
High-Throughput Small-Molecule Crystallography at the ‘Belok’ Beamline of the Kurchatov Synchrotron Radiation Source: Transition Metal Complexes with Azomethine Ligands as a Case Study

 ,
,  , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Description of the ‘Belok’ Beamline at the Kurchatov Synchrotron Radiation Source
- -
- a Si double-crystal monochromator in the (1,−1) configuration providing a fixed-exit beam position. The second crystal is sagittally bent to focus the beam in the horizontal plane with the focal spot coinciding with the 2D position-sensitive detector;
- -
- a multi-segment total reflection mirror providing focusing in the vertical plane and higher harmonics suppression;
- -
- a diffractometer.
- -
- a Rayonix SX-165 position-sensitive CCD detector with a dynamic range of 16 bits;
- -
- a MARdtb single-axis goniometer. The goniometer provides a high-precision ϕ-scanning; the sample-to-detector distance can be automatically selected over a range 43–380 mm and the detector plane can be tilted up to 29.5° to extend the 2θ scattering angle range.
- -
- LN2 low-temperature device by Oxford Cryosystems.
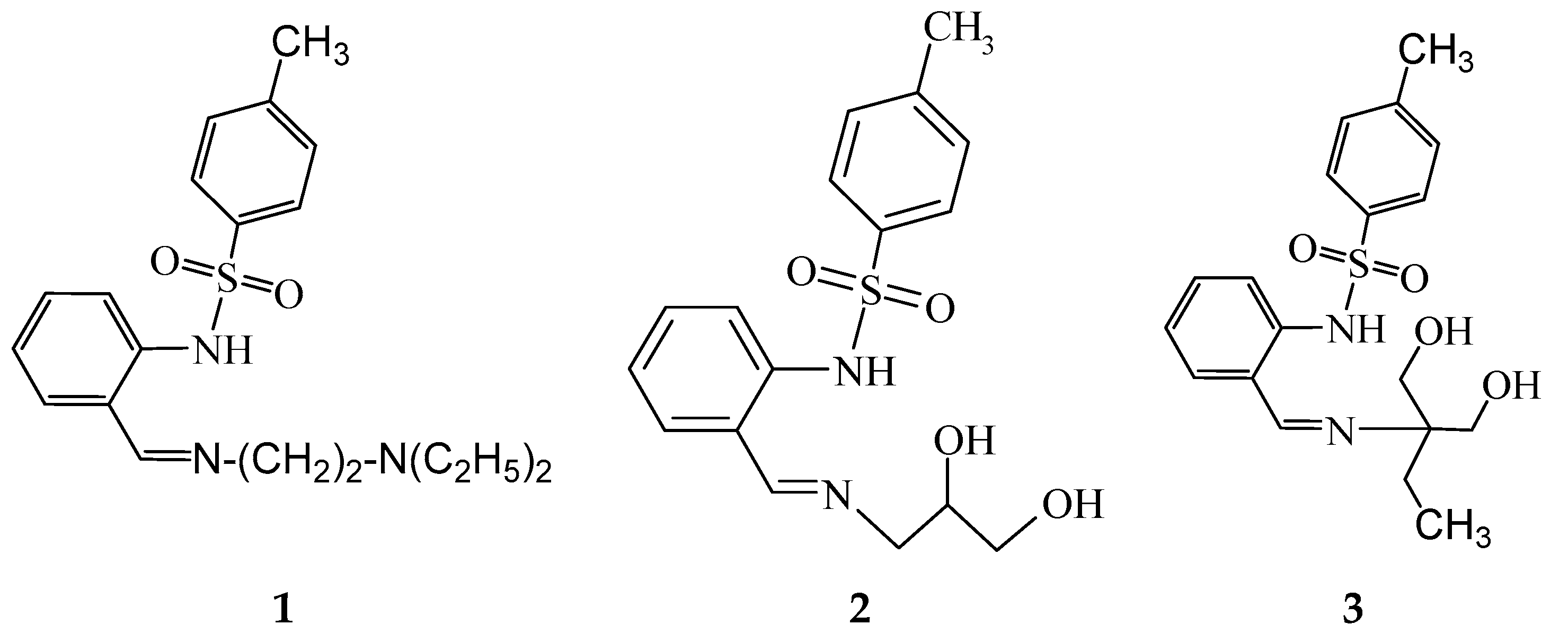
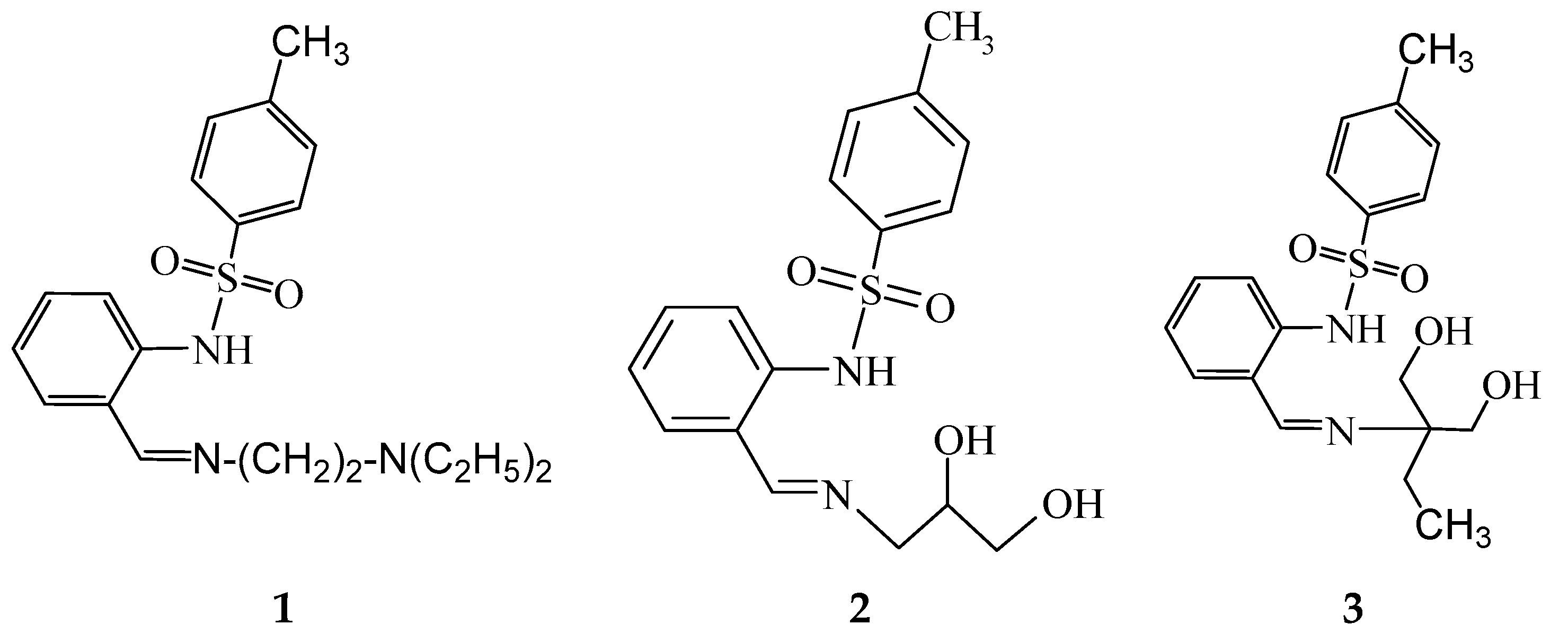
2.2. Synthesis of Free Ligands and Their Derived Metal Complexes
N-[2-(2-diethylaminoethyliminomethyl)phenyl]-4-methylbenzenesulfonamide (1)
Bis-[2-(2-diethylaminoethyliminomethyl)phenyl]-4-methylbenzenesulfonamidato}iron(II) (1-Fe)
N-[2-[(E)-2,3-dihydroxypropyliminomethyl]phenyl]-4-methylbenzenesulfonamide (2)
Bis{N-[2-[(E)-2,3-dihydroxypropyliminomethyl]phenyl]-4-methylbenzenesulfonamidato} zinc(II), DMF solvate (2-Zn)
Bis{N-[2-[(E)-2,2-bis(hydroxymethyl)butyliminomethyl]phenyl]-4-methylbenzenesulfonamide}dicopper(II) (3-Cu)
Tris{μ4-[2-((E)-2,2-bis(hydroxymethyl)butyliminomethyl)phenyl]-4-methylbenzenesulfonamide-κN,N′,κ3O}ethanolato)-(μ3-oxo)-tris(aqua)-tetra-nickel, adduct with KOAc, (HNEt3)OAc, acetic acid solvate (3-Ni)
2.3. Chemical and Spectroscopic Characterization
2.4. Crystal Structure Determinations
3. Results and Discussion
3.1. Spectroscopic Properties
3.2. Structure Description
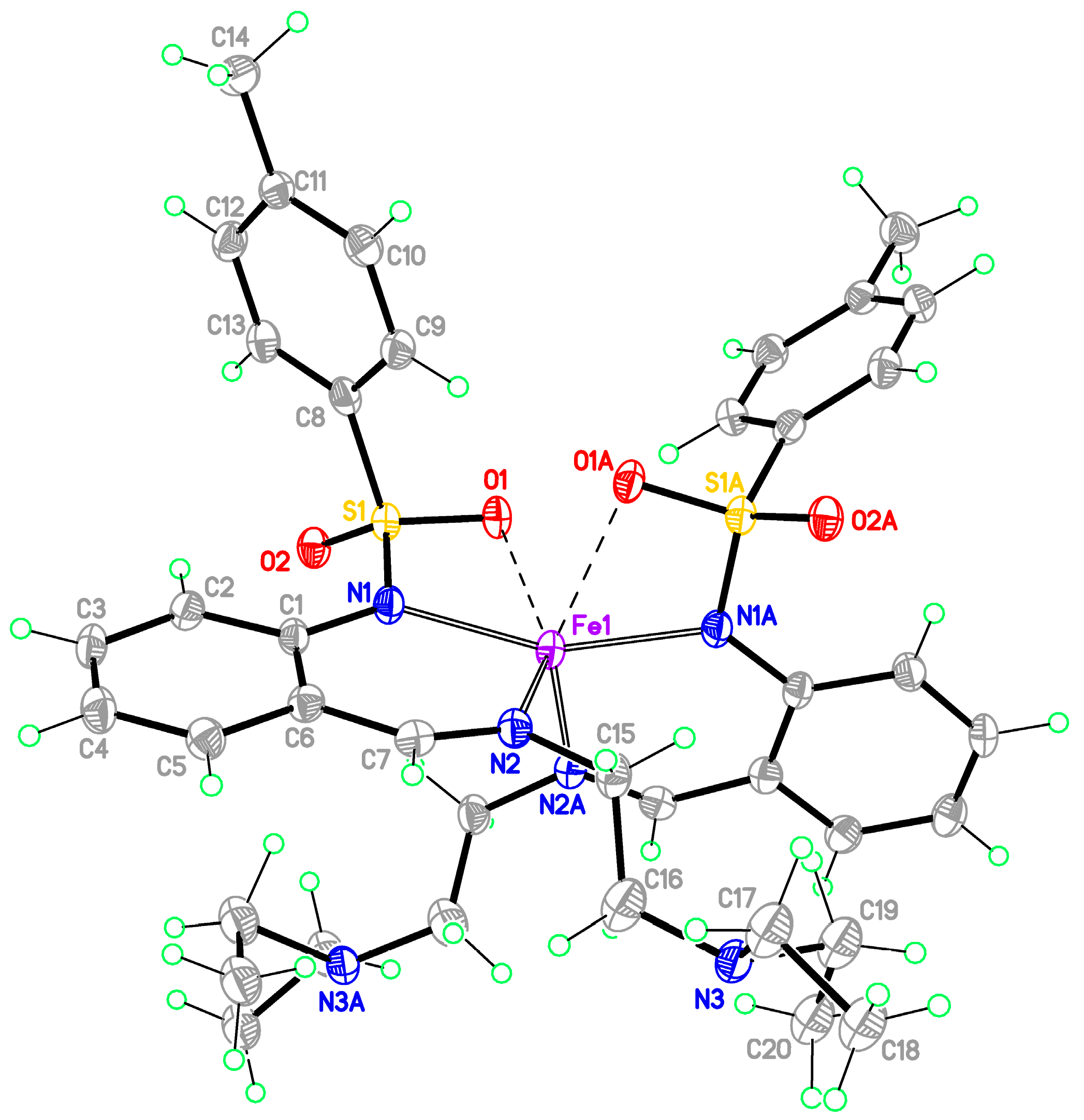
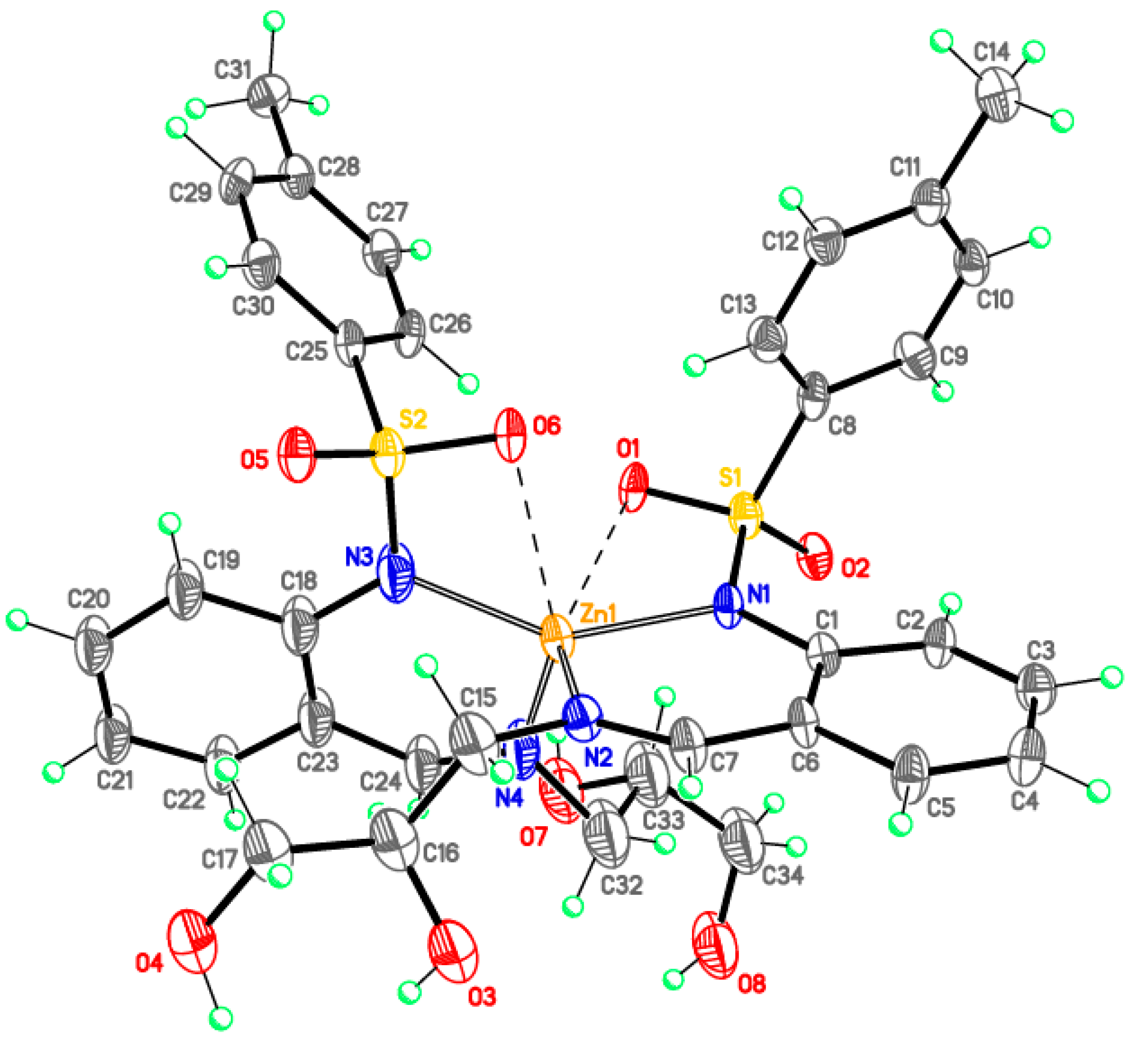
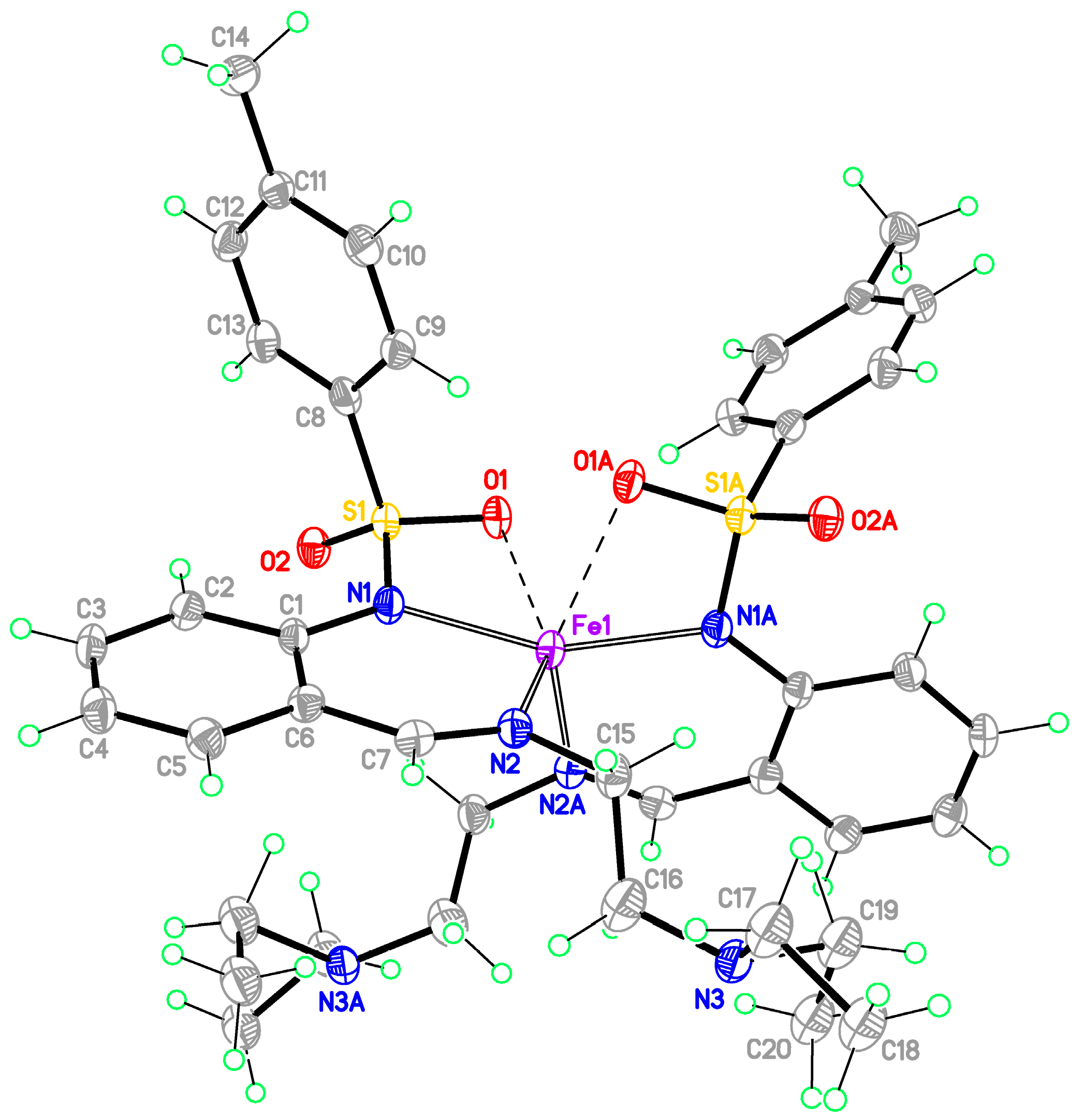
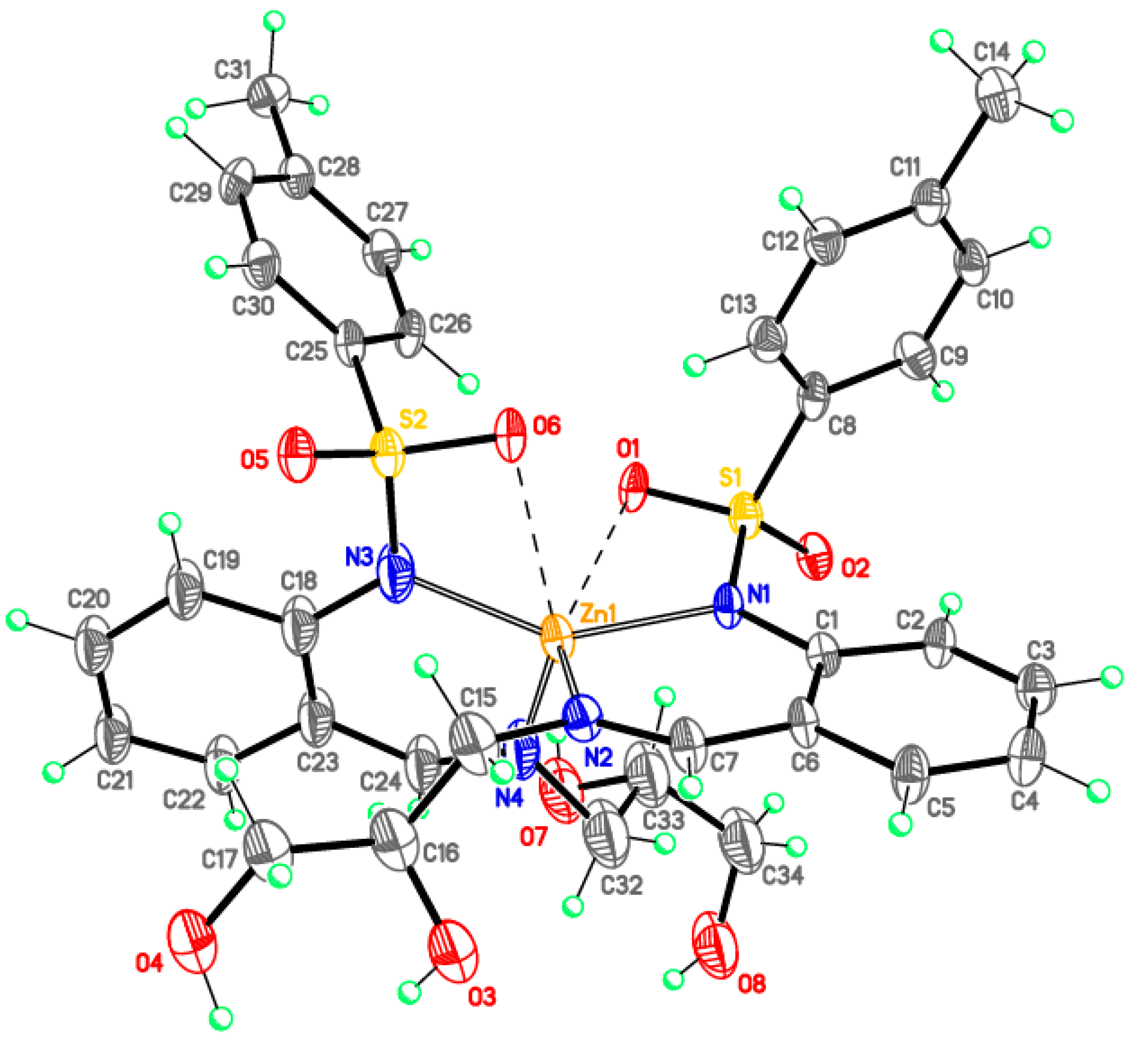
Monomeric complexes 1-Fe and 2-Zn
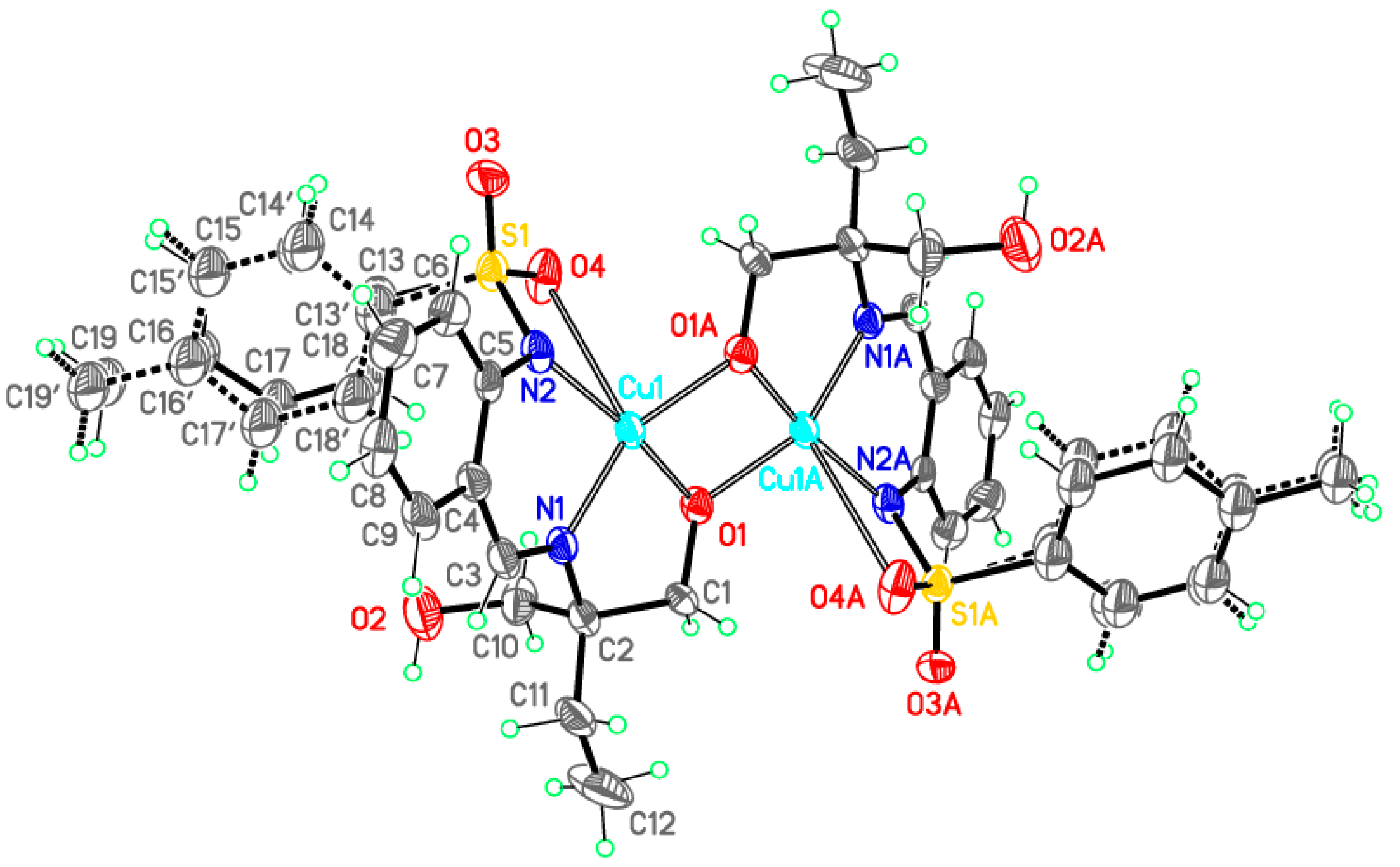
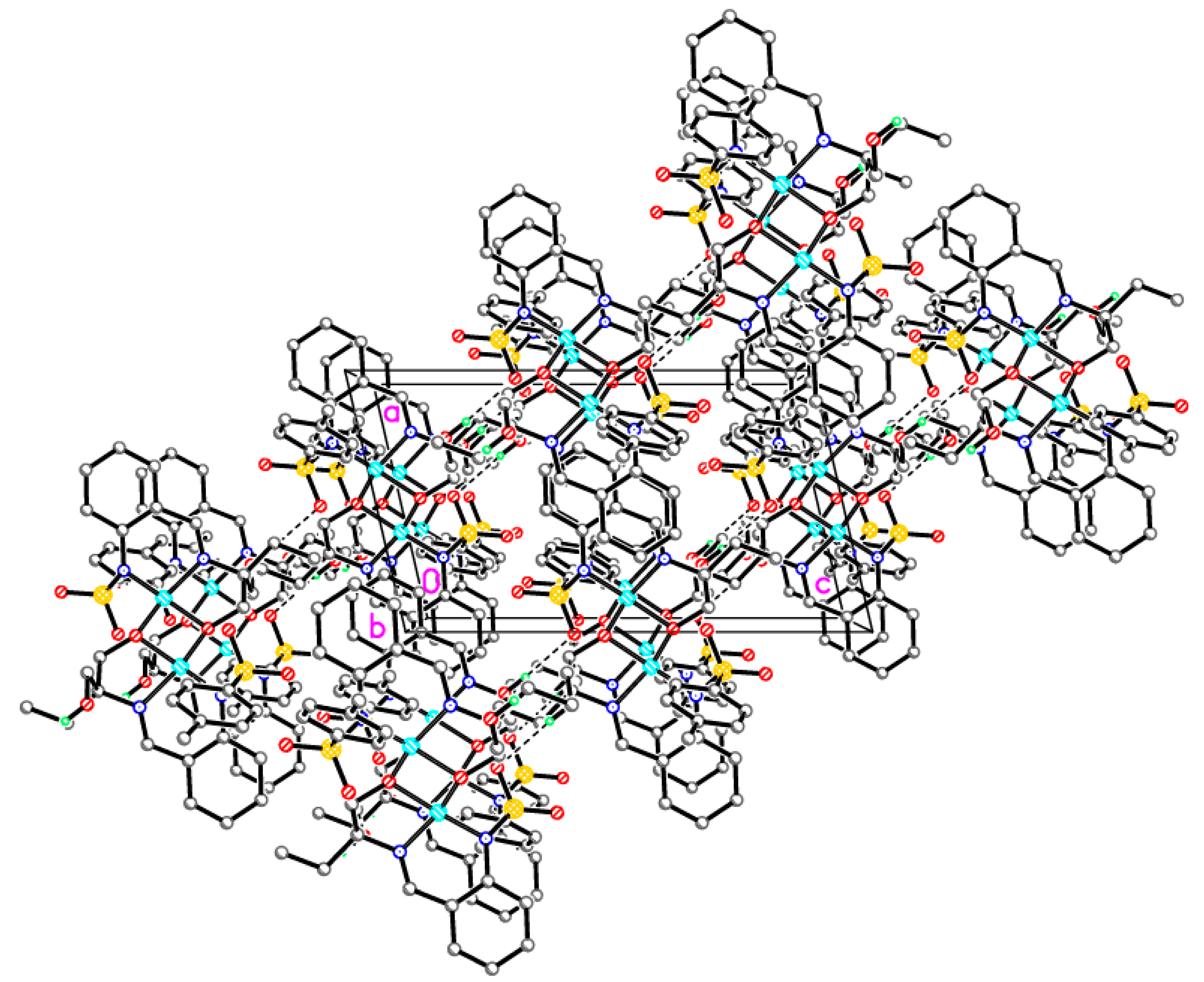
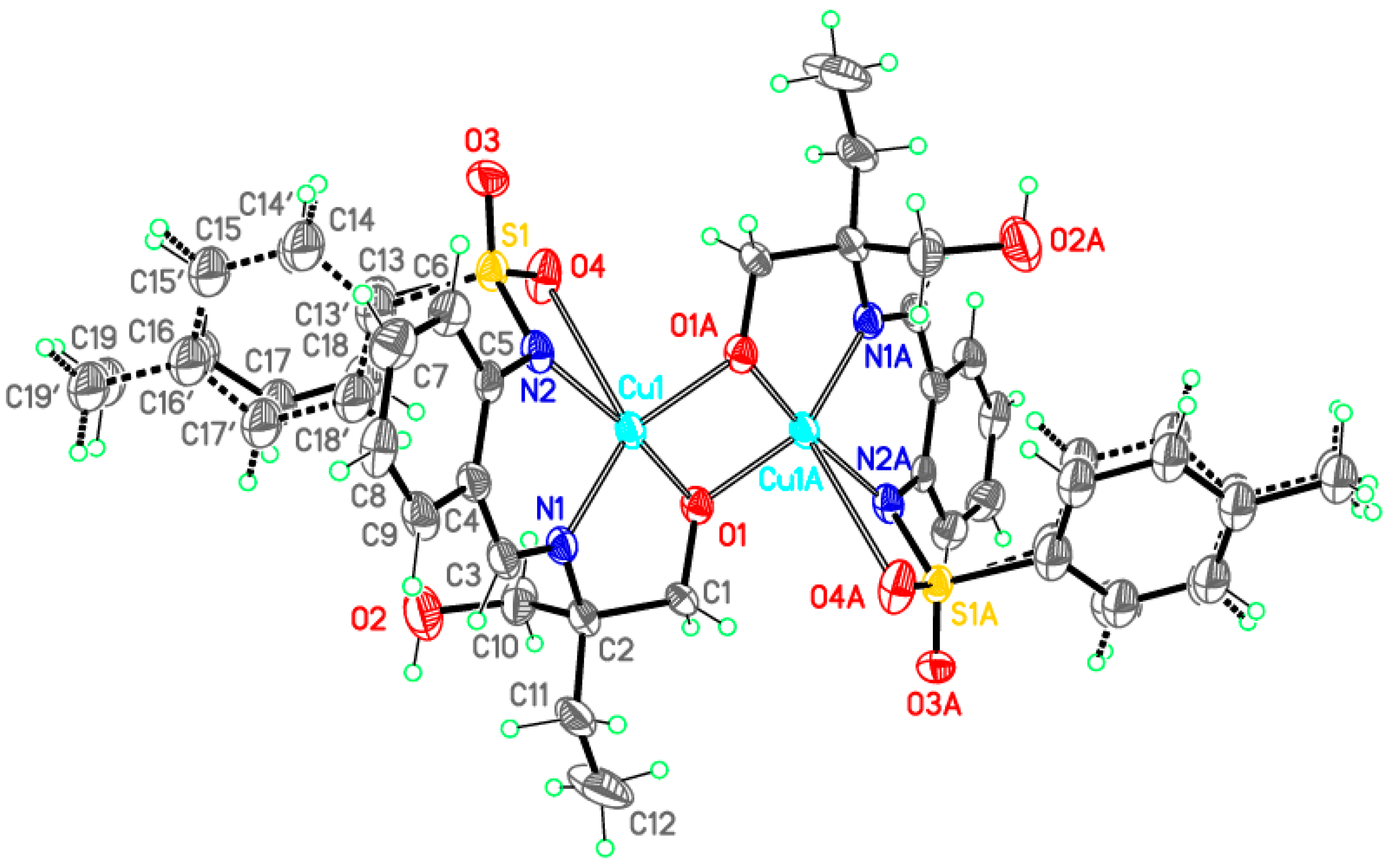
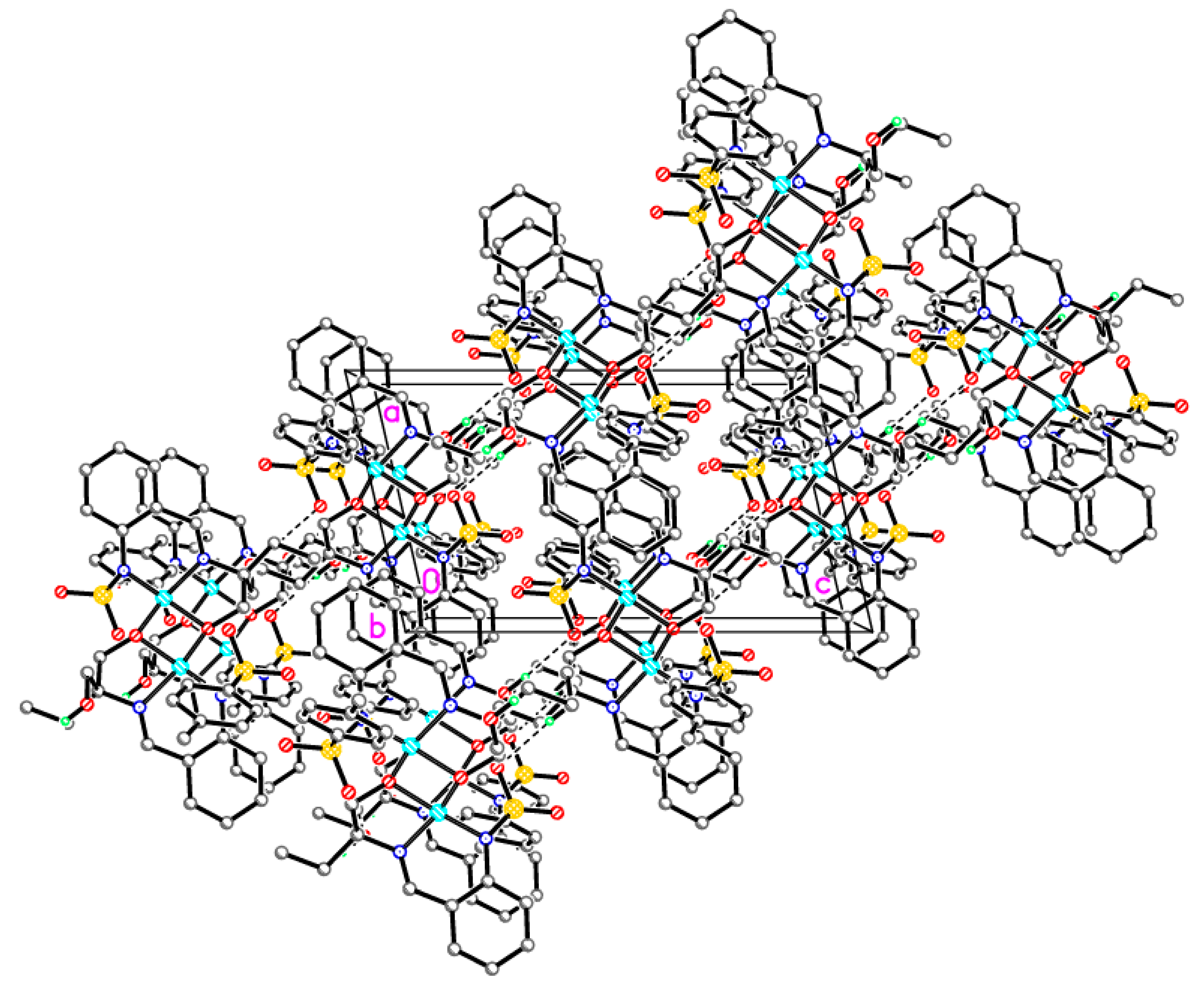
Dimeric complex 3-Cu
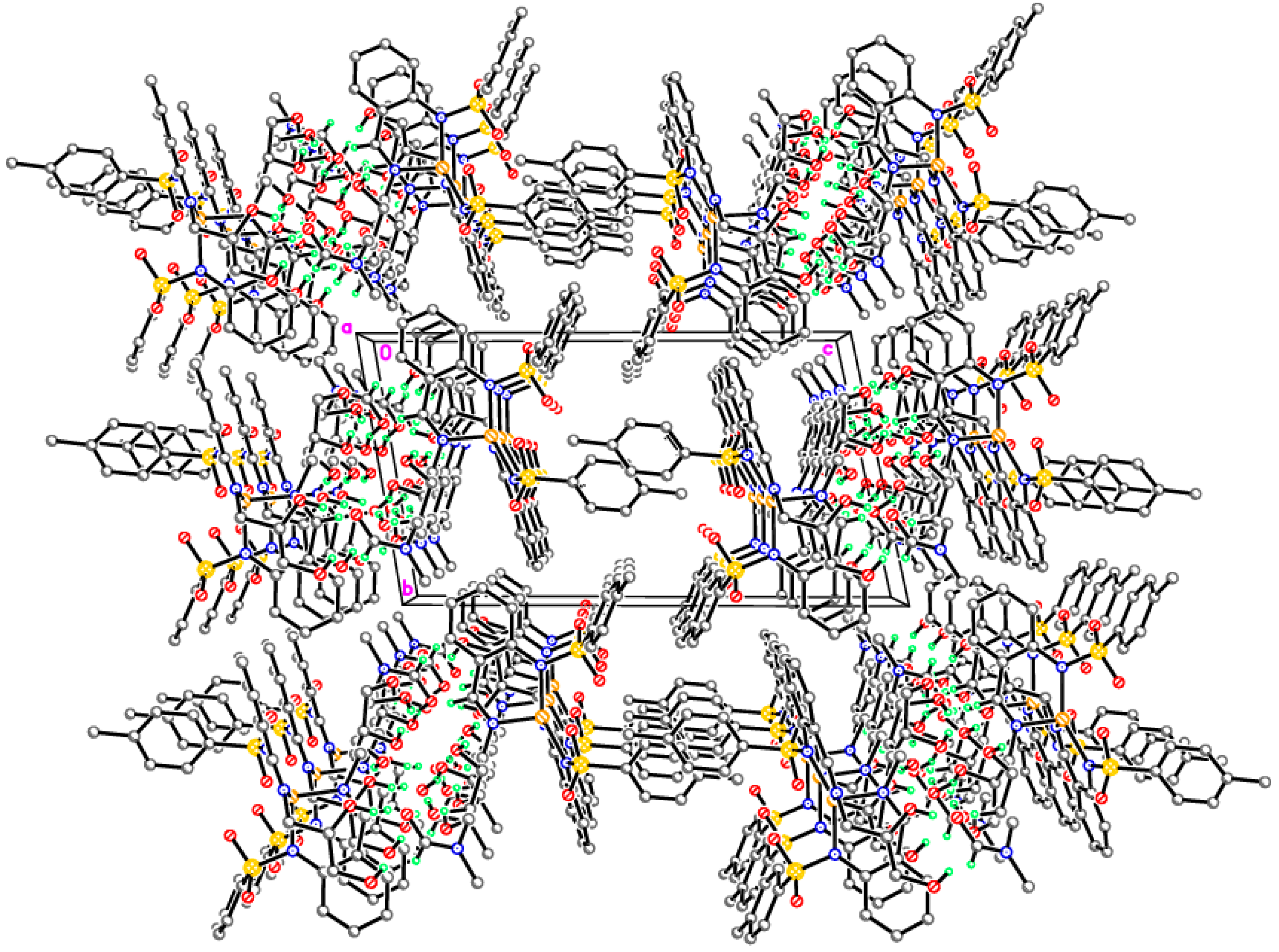
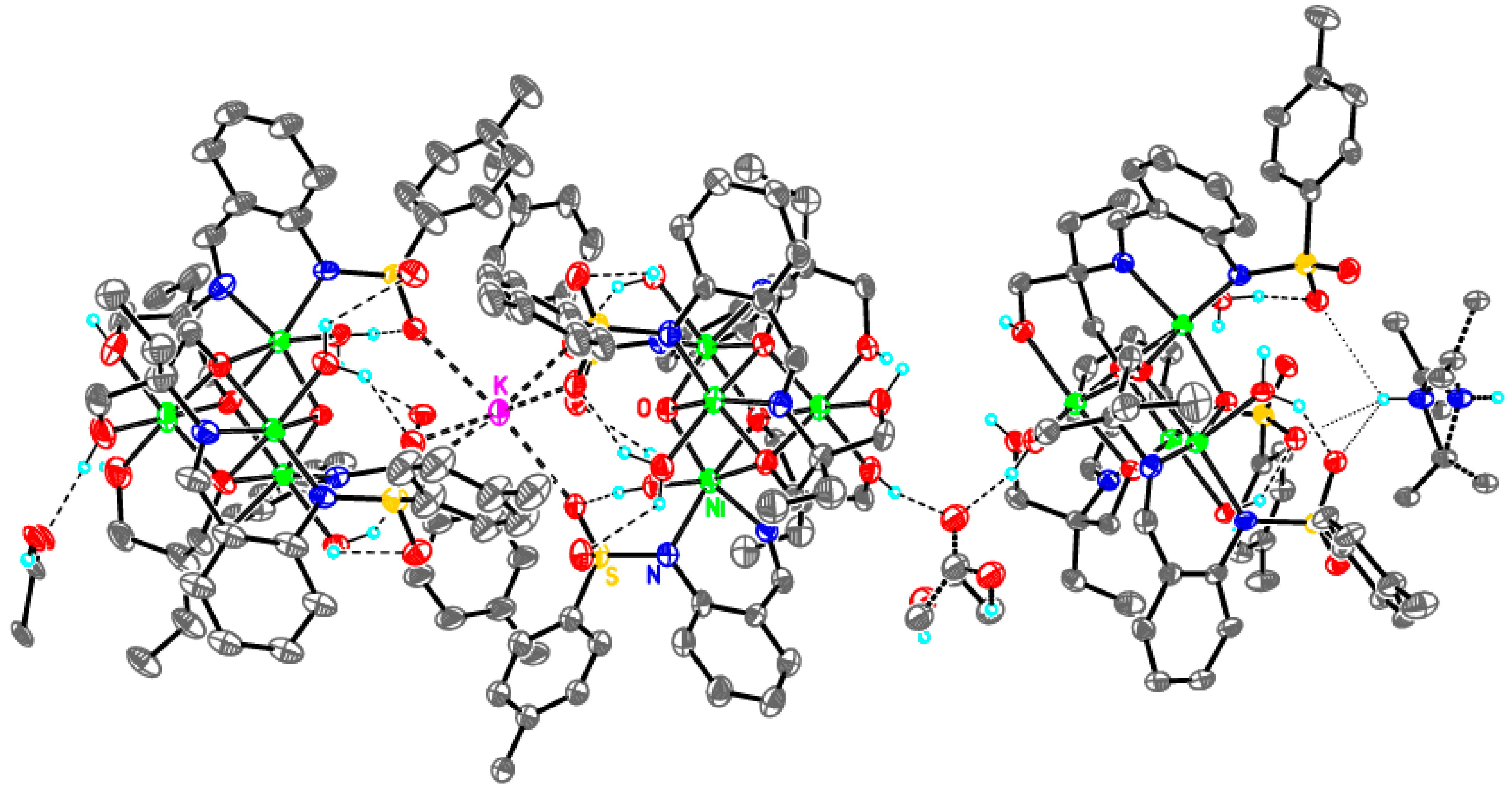
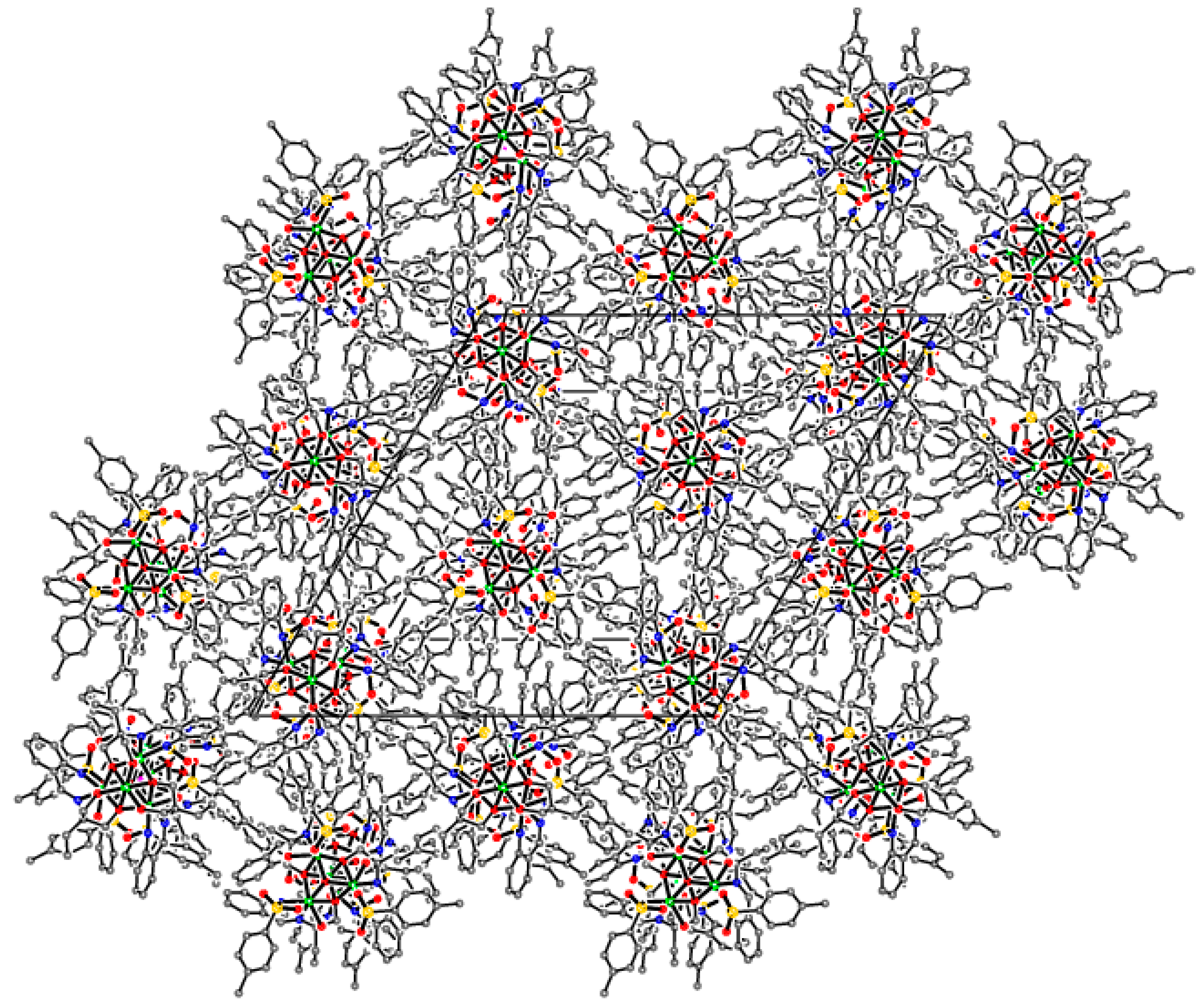
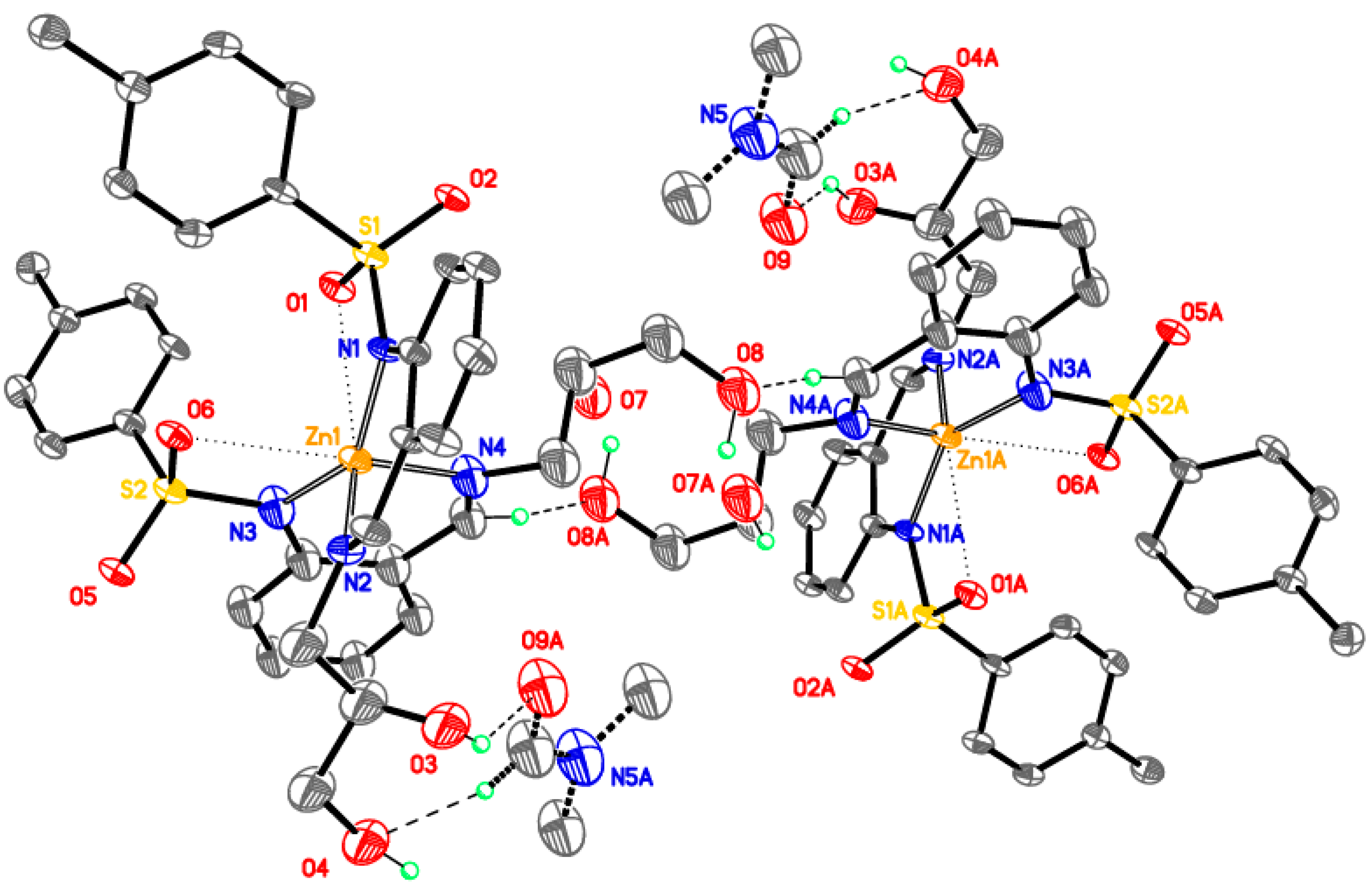
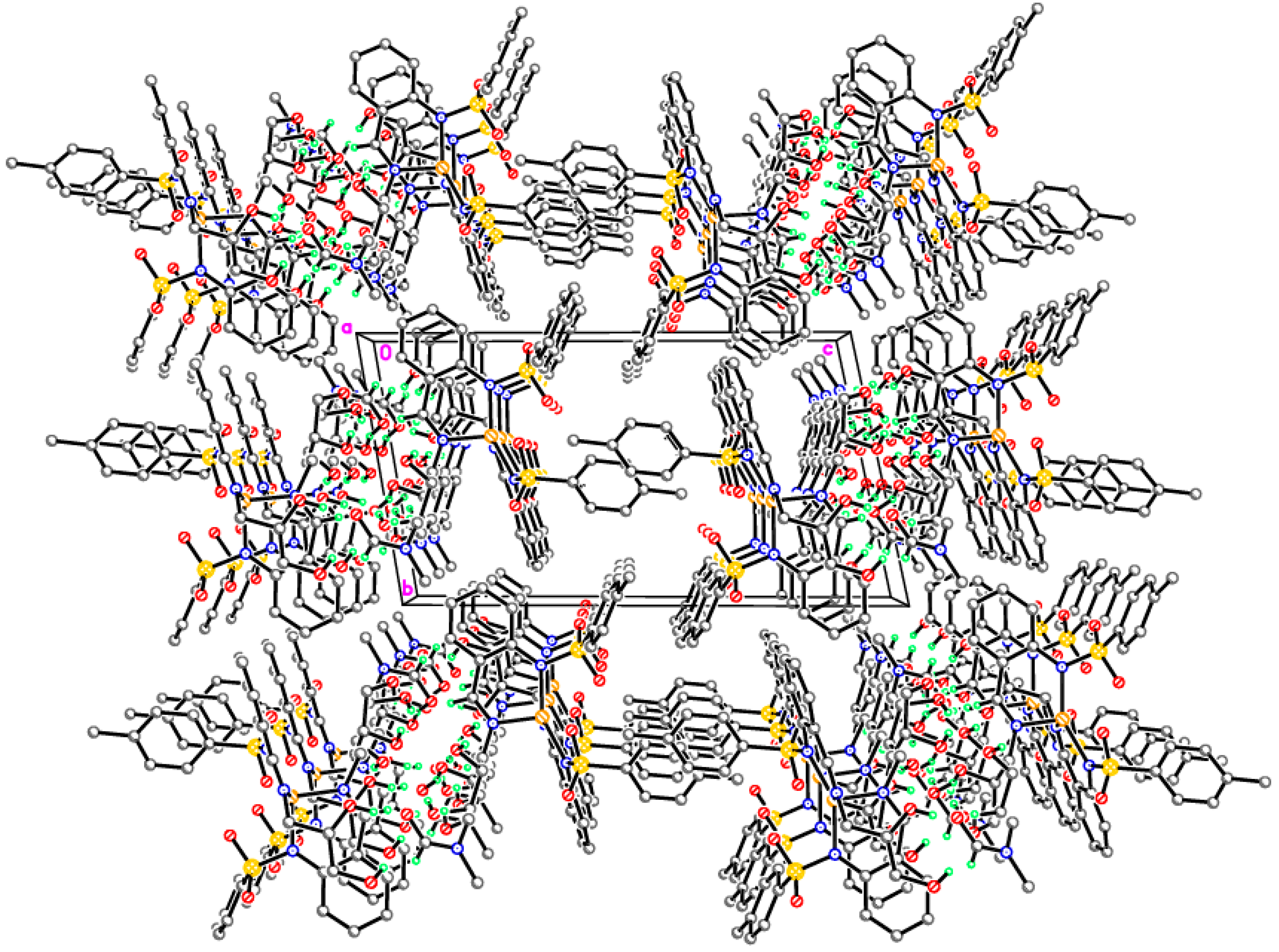
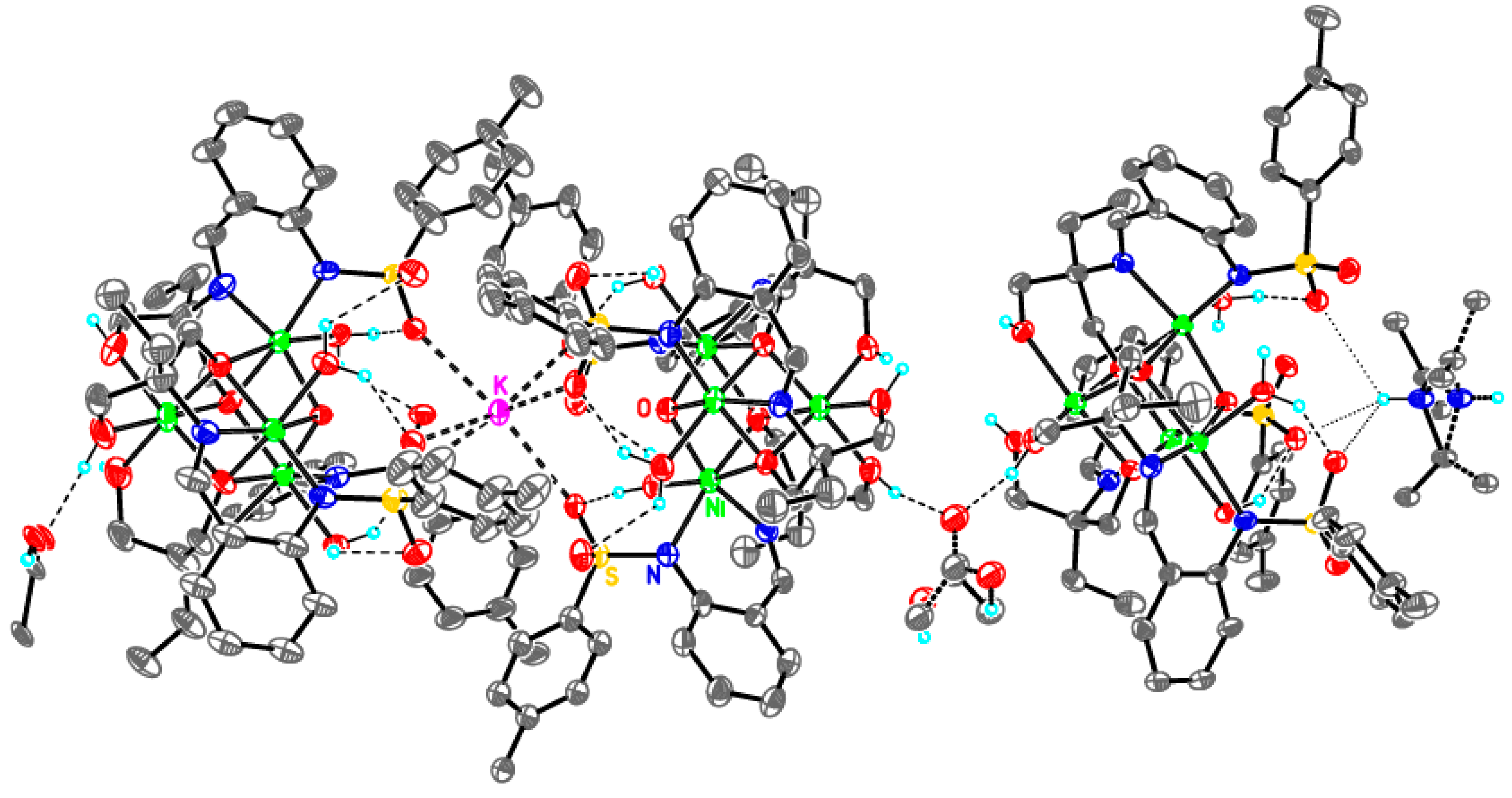
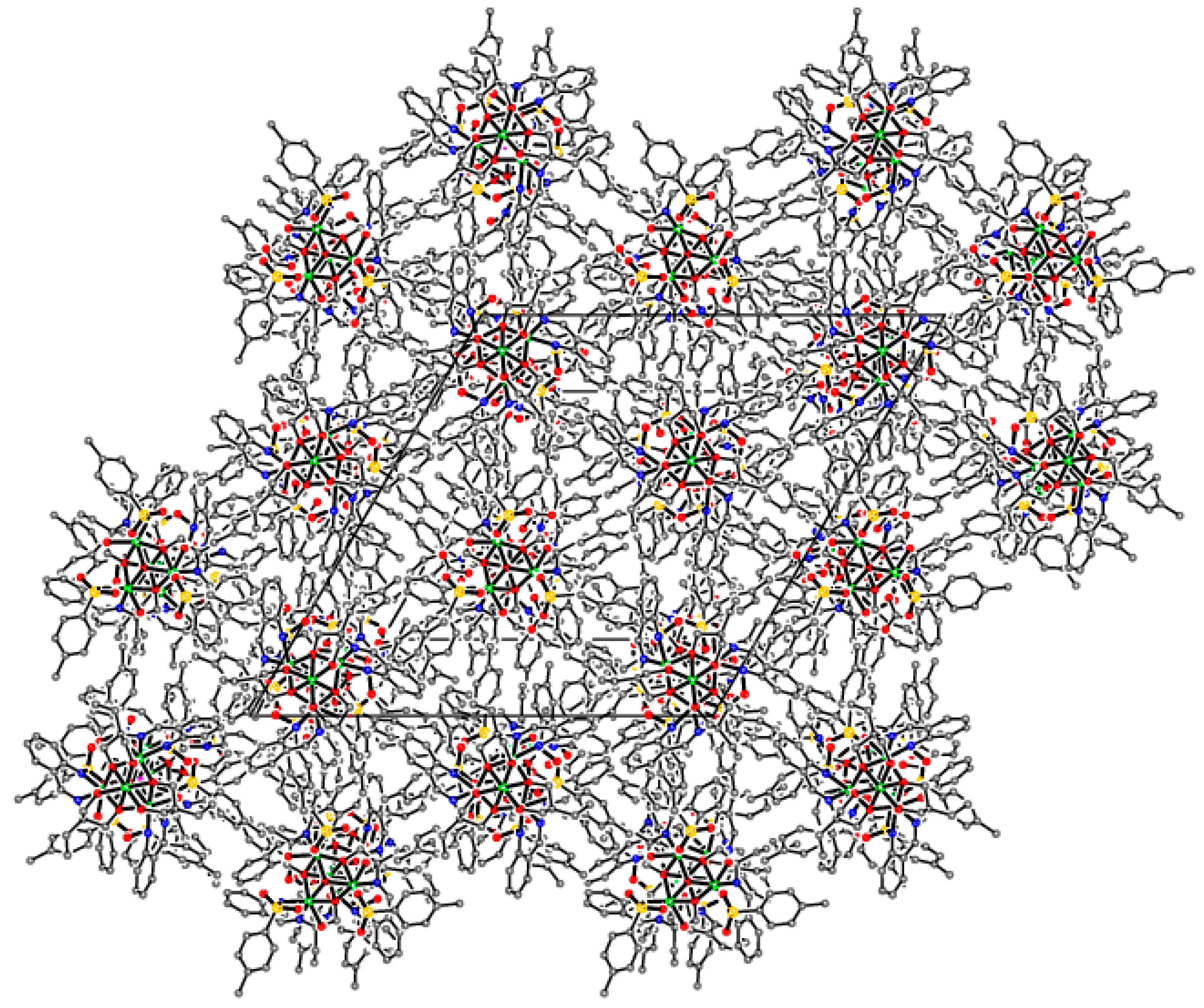
Supramolecular assembly of tetrameric clusters in 3-Ni
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Helliwell, J.R. The evolution of synchrotron radiation and the growth of its importance in crystallography. Crystallogr. Rev. 2013, 18, 32–93. [Google Scholar] [CrossRef]
- Clegg, W. Synchrotron chemical crystallography. J. Chem. Soc. Dalton Trans. 2000, 0, 3223–3232. [Google Scholar] [CrossRef]
