
Isomorphous Crystals from Diynes and Bromodiynes Involved in Hydrogen and Halogen Bonds

 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
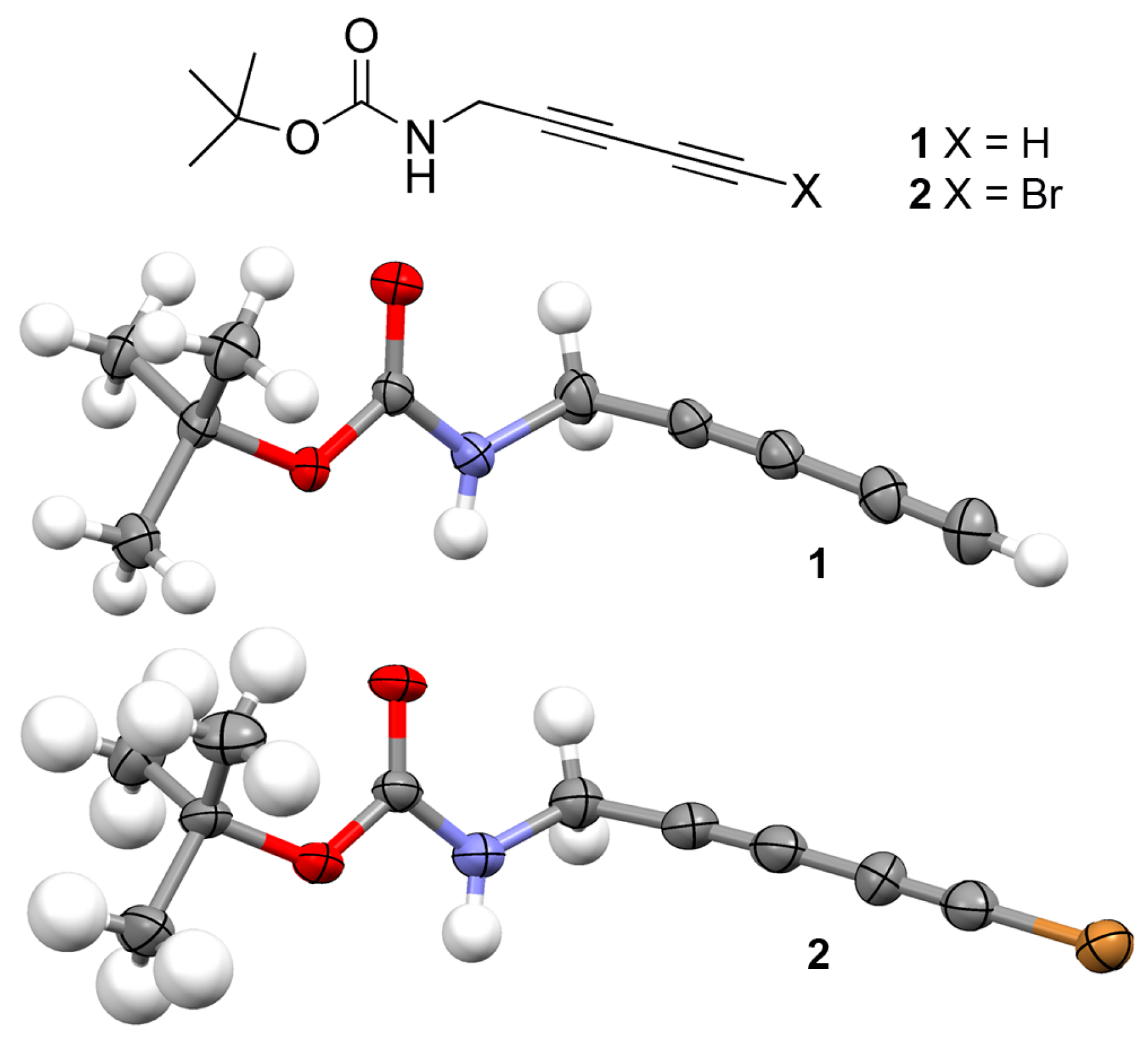
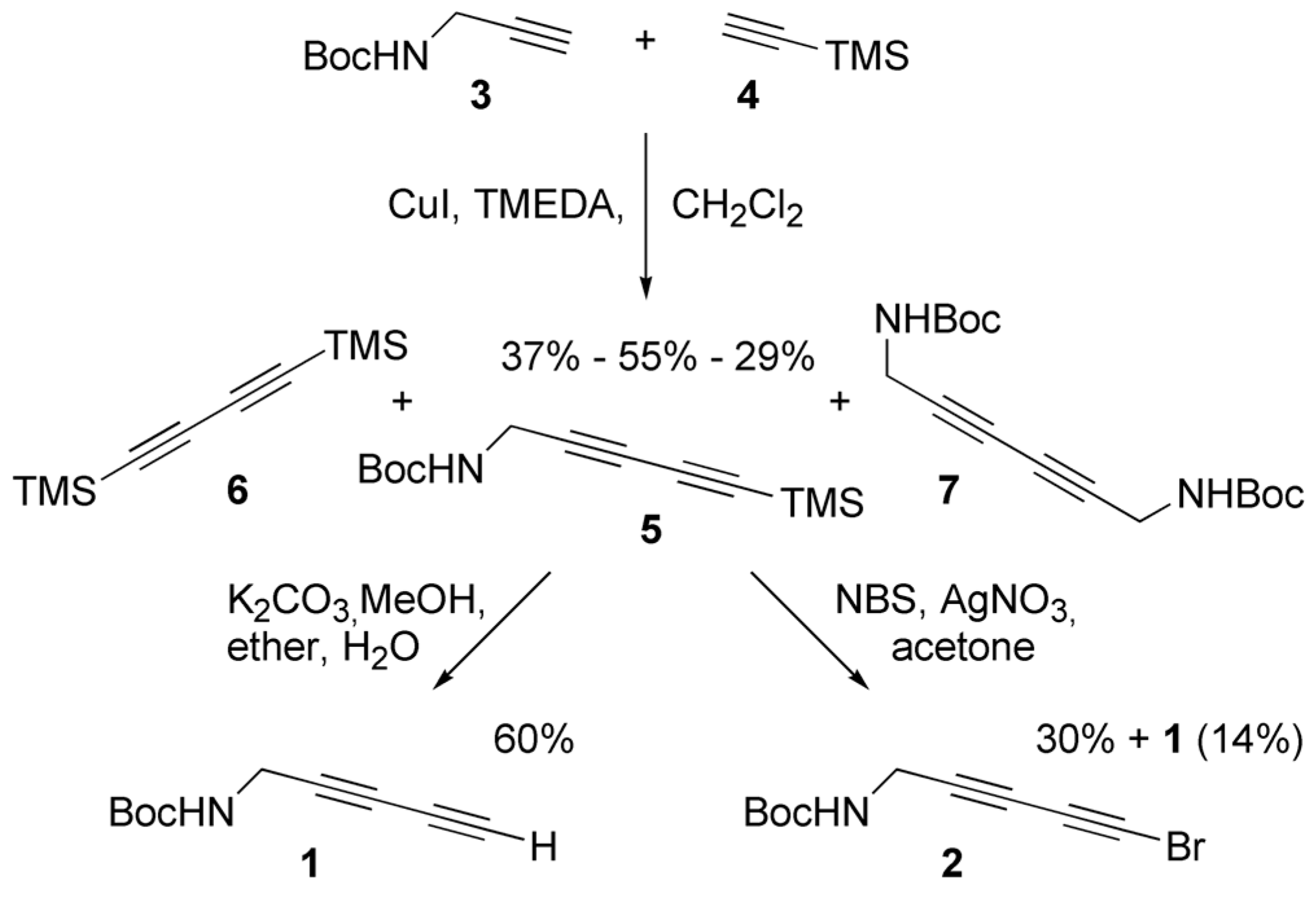
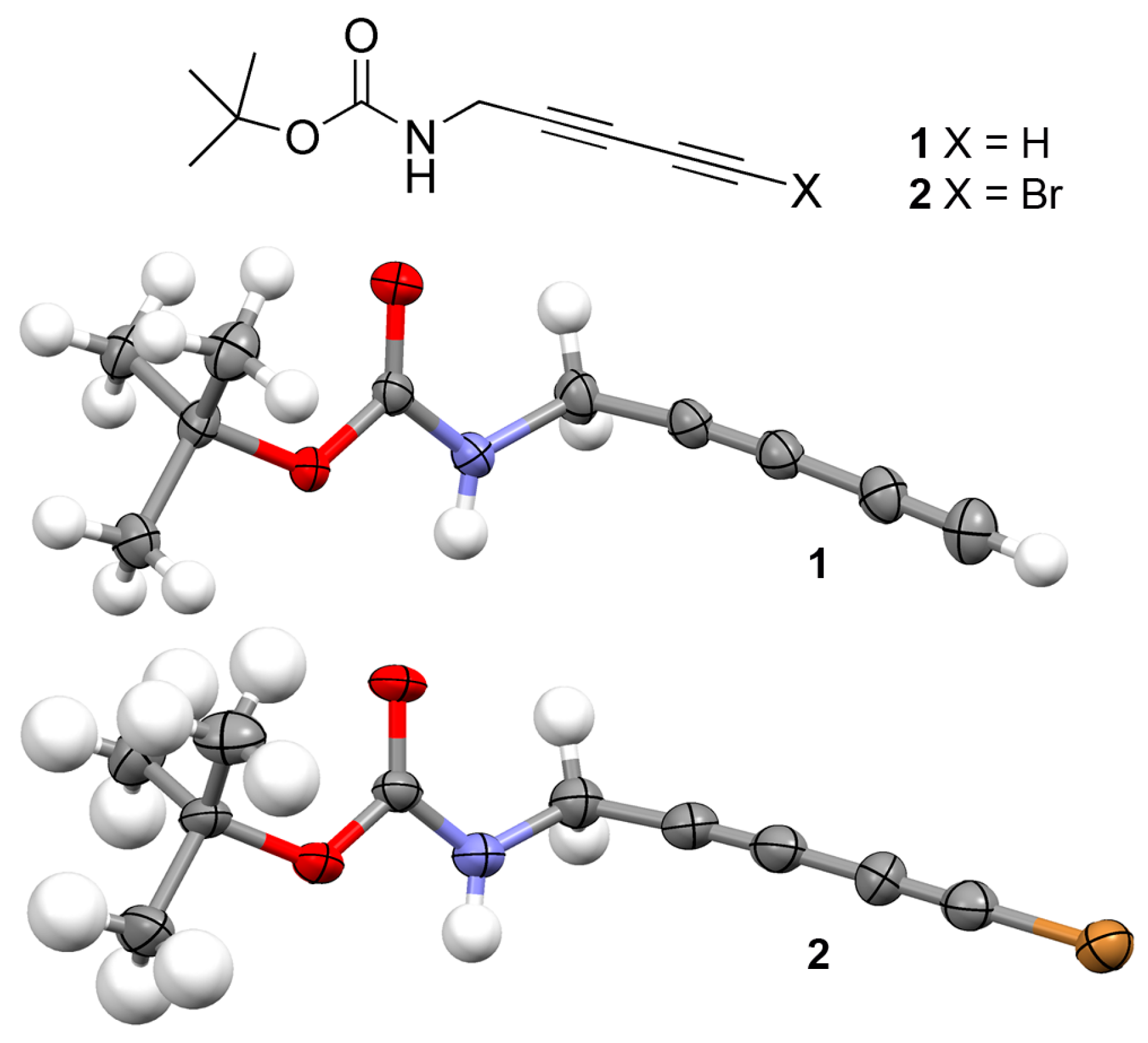
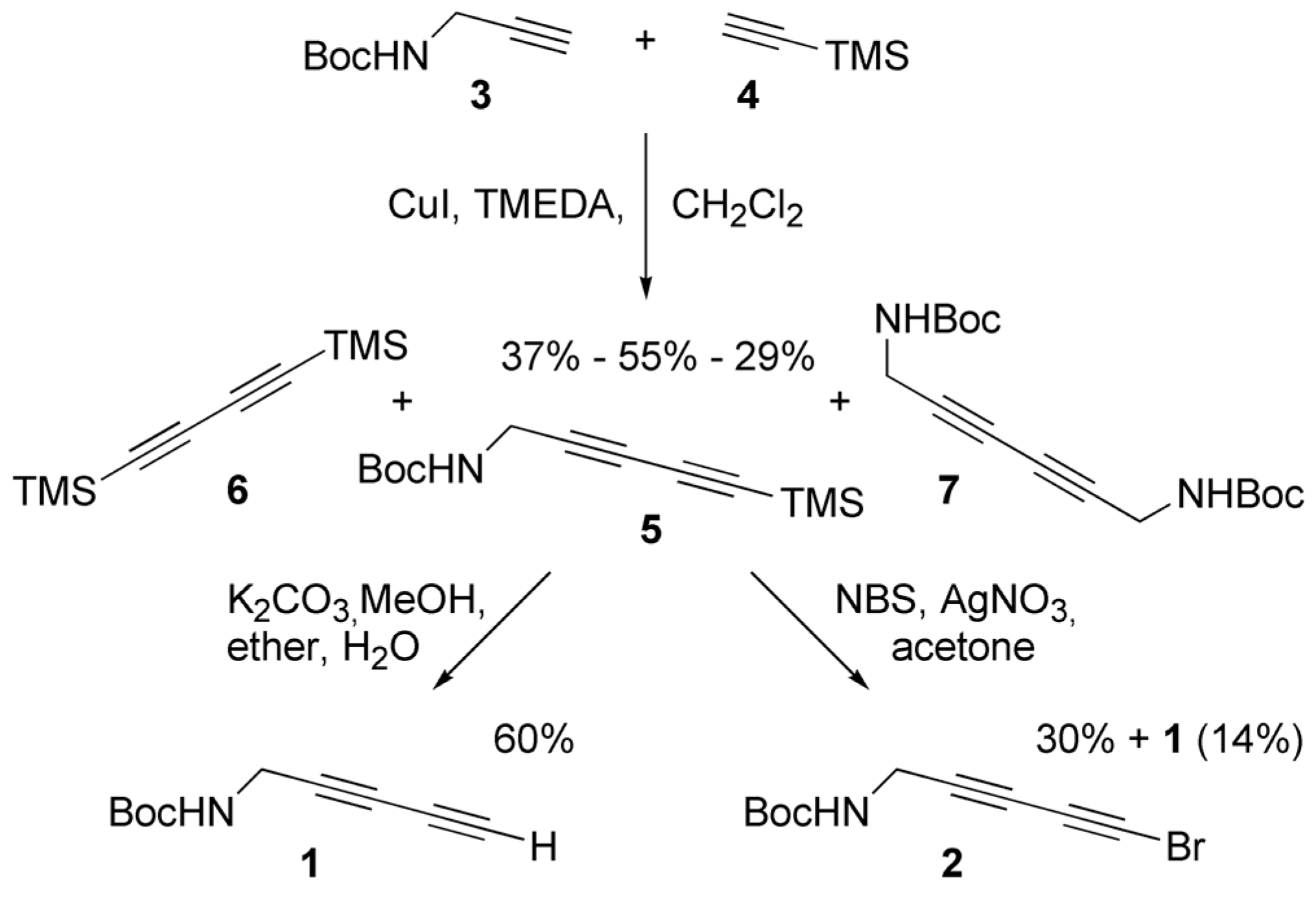
2.1. Synthesis
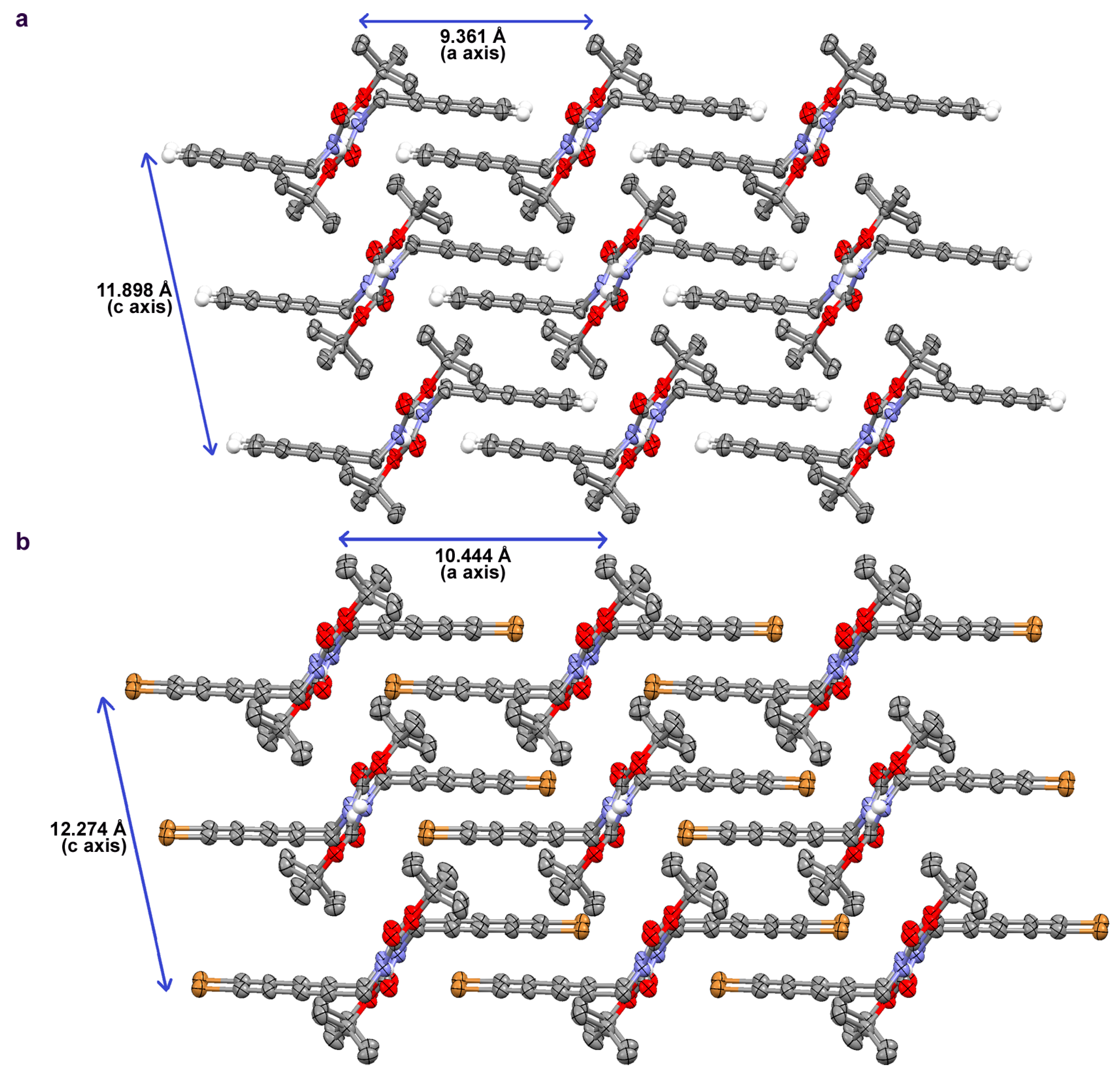
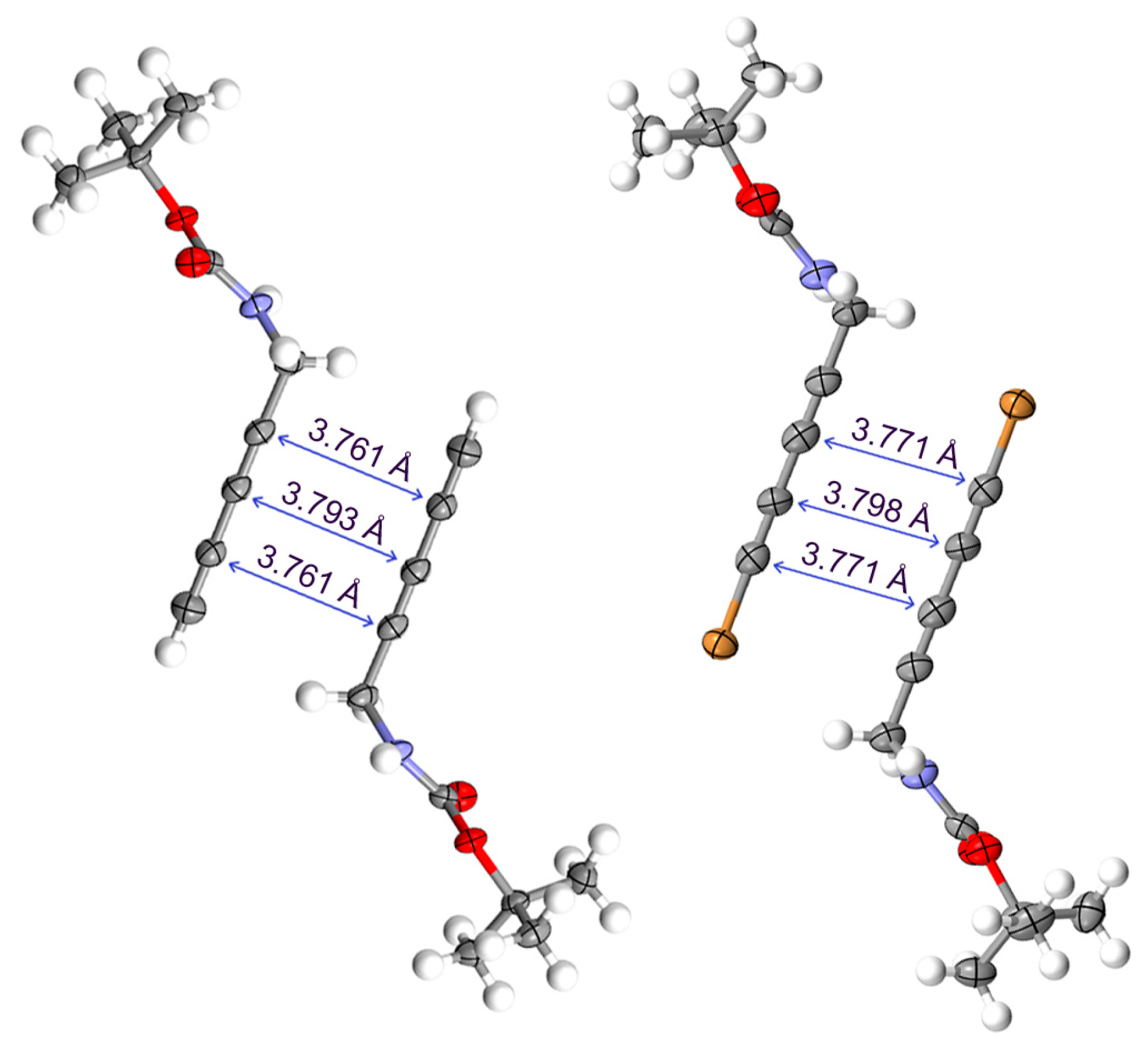
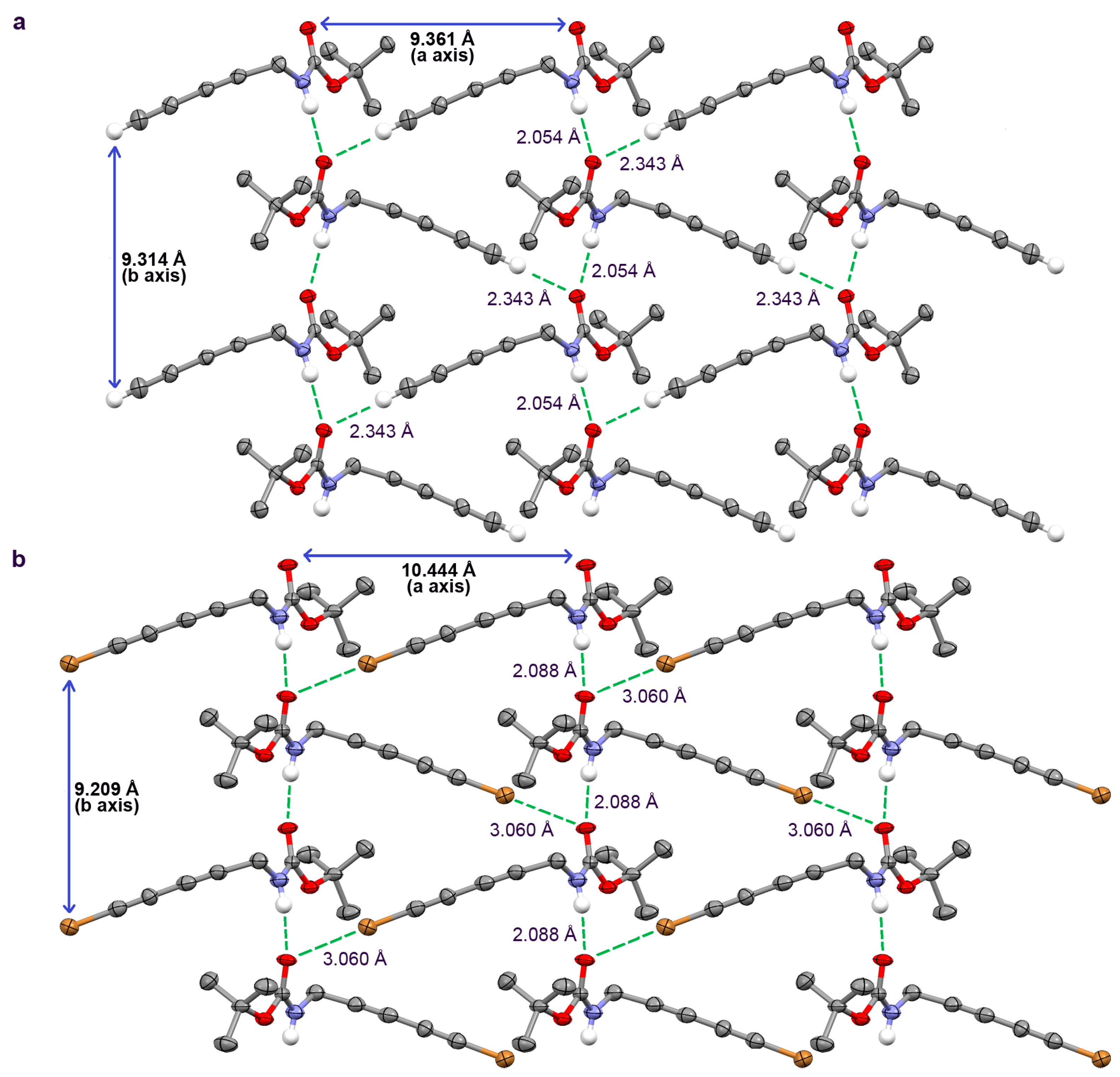
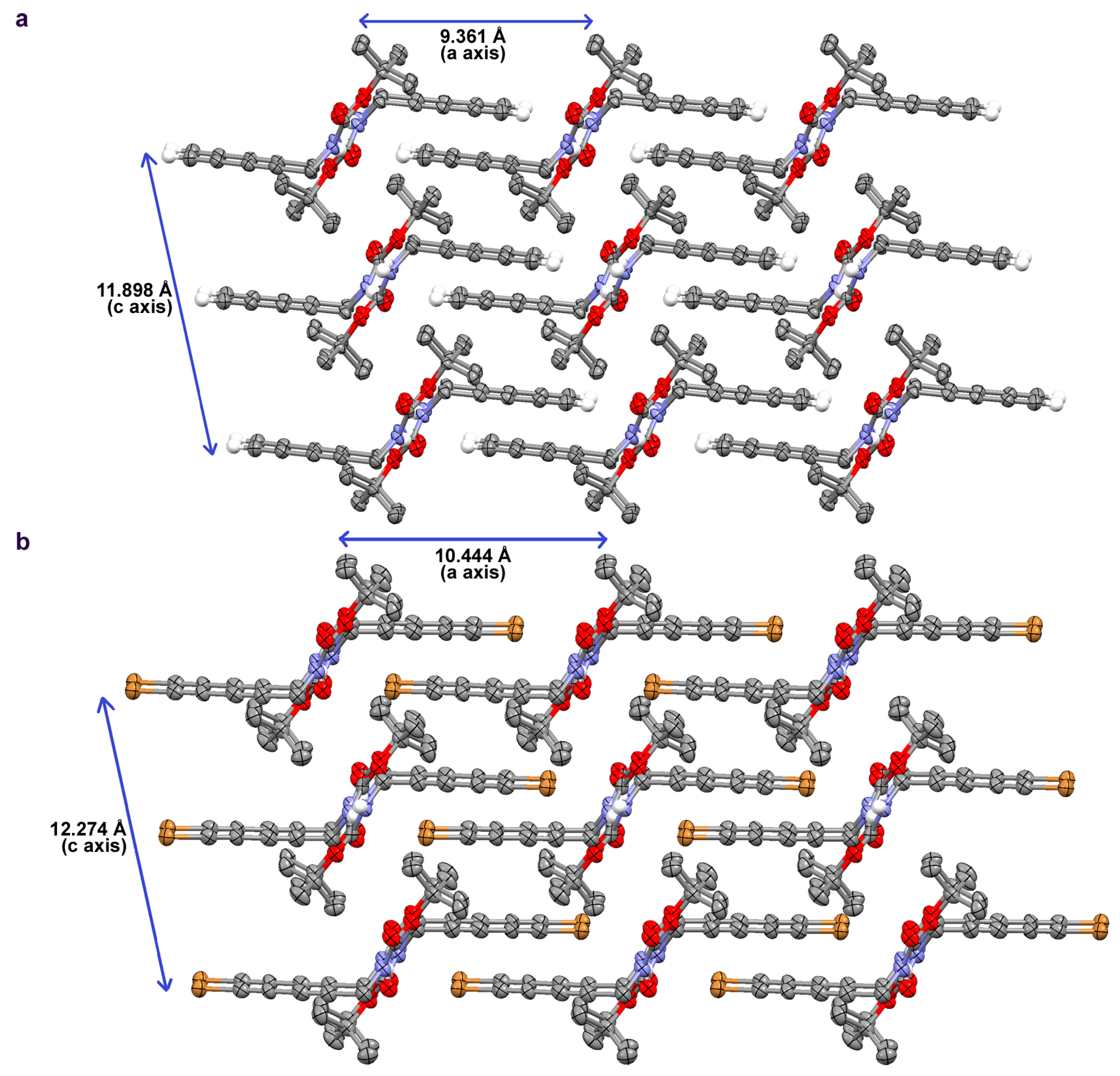
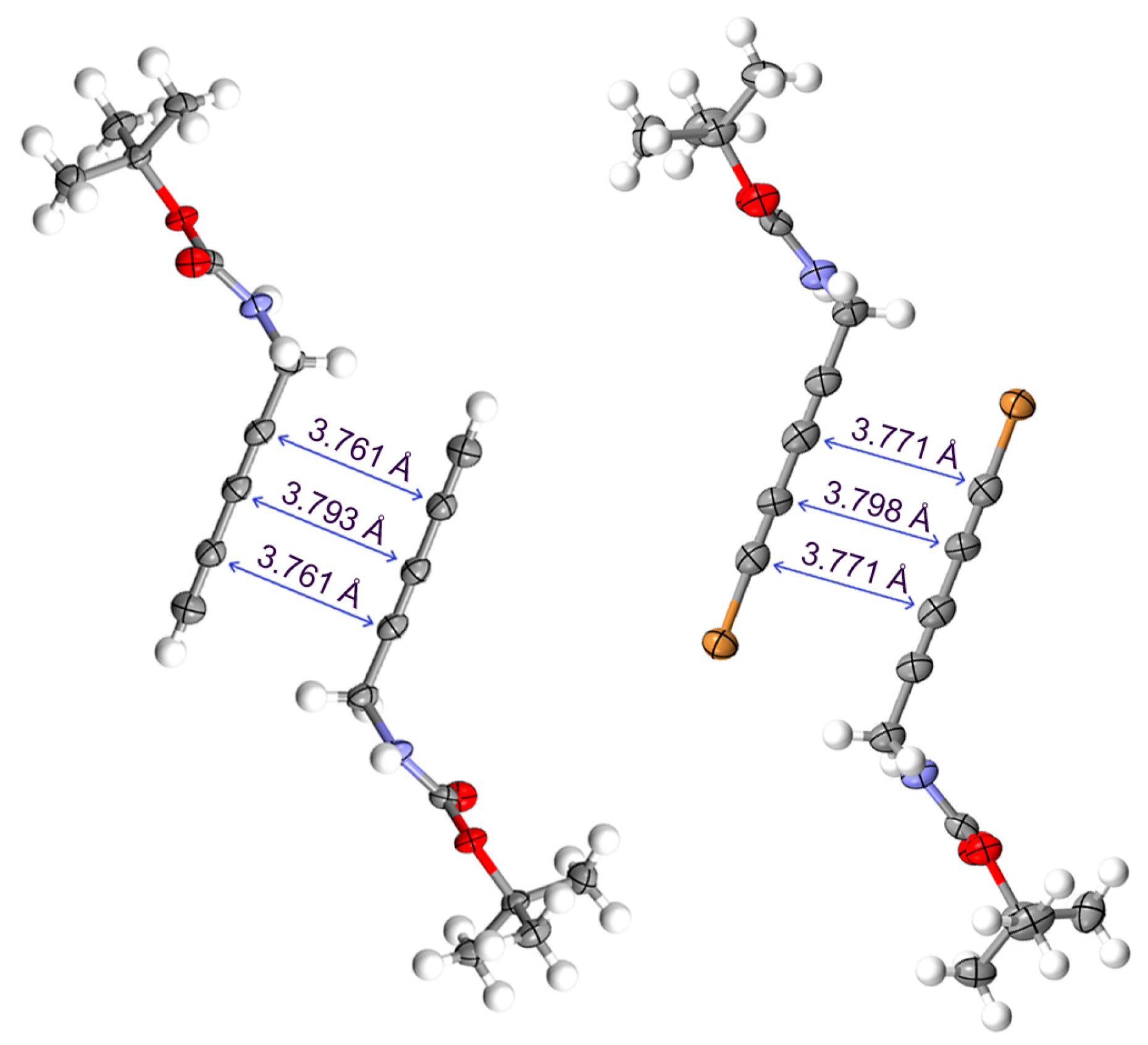
2.2. Crystallographic Studies
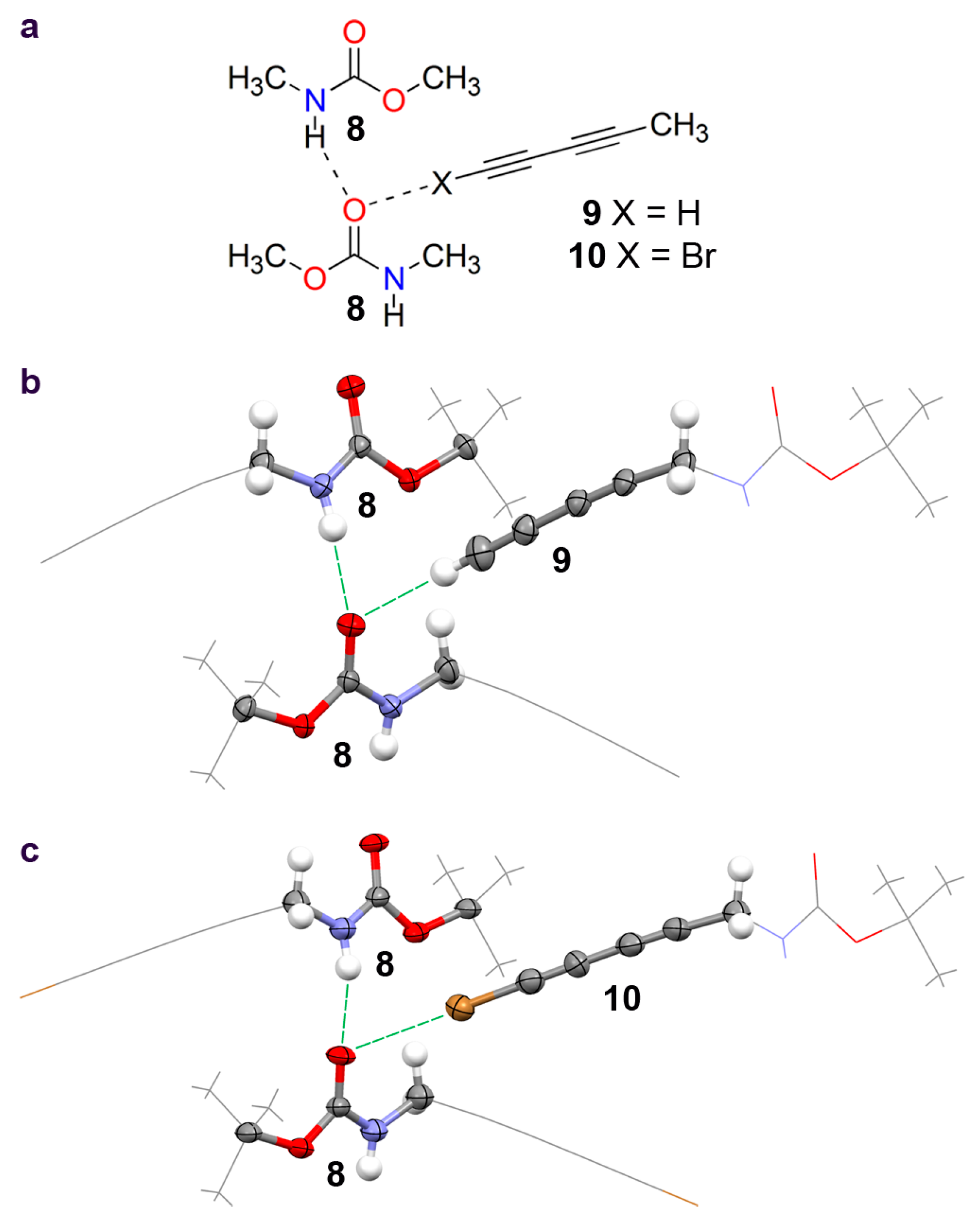
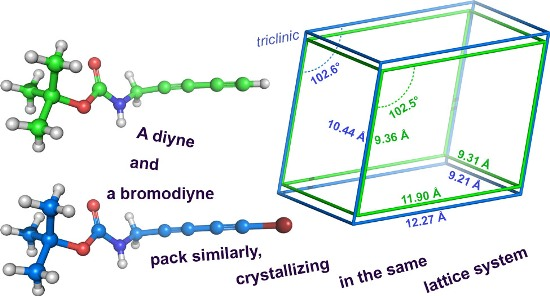
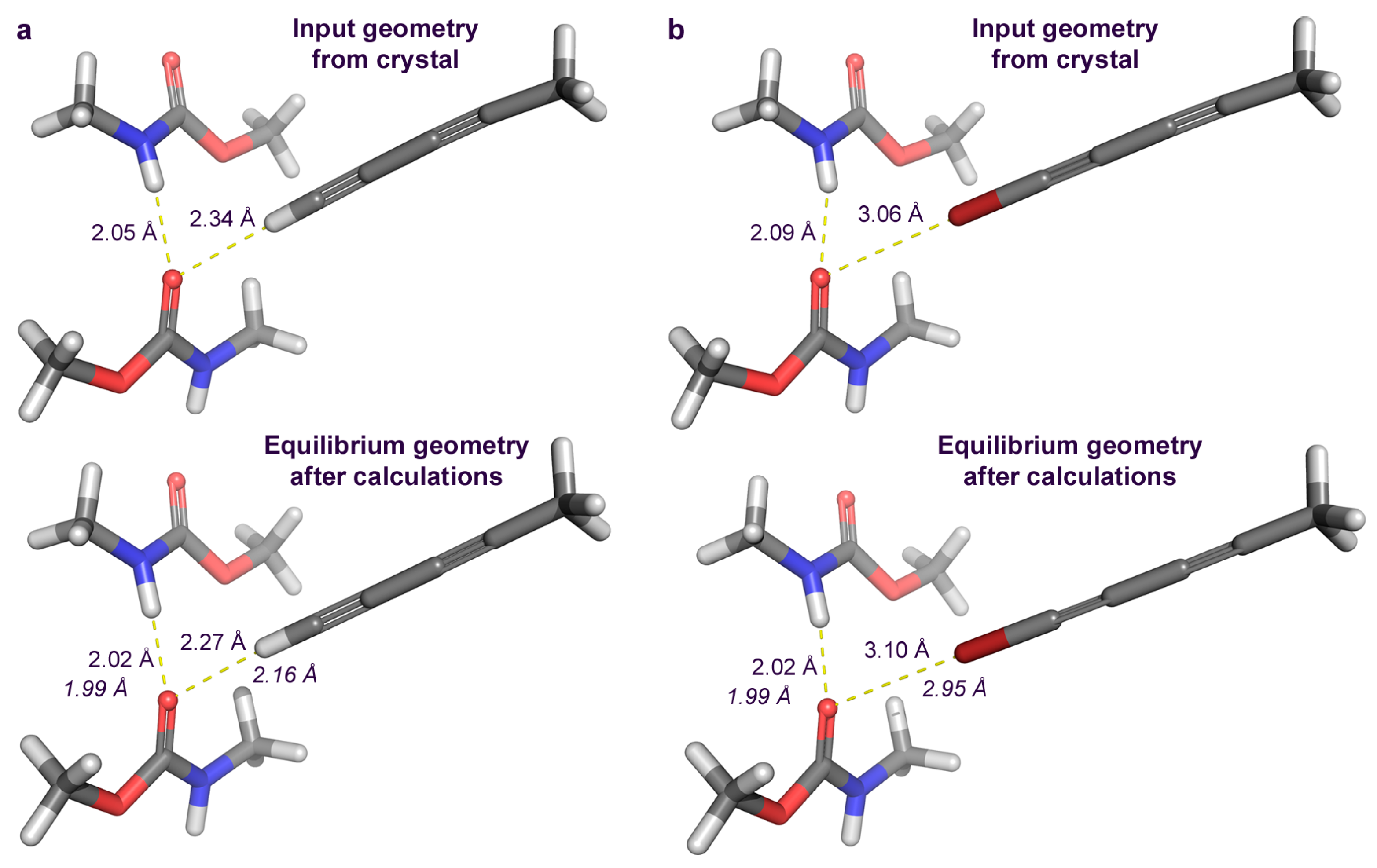
2.3. Computational Studies
3. Experimental Section
3.1. Synthesis
3.2. Crystallizations
3.3. X-Ray Crystallography
3.4. Computational Details
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| NBS | N-BromoSuccinimide |
