The Neutron Macromolecular Crystallography Instruments at Oak Ridge National Laboratory: Advances, Challenges, and Opportunities



Abstract
1. Introduction
2. High Flux Isotope Reactor Cold Guide 4D, IMAGINE
3. Spallation Neutron Source Beam Line 11B, MaNDi
4. Software
5. Protein Preparation and Crystal Growth
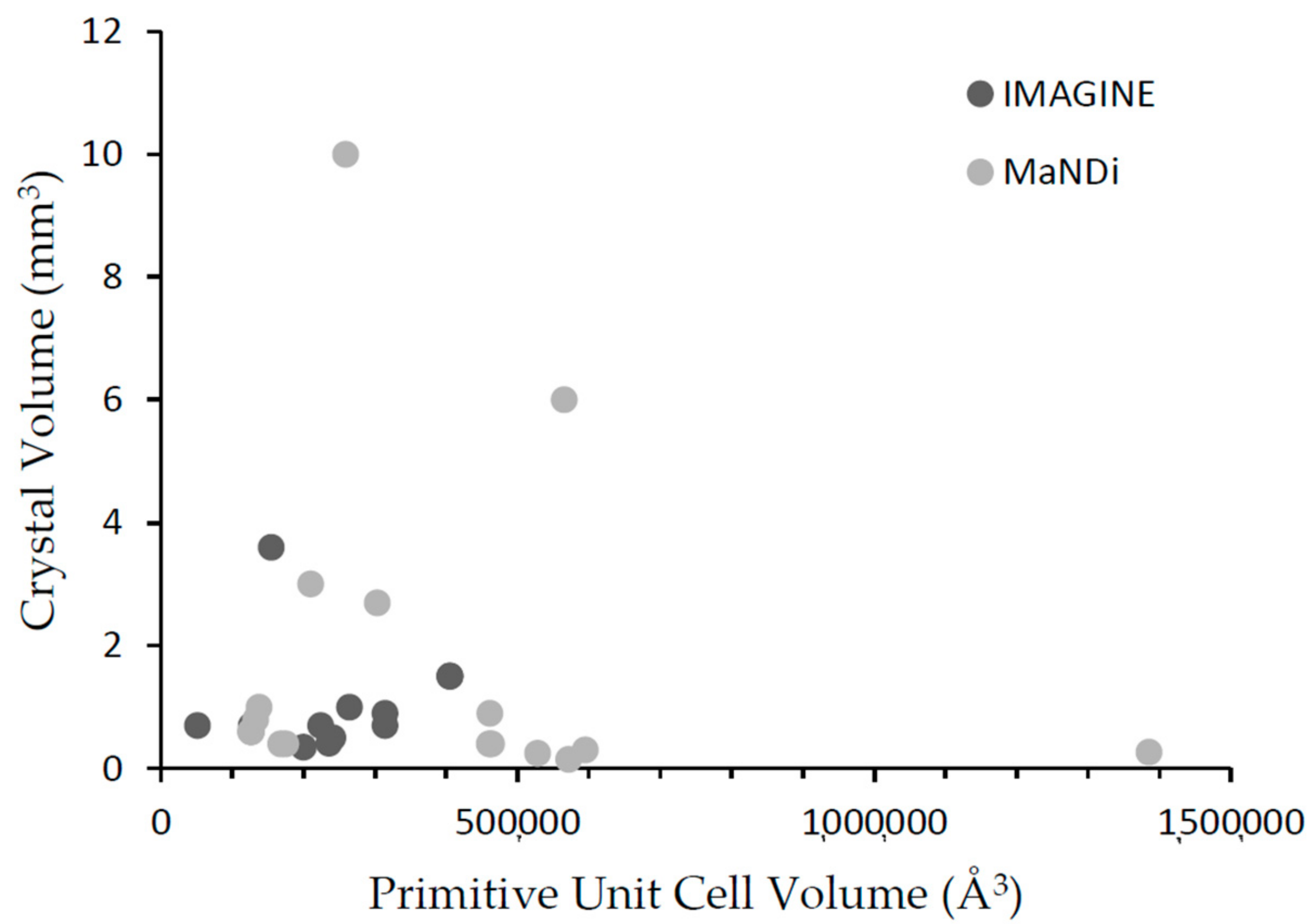
6. IMAGINE and MaNDi Complementarity
7. Future Developments
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Neutron Scattering Lengths and Cross Sections. Available online: https://www.nist.gov/ncnr/planning-your-experiment/sld-periodic-table accessed on (accessed on 10 October 2018).