- Helliwell, M. Anomalous scattering for small-molecule crystallography. J. Synchrotron Rad. 2000, 7, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Harding, M.M. Synchrotron radiation - new opportunities for chemical crystallography. Acta Cryst. 1995, B51, 432–446. [Google Scholar] [CrossRef]
- Coppens, P.; Cox, D.; Vlieg, E.; Robinson, I.K.; Paufler, P. Synchrotron Radiation Crystallography; Academic Press: London, UK, 1992; 316p, ISBN 0-12-188080-X. [Google Scholar]
- Nowell, H.; Barnett, S.A.; Christensen, K.E.; Teat, S.J.; Allan, D.R. I19, the small-molecule single-crystal diffraction beamline at diamond light source. J. Synchrotron Rad. 2012, 19, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Allan, D.R.; Collins, S.P.; Evans, G.; Hall, D.; McAuley, K.; Owen, R.L.; Sorensen, T.; Tang, C.C.; von Delft, F.; Wagner, A.; et al. Status of the crystallography beamlines at diamond light source. Eur. Phys. J. Plus 2015, 130, 56. [Google Scholar] [CrossRef]
- Cowieson, N.P.; Aragao, D.; Clift, M.; Ericsson, D.J.; Gee, C.; Harrop, S.J.; Mudie, N.; Panjikar, S.; Price, J.R.; Riboldi-Tunnicliffe, A.; et al. MX1: A bending-magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Rad. 2015, 22, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Eom, K.; Moon, D. BL2D-SMC, the supramolecular crystallography beamline at the Pohang Light Source II, Korea. J. Synchrotron Rad. 2016, 23, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Teat, S.J.; McCormick, L.J. Beamline 11.3.1. Small-Molecule Crystallography. Available online: https://als.lbl.gov/beamlines/11-3-1 (accessed on 10 October 2017).
- Kunz, M.; Beavers, C. Beamline 12.2.2. Diffraction Under Non-Ambient Conditions. Available online: https://als.lbl.gov/beamlines/12-2-2 (accessed on 10 October 2017).
- Raithby, P.R. Small-molecule chemical crystallography–from three to four dimensions: A personal perspective. Crystallogr. Rev. 2007, 13, 121–142. [Google Scholar] [CrossRef]
- Hoshino, M.; Adachi, S.; Koshihara, S. Crystal structure analysis of molecular dynamics using synchrotron X-rays. CrystEngComm 2015, 17, 8786–8795. [Google Scholar] [CrossRef]
- Cole, J.M. Photocrystallogaphy. Acta Cryst. 2008, A64, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, L.E.; Raithby, P.R. Dynamic single-crystal diffraction studies using synchrotron radiation. Coord. Chem. Rev. 2014, 277–278, 69–79. [Google Scholar] [CrossRef]
- Burlov, A.S.; Mal’tsev, E.I.; Vlasenko, V.G.; Garnovskii, D.A.; Dmitriev, A.V.; Lypenko, D.A.; Vannikov, A.V.; Dorovatovskii, P.V.; Lazarenko, V.A.; Zubavichus, Y.V.; et al. Synthesis, structure, photo- and electroluminescent properties of bis{(4-methyl-N-[2-[(E)-2-pyridyliminomethyl]phenyl)]benzenesulfonamide}zinc(II). Polyhedron 2017, 133, 231–237. [Google Scholar] [CrossRef]
- Burlov, A.S.; Chesnokov, V.V.; Uraev, A.I.; Garnovskii, D.A.; Vasilchenko, I.S.; Borodkin, G.S.; Vlasenko, V.G.; Dmitriev, A.V.; Lypenko, D.A.; Maltsev, E.I.; et al. Synthesis, structure, photo- and electroluminescent properties of zinc(II) complexes with aminomethylene derivatives of 1-phenyl-3-methyl-4-formylpyrazol-5-one and 3- and 6-aminoquinolines. Synth. Met. 2015, 203, 156–163. [Google Scholar] [CrossRef]