| DFT | Density-Functional Theory |
| DCM | DiChloroMethane |
| TMEDA | TetraMethylEthyleneDiAmine |
| Rf | Retardation Factor |
| IR | InfraRed |
| NMR | Nuclear Magnetic Resonance |
| HRMS | High-Resolution Mass Spectrometry |
| GAMESS | General Atomic and Molecular Electronic Structure System |
References
- Rao, C.N.R.; Cheetham, A.K. Science and technology of nanomaterials: Current status and future prospects. J. Mater. Chem. 2001, 11, 2887–2894. [Google Scholar] [CrossRef]
- Gauthier, D.; Baillargeon, P.; Drouin, M.; Dory, Y.L. Self-Assembly of Cyclic Peptides into Nanotubes and Then into Highly Anisotropic Crystalline Materials. Angew. Chem. Int. Ed. 2001, 40, 4635–4638. [Google Scholar] [CrossRef]
- Pasini, D.; Ricci, M. Macrocycles as Precursors for Organic Nanotubes. Curr. Org. Synth. 2007, 4, 59–80. [Google Scholar] [CrossRef]
- Baillargeon, P.; Bernard, S.; Gauthier, D.; Skouta, R.; Dory, Y.L. Efficient Synthesis and Astonishing Supramolecular Architectures of Several Symmetric Macrolactams. Chem. Eur. J. 2007, 13, 9223–9235. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R. Polar Order by Rational Design: Crystal Engineering with Parallel Beloamphiphile Monolayers. Acc. Chem. Res. 2007, 40, 9–17. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Nangia, A.; Bagieu-Beucher, M.; Masse, R.; Nicoud, J.-F. Crystal engineering of two-dimensional polar layer structures: Hydrogen bond networks in some N-meta-phenylpyrimidinones. New J. Chem. 2003, 27, 568–576. [Google Scholar] [CrossRef]
- Stone, A.J.; Tsuzuki, S. Intermolecular Interactions in Strongly Polar Crystals with Layer Structures. J. Phys. Chem. B 1997, 101, 10178–10183. [Google Scholar] [CrossRef]
