline contacts are due to van der Waals, salt bridging, and hydration interactions. These data show that the patches on the protein molecule surface, which can create crystal lattice contacts, are also selected. Observed across many proteins, the most frequently-found residues in crystal contacts are arginine (Arg) and glutamine (Gln) residues, while the most infrequent participation in protein crystal contacts is that of lysine (Lys) and glutamate residues (i.e., the carboxylate anion of glutamic acid, Glu) [
32], although both are situated almost exclusively on the surface of the proteins. The hypothesis of Doye et al. [
37,
38] for an evolutionary negative design (i.e., the evolutionary selection of the surface properties of proteins to prevent any arbitrary association) is that lysine helps to prevent unwanted protein-protein interactions. This protein ability is compulsory because proteins operate within the cellular context, with typical concentrations of up to 300 mg/mL. Therefore, because of millions of years of natural selection, physiological protein-protein bonds are highly specific; any non-specific inter-protein interaction may be fatal.
Information about specific residues participating in the crystal contacts was reported by Gillespie et al. [
39]. The contacting residues were identified as participating in either direct residue-residue interactions or in water-mediated interactions, with the interaction free energy of crystal contacts being calculated as well. (For a thorough consideration of the role which play water molecules found at interfaces see [
40].) The authors confirmed that the non-specific, long-range electrostatics are not significant in crystal contacts, and that the interactions in crystals are likely dominated by short-range interactions, such as van der Waals, salt bridging, and hydration interactions, which are highly specific.
The big success of the rational site-directed mutagenesis strategy is an (albeit indirect) argument for bond selection during protein crystal nucleation. As early as 1992, rational surface engineering was accomplished by McElroy et al. [
41]. It has been established that even single amino acid changes on the surface of the thymidylate synthase can dramatically affect the solubility of a protein and its crystallizability, while not decreasing stability. The latter is of importance because structurally-rigid proteins are more prone to crystallization. Therefore, keeping the protein folded is a prerequisite for protein crystallization ability. It is logical to conclude that the surface mutations mediate novel crystal contacts.
Site-directed mutagenesis has also been applied to crystallize other proteins that are recalcitrant to crystallization [
42,
43,
44]. Mutation of surface amino acid residues with high conformational entropy to residues with no conformational entropy lead to the enhancement of crystallization; lysine residues were systematically mutated to alanine. Also, modifying the surface properties of a protein by mutating glutamic acid residues by alanine or aspartic acid results in enhanced crystallizability. In fact, any additional CH2 group enhances the conformational entropy [
45].
Also, chemical functionalization (a modification which often retains native protein structure), e.g., acetylation of surface lysine groups, coerces bovine carbonic anhydrase to crystallize [
46]. The authors have also shown how acetylation altered the organization and composition of crystal contacts—when proteins crystallize, they bury solvent-exposed surface areas in contact regions, which accommodate charged residues through salt bridges and/or hydrogen bonds. Acetylation has little influence on the size and geometry of crystal contacts, but reduces their charge complementarity. Also, contact regions generate and define adjacent non-contact regions. It was concluded that crystal contacts appear to be the principal determinant of the quality of the crystals, as measured by the X-ray diffraction resolution, and that the probability of obtaining crystals with adequate diffraction has been increased substantially due to acetylation of surface lysine groups [
46].
Studying the role of charges in protein-protein interactions, Kang et al. [
46] show why lysine residues are often excluded from the crystal lattice contacts. The authors conclude that the charge of Lys, but not its conformational flexibility, reduces its propensity to participate in the contact regions of proteins [
47]. Kang et al. [
46] also concluded that the protein-protein interactions that give rise to crystals are generally weaker than the protein-protein interactions that permit biological function. Indeed, molecules with weak intermolecular interactions can relatively easily reorient to find the preferred orientation needed for the crystal. To compensate for the weak interaction, crystals of proteins form in vitro at concentrations of protein that are much higher than those found inside the cell.