- Metelitsa, A.V.; Burlov, A.S.; Borodkina, I.G.; Bren, V.A.; Garnovskii, A.D.; Minkin, V.I.; Bezuglyi, S.O. Luminescent complexes with ligands containing C=N bond. Russ. J. Coord. Chem. 2006, 32, 858–868. [Google Scholar] [CrossRef]
- Harding, M.M. The geometry of metal-ligand interactions relevant to proteins. II. Angles at the metal atom, additional weak metal-donor interactions. Acta Cryst. 2000, D56, 857–867. [Google Scholar] [CrossRef]
- Bertini, I.; Sigel, A. (Eds.) Handbook on metalloproteins; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G.L.; Thornton, J.M. Metal ions in biological catalysis: From enzyme databases to general principles. J. Biol. Inorg. Chem. 2008, 13, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Chohan, Z.H.; Arif, M.; Sarfraz, M. Metal-based antibacterial and antifungal amino acid derived Schiff bases: Their synthesis, characterization and in vitro biological activity. Appl. Organomet. Chem. 2007, 21, 294–302. [Google Scholar] [CrossRef]
- Kaczmarek, M.T.; Jastrzab, R.; Holderna-Kedzia, E.; Radecka-Paryzek, W. Self-assembled synthesis, characterization and antimicrobial activity of zinc(II) salicylaldimine complexes. Inorg. Chim. Acta 2009, 362, 3127–3133. [Google Scholar] [CrossRef]
- Halcrow, M.A. The spin-states and spin-transitions of mononuclear iron(II) complexes of nitrogen-donor ligands. Polyhedron 2007, 26, 3523–3576. [Google Scholar] [CrossRef]
- Harding, D.J.; Harding, P.; Phonsri, W. Spin crossover in iron(III) complexes. Coord. Chem. Rev. 2016, 313, 38–61. [Google Scholar] [CrossRef]
- Ovcharenko, V.I.; Sagdeev, R.Z. Molecular ferromagnets. Russ. Chem. Rev. 1999, 68, 345–363. [Google Scholar] [CrossRef]
- Pestov, A.V.; Slepukhin, P.A.; Charushin, V.N. Copper and nickel chelate complexes with polydentate N,O-ligands: Structure and magnetic properties of polynuclear complexes. Russ. Chem. Rev. 2015, 84, 310–333. [Google Scholar] [CrossRef]
- Kogan, V.A.; Zelentsov, V.V.; Osipov, O.A.; Burlov, A.S. Dinuclear and polynuclear complexes with azomethine ligands and their magnetic properties. Russ. Chem. Rev. 1979, 48, 645–656. [Google Scholar] [CrossRef]
- Garnovskii, A.D.; Vasilchenko, I.S.; Burlov, A.S.; Uraev, A.I.; Garnovskii, D.A. Binuclear and polynuclear complexes of Schiff bases. Russ. J. Gen. Chem. 2009, 79, 2776–2786. [Google Scholar] [CrossRef]
- Burlov, A.S.; Nikolaevskii, S.A.; Vasilchenko, I.S.; Koshchienko, Y.V.; Uraev, A.I.; Sennikova, E.V.; Borodkin, G.S.; Garnovskii, A.D.; Minkin, V.I.; Bogomyakov, A.S.; et al. New magnetically active metal complexes of tridentate Schiff bases of phenylazosalicylaldehyde. Russ. J. Coord. Chem. 2009, 35, 486–491. [Google Scholar] [CrossRef]
- Popov, L.D.; Tupolova, Yu.P.; Lukov, V.V.; Shcherbakov, I.N.; Burlov, A.S.; Kogan, V.A.; Ivannikova, E.V.; Levchenkov, S.I.; Lyssenko, K.A. Physico-chemical study of first row transition metal ions coordination compounds with N,N′-bis(2-tosylaminobenzylidene)-1,3-diaminopropanol. The crystal structure of bis-azomethine and its cobalt(II) complex. Inorg. Chim. Acta 2009, 362, 1673–1680. [Google Scholar] [CrossRef]
- Emami, M.; Noshiranzadeh, N.; Bikas, R.; Gutierrez, A.; Kozakiewicz, A. Synthesis, crystal structure and magnetic studies of linear and cubane-type tetranuclear Cu(II) complexes obtained by stoichiometric control of the reagents. Polyhedron 2017, 122, 137–146. [Google Scholar] [CrossRef]