- Schroder, G.C.; O’Dell, W.B.; Myles, D.A.A.; Kovalevsky, A.; Meilleur, F. Imagine: Neutrons reveal enzyme chemistry. Acta Crystallogr. D Biol. Crystallogr. 2018, 74, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Meilleur, F.; Munshi, P.; Robertson, L.; Stoica, A.D.; Crow, L.; Kovalevsky, A.; Koritsanszky, T.; Chakoumakos, B.C.; Blessing, R.; Myles, D.A. The imagine instrument: First neutron protein structure and new capabilities for neutron macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2157–2160. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, C.; Lehmann, M.S.; Meilleur, F.; Blakeley, M.P.; Myles, D.A.A.; Vogelmeier, S.; Thoms, M.; Walsh, M.; McIntyre, G.J. Characterization of image plates for neutron diffraction. J. Appl. Crystallogr. 2009, 42, 749–757. [Google Scholar] [CrossRef]
- Haberl, B.; Dissanayake, S.; Wu, Y.; Myles, D.A.A.; Dos Santos, A.M.; Loguillo, M.; Rucker, G.M.; Armitage, D.P.; Cochran, M.; Andrews, K.M.; et al. Next-generation diamond cell and applications to single-crystal neutron diffraction. Rev. Sci. Instrum. 2018, 89, 092902. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shukla, S.; Meilleur, F.; Standaert, R.F.; Pierce, J.; Myles, D.A.A.; Cuneo, M.J. Neutron crystallographic studies of t4 lysozyme at cryogenic temperature. Protein Sci. 2017, 26, 2098–2104. [Google Scholar] [CrossRef] [PubMed]
- Hiromoto, T.; Meilleur, F.; Shimizu, R.; Shibazaki, C.; Adachi, M.; Tamada, T.; Kuroki, R. Neutron structure of the T26H mutant of T4 phage lysozyme provides insight into the catalytic activity of the mutant enzyme and how it differs from that of wild type. Protein Sci. 2017, 26, 1953–1963. [Google Scholar] [CrossRef] [PubMed]
- Coates, L.; Cuneo, M.J.; Frost, M.J.; He, J.H.; Weiss, K.L.; Tomanicek, S.J.; McFeeters, H.; Vandavasi, V.G.; Langan, P.; Iverson, E.B. The macromolecular neutron diffractometer mandi at the spallation neutron source. J. Appl. Crystallogr. 2015, 48, 1302–1306. [Google Scholar] [CrossRef]
- Schultz, A.J.; Thiyagarajan, P.; Hodges, J.P.; Rehm, C.; Myles, D.A.A.; Langan, P.; Mesecar, A.D. Conceptual design of a macromolecular neutron diffractometer (MaNDi) for the SNS. J. Appl. Crystallogr. 2005, 38, 964–974. [Google Scholar] [CrossRef]
- Coates, L.; Stoica, A.D.; Hoffmann, C.; Richards, J.; Cooper, R. The macromolecular neutron diffractometer (MaNDi) at the Spallation Neutron Source, Oak Ridge: Enhanced optics design, high-resolution neutron detectors and simulated diffraction. J. Appl. Crystallogr. 2010, 43, 570–577. [Google Scholar] [CrossRef]
- Coates, L.; Tomanicek, S.; Schrader, T.E.; Weiss, K.L.; Ng, J.D.; Juttner, P.; Ostermann, A. Cryogenic neutron protein crystallography: Routine methods and potential benefits. J. Appl. Crystallogr. 2014, 47, 1431–1434. [Google Scholar] [CrossRef]
- Afonine, P.V.; Mustyakimov, M.; Grosse-Kunstleve, R.W.; Moriarty, N.W.; Langan, P.; Adams, P.D. Joint X-ray and neutron refinement with phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Gruene, T.; Hahn, H.W.; Luebben, A.V.; Meilleur, F.; Sheldrick, G.M. Refinement of macromolecular structures against neutron data with Shelxl2013. J. Appl. Crystallogr. 2014, 47, 462–466. [Google Scholar] [CrossRef] [PubMed]
- 104 Patch for CNS; nCNS an Open Source Distribution Patch for CNS Macromolecular Structure Refinement; Los Alamos National Security: Los Alamos, NM, USA, 2007.