4. Equilibration between Crystal Bonding and Destructive Energies (EBDE)
The intuitive suggestion of Garcia-Ruiz [
48] for a balance between the sum of all intra-crystal bond energies (which maintain the integrity of a crystalline cluster) and the sum of surface destructive energies (which tend to tear up it) was our starting platform for further considerations [
49], which brings us to the so-called EBDE method [
50]. The thermodynamic substantiation of EBDE was presented earlier in this Journal. Let us outline it here.
When the system is undersaturated and no crystallization is possible, the protein ‘affinity’ to water molecules prevails over the crystallization propensity. To evoke crystallization, it is necessary to impose supersaturation, and the higher the latter, the more thermodynamically-stable the crystal, with respect to the solution. Thus, it is logical to assume that the imposed supersaturation decreases the protein-to-water ‘affinity’, i.e., supersaturation diminishes the destructive energy (
ψd) per bond. In other words, the tendency to disintegrate the crystal depends on the degree of supersaturation, in contrast to the cohesive energy per bond in the crystal lattice (
ψb) which is supersaturation independent. This means that any supersaturation increase will lead to an increase in
ψb/
ψd ratio. A rigorous definition of protein-to-water ‘affinity’, which uses both nucleation process enthalpy and entropy, was provided in [
50].
The advantage of the EBDE method is that it can predict the nucleation of crystals of diverse lattice structures (and can be aided by crystallographic computer programs), while the classical mean work of the separation (MWS) method of Stranski–Kaischew [
51,
52,
53] is applicable only with a Kossel-crystal [
54]. However, because EBDE rests on an intuitive suggestion, a question may arise of whether the supersaturation dependent critical nucleus size determined using the EBDE method corresponds to the one from CNT. Proof that the two coincide is presented below.
According to CNT, a free energy change (Δ
G) is needed for a homogeneous crystal to occur, e.g., [
55]:
where
n denotes the number of molecules constituting the cluster and Δ
μ is supersaturation;
S means the total surface of the new phase, and
γ is the specific interphase energy.
Classical thermodynamics points out that the cohesive energy (Δ
Gv = −
nΔ
μ) maintaining the integrity of a new phase cluster is proportional to its volume, i.e., to
n in power three, while the sum of energies (Δ
Gs =
Sγ) tending to tear up the cluster is proportional to its surface, i.e., to
n in power two. According to EBDE, the balance between cohesive and destructive energies means −Δ
Gv + Δ
Gs = 0. Importantly, in the case of under-critically sized clusters when Δ
Gv is smaller than ΔG
s, also Δ
G is smaller than Δ
Gmax, which is the energy barrier for critical nucleus formation. However, increasing more rapidly with cluster size enlargement (in power three) than Δ
Gs, Δ
Gv approaches Δ
Gs, and thus also Δ
Gmax. Afterwards, when super-critically sized clusters are growing, Δ
Gv becomes larger than Δ
Gs. Thus, Δ
G becomes again smaller than Δ
Gmax. So, it is seen that Δ
G is smaller on both sides of Δ
Gmax. This is exactly the mathematical definition for maximum of a function
y =
f(
x), according to which the function maximum appears at the argument value
xo for which:
This is an inequality that holds true for every negative and positive small value of h. In other words, f(xo) is larger than all neighboring function values. In conclusion, ΔGmax coincides with the energy balance appearing at ΔGv = ΔGs.
The EBDE method is used here to consider the protein crystal nucleation in pores.
5. Protein Crystal Nucleation in Pores
Experimental studies have shown that porous materials are effective at inducing protein crystal nucleation [
56,
57,
58,
59,
60,
61,
62,
63,
64,
65]. Remarkable success has been achieved with the use of a mesoporous bioactive gel-glass (CaO–P
2O
5–SiO
2) as a nucleating reagent [
66]. The bioglass (Naomi’s Nucleant’) [
67] proved to be very successful in producing high quality crystals of both model and target proteins. So, this discovery has set a trend for the use of porous materials as nucleants [
68].