- Kheiker, D.M.; Kovalchuk, M.V.; Shilin, Yu.N.; Shishkov, V.A.; Sulyanov, S.N.; Dorovatovskii, P.V.; Rusakov, A.A. The belok station for protein crystallography on the synchrotron radiation beam from the bending magnet in the sibir-2 storage ring. Crystallogr. Rep. 2007, 52, 358–364. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. 2010, D66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. 2011, D67, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. 2011, D67, 235–242. [Google Scholar]
- Sauter, N.K.; Hattne, J.; Grosse-Kunstleve, R.W.; Echols, N. New Python-based methods for data processing. Acta Crystallogr. 2013, D69, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. 2006, D62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Safonova, T.N.; Mordkovich, N.N.; Veiko, V.P.; Okorokova, N.A.; Manuvera, V.A.; Dorovatovskii, P.V.; Popov, V.O.; Polyakov, K.M. Concerted action of two subunits of the functional dimer of Shewanella oneidensis MR-1 uridine phosphorylase derived from a comparison of the C212S mutant and the wild-type enzyme. Acta Crystallogr. 2016, D72, 203–210. [Google Scholar]
- Osipov, E.M.; Polyakov, K.M.; Tikhonova, T.V.; Kittl, R.; Dorovatovskii, P.V.; Shleev, S.V.; Popov, V.O.; Ludwig, R. Incorporation of copper ions into crystals of T2 copper-depleted laccase from Botrytis aclada. Acta Crystallogr. 2016, F71, 1465–1469. [Google Scholar]
- Glazunova, O.A.; Polyakov, K.M.; Fedorova, T.V.; Dorovatovskii, P.V.; Koroleva, O.V. Elucidation of the crystal structure of Coriolopsis caperata laccase: Restoration of the structure and activity of the native enzyme from the T2-depleted form by copper ions. Acta Crystallogr. 2015, D71, 854–861. [Google Scholar]
- Osipov., E.; Polyakov, K.; Kittl, R.; Shleev, S.; Dorovatovsky, P.; Tikhonova, T.; Hann, S.; Ludwig, R.; Popov, V. Effect of the L499M mutation of the ascomycetous Botrytis aclada laccase on redox potential and catalytic properties. Acta Crystallogr. 2013, D70, 2913–2923. [Google Scholar]
- Mitkevich, V.A.; Schulga, A.A.; Trofimov, A.A.; Dorovatovskii, P.V.; Goncharuk, D.A.; Tkach, E.N.; Makarov, A.A.; Polyakov, K.M. Structure and functional studies of the ribonuclease binase Glu43Ala/Phe81Ala mutant. Crystallogr. 2013, D69, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Tursina, A.I.; Nesterenko, S.N.; Chernyshev, V.V.; Sulyanov, S.N.; Dorovatovskii, P.V.; Britz, D.; Strydom, A.M. Synthesis, structure and physical characterization of the structural transitions in CePd3Sn and LaPd3Sn polymorphs. J. Alloys Comp. 2016, 688, 1162–1171. [Google Scholar] [CrossRef]
- Petrov, A.A.; Sokolova, I.P.; Belich, N.A.; Peters, G.S.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Khrustalev, V.N.; Petrov, A.V.; Grätzel, M.; Goodilin, E.A.; et al. Crystal structure of DMF-intermediate phases uncovers the link between CH3NH3PbI3 morphology and precursor’s stoichiometry. J. Phys. Chem. C 2017, 21, 20739–20743. [Google Scholar] [CrossRef]
- Pozhidaev, E.P.; Torgova, S.I.; Barbashov, V.A.; Minchenko, M.V.; Sulyanov, S.N.; Dorovatovskii, P.V.; Ostrovskii, B.I.; Strigazzi, A. Ferroelectric C* phase induced in a nematic liquid crystal matrix by a chiral non-mesogenic dopant. Appl. Phys. Lett. 2015, 106, 062904. [Google Scholar] [CrossRef]