- Govindaraju, T.; Avinash, M.B. Two-dimensional nanoarchitectonics: Organic and hybrid materials. Nanoscale 2012, 4, 6102–6117. [Google Scholar] [CrossRef] [PubMed]
- Palmore, G.T.R.; McBride, M.T. Engineering layers in molecular solids with the cyclic dipeptide of (S)-aspartic acid. Chem. Commun. 1998, 145–146. [Google Scholar] [CrossRef]
- Hosseini, M.W. Molecular Tectonics: From Simple Tectons to Complex Molecular Networks. Acc. Chem. Res. 2005, 38, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Wuest, J.D. Engineering crystals by the strategy of molecular tectonics. Chem. Commun. 2005, 5830–5837. [Google Scholar] [CrossRef] [PubMed]
- Baillargeon, P.; Fortin, D.; Dory, Y.L. Hierarchical Self-Assembly of Lactams into Supramolecular CO-Spiked “Sea Urchins” and Then into a Channeled Crystal. Cryst. Growth Des. 2010, 10, 4357–4362. [Google Scholar] [CrossRef]
- Gordon, M.S.; Jensen, J.H. Understanding the Hydrogen Bond Using Quantum Chemistry. Acc. Chem. Res. 1996, 29, 536–543. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Taylor, R.; Kennard, O. Hydrogen-Bond Geometry in Organic Crystals. Acc. Chem. Res. 1984, 17, 320–326. [Google Scholar] [CrossRef]
- Desiraju, G.R. The C-H···O Hydrogen Bond: Structural Implications and Supramolecular Design. Acc. Chem. Res. 1996, 29, 411–449. [Google Scholar] [CrossRef] [PubMed]
- Bella, J.; Humphries, M.J. Cα-H···O=C hydrogen bonds contribute to the specificity of RGD cell-adhesion interactions. BMC Struct. Biol. 2005, 5. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.C.; Sandretto, K.L.; Bemis, G.W. Kinase Inhibitors and the Case for CH···O Hydrogen Bonds in Protein-Ligand Binding. Proteins 2002, 49, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.S.; Brandl, M.; Sühnel, J.; Pal, D.; Hilgenfeld, R. More hydrogen bonds for the (structural) biologist. Trends Biochem. Sci. 2001, 26, 521–523. [Google Scholar] [CrossRef]