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.W. Lauegen, an X-windows-based program for the processing of laue X-ray-diffraction data. J. Appl. Crystallogr. 1995, 28, 228–236. [Google Scholar] [CrossRef]
- Campbell, J.W.; Hao, Q.; Harding, M.M.; Nguti, N.D.; Wilkinson, C. Lauegen version 6.0 and INTLDM. J. Appl. Crystallogr. 1998, 31, 496–502. [Google Scholar] [CrossRef]
- Arnold, O.; Bilheux, J.C.; Borreguero, J.M.; Buts, A.; Campbell, S.I.; Chapon, L.; Doucet, M.; Draper, N.; Leal, R.F.; Gigg, M.A.; et al. Mantid-data analysis and visualization package for neutron scattering and mu sr experiments. Nucl. Instrum. Meth. A 2014, 764, 156–166. [Google Scholar] [CrossRef]
- Schultz, A.J.; Jorgensen, M.R.V.; Wang, X.P.; Mikkelson, R.L.; Mikkelson, D.J.; Lynch, V.E.; Peterson, P.F.; Green, M.L.; Hoffmann, C.M. Integration of neutron time-of-flight single-crystal bragg peaks in reciprocal space. J. Appl. Crystallogr. 2014, 47, 915–921. [Google Scholar] [CrossRef]
- Sullivan, B.; Archibald, R.; Langan, P.S.; Dobbek, H.; Bommer, M.; McFeeters, R.L.; Coates, L.; Wang, X.P.; Gallmeier, F.; Carpenter, J.M.; et al. Improving the accuracy and resolution of neutron crystallographic data by three-dimensional profile fitting of Bragg peaks in reciprocal space. Acta Crystallogr. Sect. D 2018. Accepted. [Google Scholar]
- Arzt, S.; Campbell, J.W.; Harding, M.M.; Hao, Q.; Helliwell, J.R. Lscale—The new normalization, scaling and absorption correction program in the daresbury laue software suite. J. Appl. Crystallogr. 1999, 32, 554–562. [Google Scholar] [CrossRef]
- Helliwell, J.R.; Habash, J.; Cruickshank, D.W.J.; Harding, M.M.; Greenhough, T.J.; Campbell, J.W.; Clifton, I.J.; Elder, M.; Machin, P.A.; Papiz, M.Z.; et al. The recording and analysis of synchrotron X-radiation laue diffraction photographs. J. Appl. Crystallogr. 1989, 22, 483–497. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the ccp4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Zikovsky, J.; Peterson, P.F.; Wang, X.P.P.; Frost, M.; Hoffmann, C. Crystalplan: An experiment-planning tool for crystallography. J. Appl. Crystallogr. 2011, 44, 418–423. [Google Scholar] [CrossRef]
- O’Dell, W.B.; Bodenheimer, A.M.; Meilleur, F. Neutron protein crystallography: A complementary tool for locating hydrogens in proteins. Arch. Biochem. Biophys. 2016, 602, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Meilleur, F.; Weiss, K.L.; Myles, D.A. Deuterium labeling for neutron structure-function-dynamics analysis. Methods Mol. Biol. 2009, 544, 281–292. [Google Scholar] [PubMed]
- Kovalevsky, A.; Aggarwal, M.; Velazquez, H.; Cuneo, M.J.; Blakeley, M.P.; Weiss, K.L.; Smith, J.C.; Fisher, S.Z.; McKenna, R. “To be or not to be” protonated: Atomic details of human carbonic anhydrase-clinical drug complexes by neutron crystallography and simulation. Structure 2018, 26, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Dajnowicz, S.; Johnston, R.C.; Parks, J.M.; Blakeley, M.P.; Keen, D.A.; Weiss, K.L.; Gerlits, O.; Kovalevsky, A.; Mueser, T.C. Direct visualization of critical hydrogen atoms in a pyridoxal 5′-phosphate enzyme. Nat. Commun. 2017, 8, 955. [Google Scholar] [CrossRef] [PubMed]