- Trofimchuk, E.S.; Efimov, A.V.; Grokhovskaya, T.E.; Nikonorova, N.I.; Moskvina, M.A.; Sedush, N.G.; Dorovatovskii, P.V.; Ivanova, O.A.; Rukhlya, E.G.; Volynskii, A.L.; et al. Cold crystallization of glassy polylactide during solvent crazing. ACS Appl. Mater. Inter. 2017, 9, 34325–34336. [Google Scholar] [CrossRef] [PubMed]
- Bilyachenko, A.N.; Kulakova, A.N.; Levitsky, M.M.; Petrov, A.A.; Korlyukov, A.A.; Shul’pina, L.S.; Khrustalev, V.N.; Dorovatovskii, P.V.; Vologzhanina, A.V.; Tsareva, U.S.; et al. Unusual tri-, hexa-, and nonanuclear Cu(II) cage methylsilsesquioxanes: Synthesis, structures, and catalytic activity in oxidations with peroxides. Inorg. Chem. 2017, 56, 4093–4103. [Google Scholar] [CrossRef] [PubMed]
- Yalymov, A.I.; Bilyachenko, A.N.; Levitsky, M.M.; Korlyukov, A.A.; Khrustalev, V.N.; Shul’pina, L.S.; Dorovatovskii, P.V.; Es’kova, M.A.; Lamaty, F.; Bantreil, X.; et al. High catalytic activity of heterometallic (Fe6Na7 and Fe6Na6) cage silsesquioxanes in oxidations with peroxides. Catalysts 2017, 7, 101. [Google Scholar] [CrossRef]
- Bilyachenko, A.N.; Korlyukov, A.A.; Vologzhanina, A.V.; Khrustalev, V.N.; Kulakova, A.N.; Long, J.; Larionova, J.; Guari, Y.; Dronova, M.; Tsareva, U.; et al. Tuning linkage isomerism and magnetic properties of bi- and tri-metallic cage silsesquioxanes by cation and solvent effects. Dalt. Trans. 2017, 46, 12935–12949. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, K.A.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Timofeeva, T.V.; Khrustalev, V.N. Hydride transfer reactions of 5-(2-alkohybenzylidene) barbituric acids: Synthesis of 2,4,6-trioxoperhydropyrimidine-5-spiro-3′-chromanes. Tetrahedron 2017, 73, 542–549. [Google Scholar] [CrossRef]
- Zubkov, F.I.; Kvyatkovskaya, E.A.; Nikitina, E.V.; Amoyaw, P.; Kouznetsov, V.; Lazarenko, V.; Khrustalev, V.N. Comment on “An unexpected formation of the novel 7-oxa-2-azabicyclo[2.2.1]hept-5-ene skeleton during the reaction of furfurylamine with maleimides and their bioprospection using a zebrafish embryo model” by C. E. Puerto Galvis and V. V. Kouznetsov, Org. Biomol. Chem., 2013, 11, 407. Org. Biomol. Chem. 2017, 15, 6447–6450. [Google Scholar] [PubMed]
- Astakhov, A.V.; Khazipov, O.V.; Chernenko, A.Y.; Pasyukov, D.V.; Kashin, A.; Gordeev, E.G.; Khrustalev, V.N.; Chernyshev, V.M.; Ananikov, V.P. A new mode of operation of Pd-NHC systems studied in a catalytic mizoroki−heck reaction. Organometallics 2017, 36, 1981–1992. [Google Scholar] [CrossRef]
- Zubkevich, S.V.; Gagieva, S.C.; Tuskaev, V.A.; Dorovatovskii, P.V.; Khrustalev, V.N.; Sizov, A.I.; Bulychev, B.M. Synthesis and reactivity in ethylene oligomerization by heteroscorpionate dibromonickel (II) complexes. Inorg. Chim. Acta 2017, 458, 58–67. [Google Scholar] [CrossRef]
- Vologzhanina, A.V.; Avdeeva, V.V.; Buzin, M.I.; Dmitrienko, A.O.; Dorovatovskii, P.V.; Malinina, E.A.; Kuznetsov, N.T.; Voronova, E.D.; Zubavichus, Y.V. Solid-State reactions of eicosaborate [B20H18]2− salts and complexes. Chem. Eur. J. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Chernova, N.I.; Ryabokobylko, Y.S.; Brudz, V.G.; Bolotin, B.M. 2-Tosylaminobenzaldehyde and its substitutes. Zh. Org. Khim. 1971, 7, 1680–1687. [Google Scholar]