Theoretical considerations [
69] have shown that a combined diffusion-adsorption effect can increase protein concentration inside pores (and crevices) to a level that is enough for crystal nucleation onset [
70]. The reason is that molecular diffusion is the sole mass-transfer mechanism working in pores, and due to translational Brownian motion, which is equally probable in all directions, the probability that large protein molecules will land on pore walls is several times greater than the probability of their escape [
71].
The quantitative estimation of the size of a pore which can induce protein crystal nucleation is based on the mean squared diffusion displacement, <
x2>:
where
D is the diffusion coefficient; a typical value for proteins being
Dprot = 10
−6 cm
2 s
−1.
Regarding the adsorption effect, it was shown that, provided the pore is sufficiently narrow, protein molecules approach their walls and adsorb there more frequently than they can escape. The time span during which protein molecules remain adsorbed at the pore walls is calculated from the desorption rate (
Rd), which is an activated first-order rate process:
where
ca is the surface concentration of the adsorbed molecules and
t is time;
kd being the rate constant for desorption. Integration of Equation (4) gives:
where
ca0 is the initial concentration of the adsorbed molecules.
The rate constant for desorption (
kd) is
kd =
θexp(−
Ed/
kBT), where
Ed is the desorption activation energy,
θ an “attempt frequency” for desorption, k
B is Boltzmann’s constant, and T is temperature. On the other hand, the half-life (
τ1/2) for adsorption of protein molecules at the pore wall is defined as the time when
ca =
ca0/2. Thus:
It has been found [
72] that the apparent activation energy,
Ed, for desorption of an isolated protein molecule is extremely low, i.e., within the range of 2–4 kJ/mol, or less than 2
kBT per molecule. Therefore, most individual protein molecules exhibited short residence times of about 1 s and even less.
Equation (3) shows that during adsorption time
t = 0.5 s, a protein molecule can diffuse from wall to wall of a 10 μm sized pore and adsorb there (the tacit assumption being that the molecule would follow the shortest path). So, the total time during which the protein molecules are in the adsorbed state in pores narrower than several micrometers thus becomes greater than the total time during which they are in the desorbed state. Therefore, despite the increasing amounts of protein in the pores, no concentration gradient favoring back diffusion (from the pores towards bulk solution) can arise. Hence, due to adsorption being more frequent than desorption, sufficiently narrow pores (presumably about 1 μm in size) can become quasi-permanent traps for macromolecules, and thus, accumulate protein [
73]. The conclusion is that protein crystal nucleation may be enabled in sufficiently narrow pores, even under conditions where heterogeneous nucleation on flat surfaces (and even more so in bulk solution) is absent [
69].
Using the MWS method of Stranski and Kaischew [
51,
52,
53] and a Kossel-crystal as a model, Nanev et al. [
69] have calculated the crystal nucleation energy barriers resulting from pore space confinement and interaction with pore walls; pores having the shape of rectangular prism were considered. Also considered was the case in which the size of the pore opening is large enough to allow a critical nucleus smaller than the pore opening to form inside the pore. It was found, however, that such a crystal nucleus would be larger, and therefore, a smaller pore that is completely filled with the nucleus is more effective [
69]. Bearing this in mind, only the most closely-packed monomolecular crystalline layers filling the entire pore orifices are considered here; once formed, the nuclei continue their growth (with a probability 1/2) outside the pore. Evidently, to enable selection of a pore size, which is suitable for crystallization of a given protein molecule, the optimal porous material should possess a broad distribution of pore sizes [
69].