- Gerlits, O.; Wymore, T.; Das, A.; Shen, C.H.; Parks, J.M.; Smith, J.C.; Weiss, K.L.; Keen, D.A.; Blakeley, M.P.; Louis, J.M.; et al. Long-range electrostatics-induced two-proton transfer captured by neutron crystallography in an enzyme catalytic site. Angew. Chem. Int. Ed. 2016, 55, 4924–4927. [Google Scholar] [CrossRef] [PubMed]
- Bio Deuteration Laboratory. Available online: https://www.ornl.gov/facility/csmb/subpage/bio-deuteration-laboratory (accessed on 10 October 2018).
- Karen, V.L.; Mighell, A.D. Perform NIST*LATTICE Computations. Available online: https://services.mbi.ucla.edu/nist/ (accessed on 25 September 2018).
- Wan, Q.; Bennett, B.C.; Wilson, M.A.; Kovalevsky, A.; Langan, P.; Howell, E.E.; Dealwis, C. Toward resolving the catalytic mechanism of dihydrofolate reductase using neutron and ultrahigh-resolution X-ray crystallography. Proc. Natl. Acad. Sci. USA 2014, 111, 18225–18230. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Kovalevsky, A.Y.; Wilson, M.A.; Bennett, B.C.; Langan, P.; Dealwis, C. Preliminary joint X-ray and neutron protein crystallographic studies of ecDHFR complexed with folate and NADP+. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2014, 70, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Knihtila, R.; Holzapfel, G.; Weiss, K.; Meilleur, F.; Mattos, C. Neutron crystal structure of RAS GTPase puts in question the protonation state of the GTP gamma-phosphate. J. Biol. Chem. 2015, 290, 31025–31036. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Kovalevsky, A.Y.; Velazquez, H.; Fisher, S.Z.; Smith, J.C.; McKenna, R. Neutron structure of human carbonic anhydrase II in complex with methazolamide: Mapping the solvent and hydrogen-bonding patterns of an effective clinical drug. IUCrJ 2016, 3, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.; Yu, L.J.; Meilleur, F.; Blakeley, M.P.; Duff, A.P.; Karton, A.; Vrielink, A. An extended N-H bond, driven by a conserved second-order interaction, orients the flavin N5 orbital in cholesterol oxidase. Sci. Rep. 2017, 7, 40517. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, W.B.; Agarwal, P.K.; Meilleur, F. Oxygen activation at the active site of a fungal lytic polysaccharide monooxygenase. Angew. Chem. Int. Ed. 2017, 56, 767–770. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, W.B.; Swartz, P.D.; Weiss, K.L.; Meilleur, F. Crystallization of a fungal lytic polysaccharide monooxygenase expressed from glycoengineered Pichia pastoris for X-ray and neutron diffraction. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2017, 73, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Gerlits, O.O.; Coates, L.; Woods, R.J.; Kovalevsky, A. Mannobiose binding induces changes in hydrogen bonding and protonation states of acidic residues in concanavalin a as revealed by neutron crystallography. Biochemistry 2017, 56, 4747–4750. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Parks, J.M.; Hanson, B.L.; Fisher, S.Z.; Ostermann, A.; Schrader, T.E.; Graham, D.E.; Coates, L.; Langan, P.; Kovalevsky, A. Direct determination of protonation states and visualization of hydrogen bonding in a glycoside hydrolase with neutron crystallography. Proc. Natl. Acad. Sci. USA 2015, 112, 12384–12389. [Google Scholar] [CrossRef] [PubMed]