- Sousa-Pedrares, A.; Viqueira, J.A.; Antelo, J.; Labisbal, E.; Romero, J.; Sousa, A.; Nascimento, O.R.; García-Vázquez, J.A. Synthesis and characterization of homoleptic and heteroleptic cobalt, nickel, copper, zinc and cadmium compounds with the 2-(Tosylamino)-N-[2-(tosylamino)benzylidene]aniline ligand. Eur. J. Inorg. Chem. 2011, 2011, 2273–2287. [Google Scholar] [CrossRef]
- Labisbal, E.; Rodríguez, L.; Sousa-Pedrares, A.; Alonso, M.; Vizoso, A.; Romero, J.; García-Vázquez, J.A.; Sousa, A. Synthesis, characterisation and X-ray structures of diorganotin(IV) and iron(III) complexes of dianionic terdentate Schiff base ligands. J. Organomet. Chem. 2006, 691, 1321–1332. [Google Scholar] [CrossRef]
- Burlov, A.S.; Vlasenko, V.G.; Makarova, N.I.; Lyssenko, K.A.; Chesnokov, V.V.; Borodkin, G.S.; Vasilchenko, I.S.; Uraev, A.I.; Garnovskii, D.A.; Metelitsa, A.V. Chemical and electrochemical synthesis, molecular structures, DFT calculations and optical properties of metal-chelates of 8-(2-tosylaminobenzilideneimino)quinolone. Polyhedron 2016, 107, 153–162. [Google Scholar] [CrossRef]
- Martin, D.; Rouffet, M.; Cohen, S.M. Illuminating metal-ion sensors: Benzimidazolesulfonamide metal complexes. Inorg. Chem. 2010, 49, 10226–10228. [Google Scholar] [CrossRef] [PubMed]
- Cabaleiro, S.; Perez-Lourido, P.; Castro, J.; Romero, J.; Garcia-Vazquez, J.A.; Sousa, A. Copper(II) [(4-methylphenyl)sulfonyl]-1H-imino-(2-phenyl-2-oxazoline) complexes. Transition Met. Chem. 2001, 26, 709–716. [Google Scholar] [CrossRef]
- Dorokhov, A.V.; Chernyshov, D.Yu.; Burlov, A.S.; Garnovskii, A.D.; Ivanova, I.S.; Pyatova, E.N.; Tsivadze, A.Yu.; Aslanov, L.A.; Chernyshev, V.V. Synchrotron powder diffraction in a systematic study of 4′-[2-(tosylamino)benzylideneamino]-2,3-benzo-15-crown-5 complexes. Acta Crystallogr. 2007, B63, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Minacheva, L.K.; Ivanova, I.S.; Dorokhov, A.V.; Burlov, A.S.; Garnovsky, A.D.; Sergienko, V.S.; Tsivadze, A.Y. Zn complex with N-(4′-Benzo-15-crown-5)-2-(amino-N-tosyl)-phenylaldimine: Synthesis, crystal structure, and vibration spectrum. Russ. J. Coord. Chem. 2006, 32, 166–172. [Google Scholar] [CrossRef]
- Garcia-Vazquez, J.A.; Romero, J.; Duran, M.L.; Sousa, A.; Garnovskii, A.D.; Burlov, A.S.; Garnovskii, D.A. Electrochemical synthesis of metal (II) complexes of Schiff base 2-tosylamino (2′-pyridyl)aniline: The crystal structure of bis-[2-tosylamino(2′-pyridyl)anilinato]cobalt(II). Polyhedron 1998, 17, 1547–1552. [Google Scholar] [CrossRef]
- Castro, J.; Cabaleiro, S.; Perez-Lourido, P.; Romero, J.; Garcia-Vazquez, J.A.; Sousa, A. Electrochemical synthesis, X-ray diffraction and spectroscopic characterisation of Co(II) compounds with [(4-methylphenyl)sulfonyl]-1H-imino-2-phenyl-2-oxazolines. Polyhedron 2001, 20, 2329–2337. [Google Scholar] [CrossRef]
- Castro, J.; Cabaleiro, S.; Perez-Lourido, P.; Romero, J.; Garcia-Vazquez, J.A.; Sousa, A. Electrochemical synthesis and characterization of Zinc(II) complexes with [(4-methylphenyl)sulfonyl]-1h-amido-2-phenyl-2-oxazolines. Z. Anorg. Allg. Chem. 2002, 628, 1210–1217. [Google Scholar] [CrossRef]