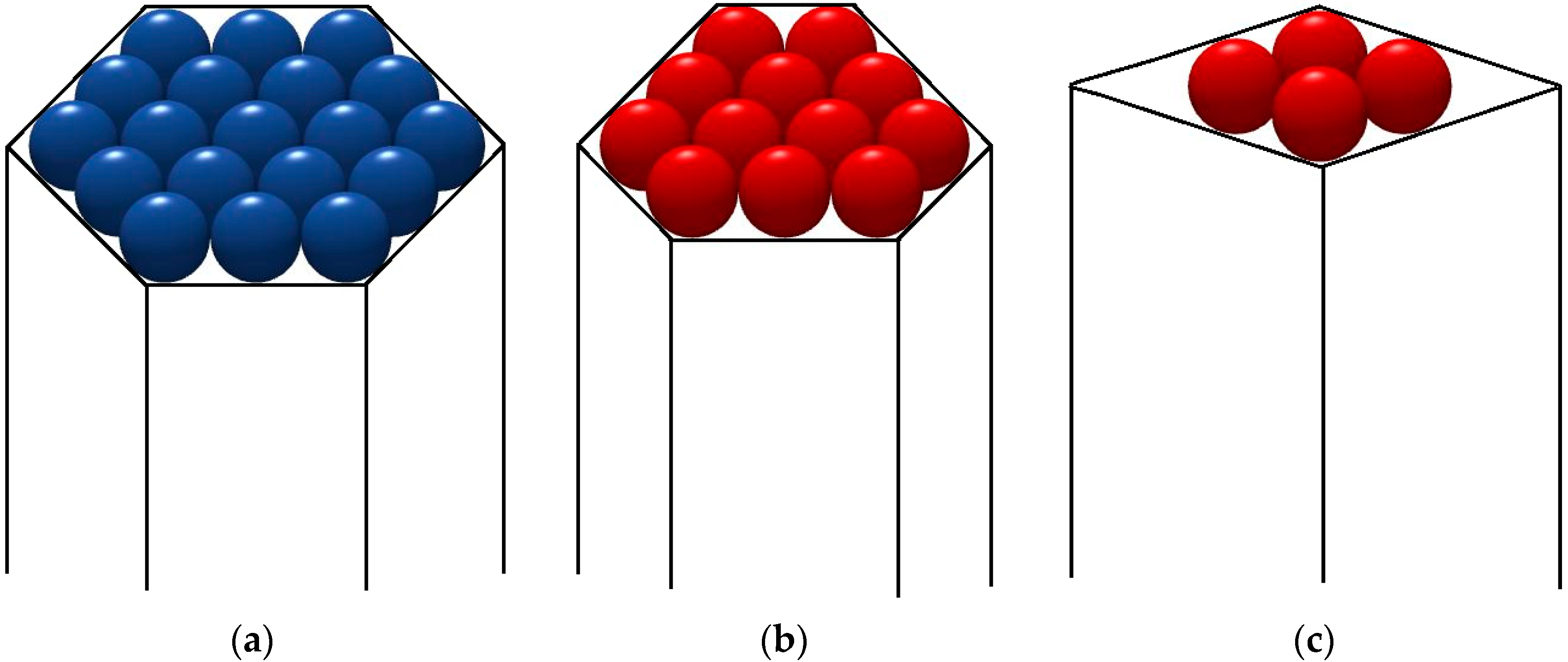
Because only idealized pore-shapes are readily liable to be brought to quantitative account, the protein crystallization promoting effect of pores, having hexagonal shapes (
Figure 1a), trigonal (
Figure 1b) and a rhombic (
Figure 1c) prisms, is calculated here by using the EBDE method. Instead of Kossel crystals (existing extremely rarely in nature), closest packings of equal spheres are considered:
1. Crystallography equations for hexagonal crystal structures (see [
50]) are applied for EBDE calculations using pore model of a hexagonal prism (
Figure 1a). By denoting the total number of molecules in the hexagonal monolayer by (
z), and the number of molecules in its edge by (
λ), we have:
which gives
z = 7, 19, 37, 61, 91, 127... for
λ = 2, 3, 4, 5, 6, 7… respectively. Using the crystallographic formula concerning the number of bonds in the hexagonal crystal layer:
we obtain the balance between cohesive and destructive energies, −Δ
Gv + Δ
Gs = 0:
where
ψ is the work of separation of one protein molecule from a cavity wall;
ψ ≈ E
d. Note that the molecules at the six crystal apexes are bond to the pore walls by energy of 2
ψ. Simultaneously, no crystal apexes and edges (which can be exposed to the enhanced destructive action of water molecules) exist on such crystalline monolayers.