- Vandavasi, V.G.; Weiss, K.L.; Cooper, J.B.; Erskine, P.T.; Tomanicek, S.J.; Ostermann, A.; Schrader, T.E.; Ginell, S.L.; Coates, L. Exploring the mechanism of beta-lactam ring protonation in the class a beta-lactamase acylation mechanism using neutron and X-ray crystallography. J. Med. Chem. 2016, 59, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Langan, P.S.; Close, D.W.; Coates, L.; Rocha, R.C.; Ghosh, K.; Kiss, C.; Waldo, G.; Freyer, J.; Kovalevsky, A.; Bradbury, A.R. Evolution and characterization of a new reversibly photoswitching chromogenic protein, dathail. J. Mol. Biol. 2016, 428, 1776–1789. [Google Scholar] [CrossRef] [PubMed]
- Vandavasi, V.G.; Langan, P.S.; Weiss, K.L.; Parks, J.M.; Cooper, J.B.; Ginell, S.L.; Coates, L. Active-site protonation states in an acyl-enzyme intermediate of a class a beta-lactamase with a monobactam substrate. Antimicrob. Agents Chemother. 2017, 61, e01636-16. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, I.; Mlynek, G.; Flego, N.; Puhringer, D.; Libiseller-Egger, J.; Coates, L.; Hofbauer, S.; Bellei, M.; Furtmuller, P.G.; Battistuzzi, G.; et al. Molecular mechanism of enzymatic chlorite detoxification: Insights from structural and kinetic studies. ACS Catal. 2017, 7, 7962–7976. [Google Scholar] [CrossRef] [PubMed]
- Bacik, J.P.; Mekasha, S.; Forsberg, Z.; Kovalevsky, A.Y.; Vaaje-Kolstad, G.; Eijsink, V.G.H.; Nix, J.C.; Coates, L.; Cuneo, M.J.; Unkefer, C.J.; et al. Neutron and atomic resolution X-ray structures of a lytic polysaccharide monooxygenase reveal copper-mediated dioxygen binding and evidence for N-terminal deprotonation. Biochemistry 2017, 56, 2529–2532. [Google Scholar] [CrossRef] [PubMed]
- Bacik, J.P.; Mekasha, S.; Forsberg, Z.; Kovalevsky, A.; Nix, J.C.; Cuneo, M.J.; Coates, L.; Vaaje-Kolstad, G.; Chen, J.C.; Eijsink, V.G.; et al. Neutron and high-resolution room-temperature X-ray data collection from crystallized lytic polysaccharide monooxygenase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2015, 71, 1448–1452. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Serpersu, E.H.; Cuneo, M.J. A low-barrier hydrogen bond mediates antibiotic resistance in a noncanonical catalytic triad. Sci. Adv. 2018, 4, eaas8667. [Google Scholar] [CrossRef] [PubMed]
- Langan, P.S.; Vandavasi, V.G.; Cooper, S.J.; Weiss, K.L.; Ginell, S.L.; Parks, J.M.; Coates, L. Substrate binding induces conformational changes in a class a beta-lactamase that prime it for catalysis. ACS Catal. 2018, 8, 2428–2437. [Google Scholar] [CrossRef]