- Li, J.; Tian, D.; Song, H.; Wang, C.; Zhu, X.; Cui, C.; Cheng, J.P. Synthesis, structures, and reactivity of nickel complexes incorporating sulfonamido-imine ligands. Organometallics 2008, 27, 1605–1611. [Google Scholar] [CrossRef]
- Garnovskii, A.D.; Burlov, A.S.; Garnovskii, D.A.; Vasilchenko, I.S.; Antsichkina, A.S.; Sadikov, G.G.; Sousa, A.; Garcia-Vazquez, J.A.; Romero, J.; Duran, M.L.; et al. Synthesis of cobalt(II), nickel(II) and copper(II) complexes with 2-(2-hydroxyphenyliminomethyl)-1-(4-methyl-phenylsulfonamido)benzene and crystal structures of {bis(methanol)[2-(2′-N-tosylaminophenyl)iminomethyl]phenolato}nickel(II) and {bis(2,2′-bipyridine)[2-(2′-N-tosylaminophenyl)iminomethyl]phenolato}copper(II). Polyhedron 1999, 18, 863–869. [Google Scholar]
- Antsyshkina, A.S.; Sadikov, G.G.; Burlov, A.S.; Vasil’chenko, I.S.; Garnovskii, A.D.; Sergienko, V.S. Synthesis and crystal and molecular structure of the solvate (1:4) of Bis {(methanol)(2-[2′-(N-tosylaminophenyl) iminomethyl] phenolato) cobalt (II)} with Methanol. Russ. J. Inorg. Chem. 2000, 45, 1666–1670. [Google Scholar]
- Uraev, A.I.; Vasilchenko, I.S.; Ikorskii, V.N.; Shestakova, T.A.; Burlov, A.S.; Lyssenko, K.A.; Vlasenko, V.G.; Kuz’menko, T.A.; Divaeva, L.N.; Pirog, I.V.; et al. Copper(II) dimers with ferromagnetic intra- and intermolecular exchange interactions. Mendeleev Commun. 2005, 15, 133–135. [Google Scholar] [CrossRef]
- Sanmartin, J.; Novio, F.; Garcia-Deibe, A.M.; Fondo, M.; Ocampo, N.; Bermejo, M.R. Dimeric complexes of a tridentate schiff base ligand – crystal structure of a cuii complex with uncommon μ2-Nsulfonamido bridges and ferromagnetic behaviour. Eur. J. Inorg. Chem. 2008, 2008, 1719–1726. [Google Scholar] [CrossRef]
- Jana, M.S.; Priego, J.L.; Jimenez-Aparicio, R.; Mondal, T.K. Novel tetranuclear Ni(II) schiff base complex containing Ni4O4 cubane core: Synthesis, X-ray structure, spectra and magnetic properties. Spectrochim. Acta A 2014, 133, 714–719. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1-Fe | 2-Zn | 3-Cu | 3-Ni |
|---|---|---|---|---|
| Empirical formula | C40H52FeN6O4S2 | C35.5H41.5N4.5O8.5S2Zn | C38H44Cu2N4O8S2 | C718H931K4N73Ni48O220S36 |
| fw | 800.84 | 796.72 | 875.97 | 18232.06 |
| T, K | 100 (2) | 100 (2) | 100 (2) | 100 (2) |
| Crystal size, mm | 0.20 × 0.15 × 0.02 | 0.20 × 0.15 × 0.05 | 0.20 × 0.15 × 0.10 | 0.12 × 0.10 × 0.02 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Trigonal |
| Space group | C2/c | P | P21/n | R c |
| a, Å | 17.231 (3) | 8.4379 (17) | 9.1000 (18) | 23.754 (3) |
| b, Å | 23.699 (5) | 11.489 (2) | 13.647 (3) | 23.754 (3) |
| c, Å | 11.442 (2) | 20.085 (4) | 15.877 (3) | 139.71 (3) |
| α, ° | 90 | 78.68 (3) | 90 | 90 |
| β, ° | 118.93 (3) | 86.99 (3) | 103.10 (3) | 90 |
| γ, ° | 90 | 75.58 (3) | 90 | 120 |
| V, Å3 | 4089.7 (17) | 1849.1 (7) | 1920.3 (7) | 68270 (26) |
| Z | 4 | 2 | 2 | 3 |
| dc, g cm−3 | 1.301 | 1.431 | 1.515 | 1.330 |
| F(0 0 0) | 1696 | 832 | 908 | 28518 |
| μ, mm−1 | 1.200 | 1.920 | 2.945 | 2.651 |
| θmax, ° | 38.40 | 38.44 | 38.20 | 38.54 |
| Index range | −20 < h < 20 −28 < k < 28 −14 < l < 4 | −10 < h < 8 −12 < k < 14 −25 < l < 25 | −9 < h < 10 −17 < k < 11 −19 < l < 19 | −25 < h < 30 −29 < k < 25 −176 < l < 176 |