Three different ratios between
ψ and
ψb are used to form an idea of how the energetic interactions between protein and pore materials influence the supersaturation dependence of critical nucleus size:
The results are presented in
Table 1.
The results in
Table 1 confirm the intuitive expectation that the closer the energetic interaction between protein and pore material (i.e., the larger
ψ/
ψb), the lower supersaturation needed for nucleus formation; and the nucleus size becomes almost supersaturation-independent if
ψ/
ψd → 1. To all appearances, the ‘biocompatibility’ of the pore material plays a major role. Perhaps, this is the reason for the effectiveness of the bioactive gel-glass at inducing protein crystal nucleation, as described by Naomi Chayen and coworkers [
66].
2. Considering a pore’s capacity to increase supersaturation, Nanev et al. [
69] have noted that the narrower the pore, the smaller the protein molecule escape probability, and thus, the higher the concentration- (respectively, supersaturation-) increase in the pore. On the other hand, the pore opening is reached, and the protein molecules enter the pore with the same probability with which they reach an equally large flat surface area, meaning that smaller openings are less accessible. The problem is addressed here by considering the results in
Table 1 and the model of a smaller crystal (see
Figure 1c). In this case, the balance between cohesive and destructive energies, −Δ
Gv + Δ
Gs = 0, gives:
Thus, we obtain:
ψ = 0.25ψb, ψb/ψd ≈ 0.57.
ψ = 0.5ψb, ψb/ψd ≈ 0.44.
ψ = 0.75ψb, ψb/ψd ≈ 0.36.
As seen, the above
ψb/
ψd-values are larger than the data for pores having the shape of a hexagonal prism,
Figure 1a; see
Table 1. In other words, as intuitively expected, the smaller the crystal nucleus, i.e., the narrower the suitable pore-opening size, the higher supersaturation needed for the crystal to nucleate. However, though feasible from an energetic point of view, very large crystal nuclei are less likely to appear for kinetic reasons—bringing together a vast number of molecules via molecule-by-molecule assembly into a crystal nucleus involves very large fluctuations, which, in turn, requires very long waiting times. So, crystals of modest size appear most probable.
3. Besides pore-opening size, pore volume and shape are also of importance. For instance, in intricate pores with many turns and corners, protein molecules can be trapped more readily. To address this point, protein crystal nucleation in a somewhat non-regularly shaped pore (see
Figure 1b) is considered. The result for this model is:
which gives:
ψ = 0.25ψb, ψb/ψd ≈ 0.43.
ψ = 0.5ψb, ψb/ψd ≈ 0.38.
ψ = 0.75ψb, ψb/ψd ≈ 0.34.
No substantial difference between the two pore kinds (
Figure 1a,b) can be established by comparing these results with the data for
λ = 2 to 3 in
Table 1. So, we might conclude that although pores in actual disordered porous materials (e.g., such as Bioglass) are much more complex, the models considered above provide adequate clues to comprehend why pores and crevices facilitate protein crystal nucleation.
6. Protein Crystallization in Solution Flow
The interest in intensifying drug manufacturing is increasing nowadays [
75,
76]. Continuous tubular crystallizers which use crystallization in solution flow have received a growing attention. They improve efficiency and enable better product-quality control, thus showing advantages in the process intensification required in the pharmaceutical industry [
75,
77,
78]. Such crystallizers contain periodically-spaced orifice baffles, which allow strong radial solution motions (turbulences) to be created, thus securing a uniform mixing [
79,
80]. Using a combination of a laboratory-scale, continuous-stirred tank crystallizer and a cooled tubular reactor in bypass, Hekmat et al. [
81] have proposed continuous protein crystallization as a viable purification alternative for continuous preparative chromatography. Li and Lakerveld [
82] demonstrate that the induction time for protein nucleation can be reduced in continuous flow compared to batch crystallization with electric-field-assisted crystallization and for the control experiment without an electric field.