- Manzoni, F.; Saraboji, K.; Sprenger, J.; Kumar, R.; Noresson, A.L.; Nilsson, U.J.; Leffler, H.; Fisher, S.Z.; Schrader, T.E.; Ostermann, A.; et al. Perdeuteration, crystallization, data collection and comparison of five neutron diffraction data sets of complexes of human galectin-3C. Acta Crystallogr. D Biol. Crystallogr. 2016, 72, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, F.; Wallerstein, J.; Schrader, T.E.; Ostermann, A.; Coates, L.; Akke, M.; Blakeley, M.P.; Oksanen, E.; Logan, D.T. Elucidation of hydrogen bonding patterns in ligand-free, lactose- and glycerol-bound galectin-3C by neutron crystallography to guide drug design. J. Med. Chem. 2018, 61, 4412–4420. [Google Scholar] [CrossRef] [PubMed]
- Azadmanesh, J.; Trickel, S.R.; Weiss, K.L.; Coates, L.; Borgstahl, G.E. Preliminary neutron diffraction analysis of challenging human manganese superoxide dismutase crystals. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2017, 73, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Bommer, M.; Coates, L.; Dau, H.; Zouni, A.; Dobbek, H. Protein crystallization and initial neutron diffraction studies of the photosystem II subunit psbo. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2017, 73, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Coates, L.; Robertson, L. Ewald: An extended wide-angle laue diffractometer for the second target station of the spallation neutron source. J. Appl. Crystallogr. 2017, 50, 1174–1178. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| H | D | C | N | O | |
|---|---|---|---|---|---|
| Neutron coherent scattering length (10−12 cm) | −0.374 | 0.667 | 0.665 | 0.936 | 0.581 |
| Neutron incoherent cross section (Barns) | 80.27 | 2.05 | 0 | 0.49 | 0 |
| X-ray Scattering Factors (10−12 cm) sinθ/λ = 0 | 0.28 | 0.28 | 1.69 | 1.97 | 2.25 |
| X-ray Scattering Factors (10−12 cm) sinθ/λ = 0.5 Å−1 | 0.02 | 0.02 | 0.48 | 0.53 | 0.62 |
| IMAGINE (HFIR, ORNL) | MANDI (SNS, ORNL) | |
|---|---|---|
| Neutron Source | Reactor | Spallation Source |
| Moderator | Hydrogen cold source | Decoupled hydrogen |
| Source frequency | n/a | 60 Hz |
| Avg. Flux | ~3 × 107 n/s/cm2 (2.8–4.0 Å) | ~2.06 × 107 n/s/cm2 |
| Flight Path | 13 m downstream CTAX instrument | 30 m |
| Beam size | 3.2 × 2.0 mm2 | Variable diameter pin holes: 1.0–6.0 mm |
| Scattering angles | Horizontal: ±151° Vertical: ±48° | 4 π |
| Optics | Flat mirrors, wavelength selection filters, Elliptical focusing mirrors & | Bandwidth Choppers, Mirrors |
| Incident λ | 2–10 Å | 1–10 Å |
| Δ λ/λ | 25% | 2.15 Å or 4.3 Å window 0.4% resolution (TOF) |
| Detector type | Neutron Image Plate | SNS Anger Camera |
| No. of detectors | 1 ½ | 40 |
| Detector active area | 1200 × 450 mm2 | 40 detectors (150 × 150 mm2) |
| Detector solid angle | 7.85 sr | 4.1 sr |
| Crystal size (protein) | >0.3 mm3 | >0.1 mm3 |
| Unit cell dimension | <150 Å | <300 Å |
| CG-4D (IMAGINE) | MANDI (SNS, ORNL) | |
|---|---|---|
| Data collection | Arinax GUI | SNS software CSS (EPICS) |
| Data collection strategy | CrystalPlan (in 2018t) | CrystalPlan |