| No. of reflections collected | 24323 | 17994 | 12853 | 223428 |
| No. of unique reflections (Rint) | 4333 (0.077) | 7593 (0.117) | 3777 (0.080) | 16597 (0.104) |
| No. of reflections with (I > 2σ(I)) | 3329 | 4327 | 2436 | 8427 |
| Data/restraints/parameters | 4333/11/240 | 7593/22/387 | 3777/38/233 | 16597/31/714 |
| R1; wR2 (I > 2σ(I)) | 0.0568; 0.1433 | 0.1175; 0.2425 | 0.0768; 0.1762 | 0.0983; 0.1870 |
| R1; wR2 (all data) | 0.0764; 0.1568 | 0.1655; 0.2822 | 0.1124; 0.1980 | 0.1662; 0.1967 |
| Goodness-of-fit (GOF) on F2 | 1.171 | 1.046 | 1.057 | 1.084 |
| Tmin; Tmax | 0.791; 0.976 | 0.700; 0.900 | 0.550; 0.750 | 0.740; 0.940 |
| Δρmax; Δρmin, eÅ−3 | 0.613; −0.802 | 1.530; −1.319 | 0.800; −0.920 | 1.206; −0.880 |
| D–H···A | d(D–H) | d(H···A) | d(D···A) | <(DHA) |
|---|---|---|---|---|
| O3–H3A···O7A a | 0.90 | 2.08 | 2.834(17) | 140 |
| O4–H4A···O8 a | 0.90 | 1.95 | 2.84(2) | 170 |
| O4–H4A···O8A a | 0.90 | 1.94 | 2.73(2) | 145 |
| O7–H7A···O3 b | 0.90 | 1.56 | 2.44(2) | 165 |
| C24–H24···O8 c | 0.95 | 2.34 | 3.23(2) | 156 |
| O3–H3A···O9 c | 0.90 | 2.14 | 2.88(2) | 139 |
| C35–H35···O4 c | 0.95 | 2.25 | 3.11(2) | 151 |
| D–H···A | d(D–H) | d(H···A) | d(D···A) | <(DHA) |
|---|---|---|---|---|
| O3–H3O···O19 | 0.92 | 1.72 | 2.616(8) | 164 |
| O6–H6A···O4 | 0.91 | 1.96 | 2.745(7) | 145 |
| O6–H6B···O5 a | 0.91 | 2.22 | 2.916(8) | 132 |
| O9–H9O···O21 | 0.91 | 1.77 | 2.67(2) | 169 |
| O12–H12D···O10 | 0.90 | 1.98 | 2.743(7) | 142 |
| O12–H12E·· O11 b | 0.92 | 2.34 | 2.950(7) | 124 |
| O15–H15O···O19 | 0.92 | 1.69 | 2.580(8) | 163 |
| O18–H18A···O17 | 0.92 | 1.90 | 2.697(7) | 145 |
| O18–H18B···N6 b | 0.92 | 2.20 | 3.051(7) | 153 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazarenko, V.A.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Burlov, A.S.; Koshchienko, Y.V.; Vlasenko, V.G.; Khrustalev, V.N. High-Throughput Small-Molecule Crystallography at the ‘Belok’ Beamline of the Kurchatov Synchrotron Radiation Source: Transition Metal Complexes with Azomethine Ligands as a Case Study. Crystals 2017, 7, 325. https://doi.org/10.3390/cryst7110325
Lazarenko VA, Dorovatovskii PV, Zubavichus YV, Burlov AS, Koshchienko YV, Vlasenko VG, Khrustalev VN. High-Throughput Small-Molecule Crystallography at the ‘Belok’ Beamline of the Kurchatov Synchrotron Radiation Source: Transition Metal Complexes with Azomethine Ligands as a Case Study. Crystals. 2017; 7(11):325. https://doi.org/10.3390/cryst7110325
Chicago/Turabian StyleLazarenko, Vladimir A., Pavel V. Dorovatovskii, Yan V. Zubavichus, Anatolii S. Burlov, Yurii V. Koshchienko, Valery G. Vlasenko, and Victor N. Khrustalev. 2017. "High-Throughput Small-Molecule Crystallography at the ‘Belok’ Beamline of the Kurchatov Synchrotron Radiation Source: Transition Metal Complexes with Azomethine Ligands as a Case Study" Crystals 7, no. 11: 325. https://doi.org/10.3390/cryst7110325
APA StyleLazarenko, V. A., Dorovatovskii, P. V., Zubavichus, Y. V., Burlov, A. S., Koshchienko, Y. V., Vlasenko, V. G., & Khrustalev, V. N. (2017). High-Throughput Small-Molecule Crystallography at the ‘Belok’ Beamline of the Kurchatov Synchrotron Radiation Source: Transition Metal Complexes with Azomethine Ligands as a Case Study. Crystals, 7(11), 325. https://doi.org/10.3390/cryst7110325