As already seen [
83], the electric field energy affects protein crystal nucleation. A reasonable question here would be whether also the flow kinetic energy may affect protein crystal nucleation. Like any moving object, solution flow has a kinetic energy (
Ek). Considering a cylinder of a fluid that is travelling at velocity (
u), we have:
where
ρ is the fluid density,
A is the tube cross-section area, and
ℓ is the tube length.
Unfortunately, there is no evident answer to above question. Shear flow alters the rate at which crystals nucleate from solution, yet the underlying mechanisms remain poorly understood [
84]. To the best of the author’s knowledge, there are no rigorous experimental indications to the solution flow stimulating or postponing crystal nucleation. Although numerous studies have been devoted to protein crystallization in a flow, they refer predominantly to crystal growth e.g., [
85,
86,
87,
88,
89,
90], rather than nucleation [
84,
91,
92,
93]. On the other hand, however, the spontaneous (primary) crystal nucleation is technologically-unmanageable and is avoided, while being replaced by easily controllable seeding technology. However, despite the seeding strategy, a significant increase in new crystal number is observed frequently in the presence of an introduced crystalline material. Why new crystals are bred is a fundamental question. Despite its significance, the understanding of this phenomenon remains limited. It is argued that the origin of the secondary nuclei is the crystal seeds themselves. Anwar et al. [
94] have considered mechanisms of secondary crystal nucleation at a molecular level. Via a molecular dynamic simulation, the authors stipulate that under-critically sized molecule clusters forming close to the crystal seed can move towards the latter, and if/when contacting the seed‘s surface, grow like crystal particles. This explanation is quite logical, but the locally-decreased supersaturation in crystal vicinity (due to crystal growth under diffusion matter supply) limits its applicability to some supersaturation interval where sufficiently-large, under-critically sized molecule clusters can arise; the size of the heterogeneous critical nucleus, although smaller than the homogeneously formed nucleus, also depends on the supersaturation.
To assess the impact of flow kinetic energy on crystal nucleation, it is necessary to recall that this energy can be liberated/dissipated by conversion into internal energy, mainly heat, i.e., solution temperature is expected to increase because of flow energy dissipation (the energy needed to raise the temperature of 1 g of water by 1 °C is 1 calorie). Temperature increase disfavors crystallization of substances possessing normal temperature dependent solubility, while in contrast, it favors crystallization of substances possessing retrograde temperature-dependent solubility. Using the mechanical equivalent of heat equal to 4.1868 joules per calorie, the resulting temperature increase is determined simply using Ek.
Occurring at larger flow velocities, a turbulent flow dissipates more energy than a laminar flow. Therefore, the turbulent flow energy can be used as a benchmark for the significance of the
Ek-effect. The fluid velocity (
u) needed for a turbulent flow to appear is found from the Reynolds number, Re =
uL/
ν, where
L [mm] is a typical length scale in the system, and
ν is the kinematic viscosity of the fluid; for water at 20 °C,
ν = 1.0034 [mm
2/s] [
95]. For a flow in a pipe of diameter
D, experimental observations have shown that turbulence occurs when Re
D ≥ 4000 [
96]. Thus, assuming
L = 1 mm, the velocity needed for a turbulent flow to occur is
u = 4000 mm/s. Then, if A = 1 mm
2 and
ℓ = 1 mm, Equation (15) will give
Ek = 16 × 10
6 erg = 1.6 joule, i.e., a negligible temperature increase (less than 0.4 °C) in a solution of 1 mm
3. Like mass mixing however, heat transfer is further enhanced by liquid shear. So, even smaller temperature increases can be expected. Furthermore, the industrial tubular crystallizers are usually tempered.
In conclusion, although the
Ek-impact of the fluid flow is negligible, mass mixing may favor crystal nucleation kinetically, e.g., by increasing the microscopic transport rate of a molecule to the cluster in shear flow [
97]. For instance, protein crystal nucleation can benefit from enhanced rotation of the huge biomolecules, which assists their proper mutual orientation during colliding; see
Section 3.




{kind=link}
{kind=link}