| Data reduction | Lauegen, Lscale, SCALA/AIMLESS | Mantid, Lauenorm, SCALA/AIMLESS |
| Data visualization | ARINAX GUI, ImageJ | Mantid |
| Data analysis packages | Phenix.refine, SHELX, nCNS | |
| Data Modeling | Coot | |
| Computing resources | PC and Linux | |
| Protein | PDB ID | Primitive Unit Cell Volume (Å3) * | Crystal Volume (mm3) | Reference |
|---|---|---|---|---|
| Rubredoxin | 4K9F | 51,030 | 0.7 | [3] |
| DHFR | 4PDJ | 154,857 | 3.6 | [32,33] |
| RAS GTPase | 4RSG | 223,731 | 0.7 | [34] |
| MTAN | 5CCD 5JPC 5K1Z | 405,350 405,350 405,350 | 1.5 | [28] |
| Carbonic Anhydrase | 5C8I | 126,464 | 0.7 | [35] |
| HIV protease | 5E5K | 241,373 | 0.5 | [29] |
| Cholesterol Oxidase | 5KWF | 235,475 | 0.4 | [36] |
| LPMO (9) | 5TKI | 200,072 | 0.3 | [37,38] |
| T4 Lysozyme | 5VNQ | 314,129 | 0.7 | [6] |
| T4 Lysozyme | 5XPE | 314,270 | 0.9 | [7] |
| ConA | 5WEY | 264,656 | 1 | [39] |
| Protein | PDB ID | Primitive Unit Cell Volume (Å3) | Crystal Volume (mm3) | Reference |
|---|---|---|---|---|
| Xylanase | 4S2F | 210,271 | 3 | [40] |
| Toho1 β-lactamase | 5A93 | 460,246 | 0.4 | [41] |
| Dathail (RSFP) | 5EBJ | 259,041 | 10 | [42] |
| Toho1 β-lactamase | 5KSC | 461,045 | 0.9 | [43] |
| Chlorite dismutase | 5NKU | 132,679 | 0.8 | [44] |
| IPP | 5TY5 | 565,529 | 6 | Unpublished |
| LPMO (10) | 5VG1 | 303,571 | 2.7 | [45,46] |
| AAC | 6BBR 6BBZ | 174,616 167,851 | 0.4 0.4 | [47] |
| CA-EZM | 6BCC | 125,909 | 0.6 | [27] |
| CA-DZM | 6BC9 | 126,768 | 0.6 | [27] |
| Toho1 β-lactamase | 6C78 | 463,740 | Not measured | [48] |
| Galectin | 6EYM | 137,845 | 1.0 | [49,50] |
| tRNA hydrolase | Not yet deposited | 571,442 | 0.15 | [8] |
| MnSOD | Not yet deposited | 1,385,615 | 0.26 | [51] |
| FMO | Not yet deposited | 595,262 | 0.3 | Lu et al., 2018, Acta Crystallographica F, under review |
| Photosystem II subunit PsbO | Not Yet Deposited | 527,955 | 0.25 | [52] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meilleur, F.; Coates, L.; Cuneo, M.J.; Kovalevsky, A.; Myles, D.A.A. The Neutron Macromolecular Crystallography Instruments at Oak Ridge National Laboratory: Advances, Challenges, and Opportunities. Crystals 2018, 8, 388. https://doi.org/10.3390/cryst8100388
Meilleur F, Coates L, Cuneo MJ, Kovalevsky A, Myles DAA. The Neutron Macromolecular Crystallography Instruments at Oak Ridge National Laboratory: Advances, Challenges, and Opportunities. Crystals. 2018; 8(10):388. https://doi.org/10.3390/cryst8100388
Chicago/Turabian StyleMeilleur, Flora, Leighton Coates, Matthew J. Cuneo, Andrey Kovalevsky, and Dean A. A. Myles. 2018. "The Neutron Macromolecular Crystallography Instruments at Oak Ridge National Laboratory: Advances, Challenges, and Opportunities" Crystals 8, no. 10: 388. https://doi.org/10.3390/cryst8100388
APA StyleMeilleur, F., Coates, L., Cuneo, M. J., Kovalevsky, A., & Myles, D. A. A. (2018). The Neutron Macromolecular Crystallography Instruments at Oak Ridge National Laboratory: Advances, Challenges, and Opportunities. Crystals, 8(10), 388. https://doi.org/10.3390/cryst8100388