- Steiner, T.; Desiraju, G.R. Distinction between the weak hydrogen bond and the van der Waals interaction. Chem. Commun. 1998, 891–892. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydrogen Bridges in Crystal Engineering: Interactions without Borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Perlstein, J.; Steppe, K.; Vaday, S.; Ndip, E.M.N. Molecular Self-Assemblies. 5. Analysis of the Vector Properties of Hydrogen Bonding in Crystal Engineering. J. Am. Chem. Soc. 1996, 118, 8433–8443. [Google Scholar] [CrossRef]
- Ji, W.; Liu, G.; Li, Z.; Feng, C. Influence of C–H···O Hydrogen Bonds on Macroscopic Properties of Supramolecular Assembly. ACS Appl. Mater. Interfaces 2016, 8, 5188–5195. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen Bonding Based Recognition Processes: A World Parallel to Hydrogen Bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.C. Prereactive Complexes of Dihalogens XY with Lewis Bases B in the Gas Phase: A Systematic Case for the Halogen Analogue B···XY of the Hydrogen Bond B···HX. Angew. Chem. Int. Ed. 1999, 38, 2686–2714. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Awwadi, F.F.; Willett, R.D.; Peterson, K.A.; Twamley, B. The Nature of Halogen···Halogen Synthons: Crystallographic and Theoretical Studies. Chem. Eur. J. 2006, 12, 8952–8960. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Pilati, T.; Resnati, G. Halogen bonding and other noncovalent interactions involving halogens: A terminology issue. CrystEngComm 2006, 8, 946–947. [Google Scholar] [CrossRef]
- Dey, A.; Jetti, R.K.R.; Boese, R.; Desiraju, G.R. Supramolecular equivalence of halogen, ethynyl and hydroxy groups. A comparison of the crystal structures of some 4-substituted anilines. CrystEngComm 2003, 5, 248–252. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G.; Pilati, T.; Liantonio, R.; Meyer, F. Engineering Functional Materials by Halogen Bonding. J. Polym. Sci. A Polym. Chem. 2007, 45, 1–15. [Google Scholar] [CrossRef]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Janjić, G.V.; Zarić, S.D. σ-Hole Interactions of Covalently-Bonded Nitrogen, Phosphorus and Arsenic: A Survey of Crystal Structures. Crystals 2014, 4, 12–31. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Baldrighi, M.; Desper, J.; Metrangolo, P.; Resnati, G. Supramolecular Hierarchy among Halogen-Bond Donors. Chem. Eur. J. 2013, 19, 16240–16247. [Google Scholar] [CrossRef] [PubMed]
- Lieffrig, J.; Jeannin, O.; Frackowiak, A.; Olejniczak, I.; Swietlik, R.; Dahaoui, S.; Aubert, E.; Espinosa, E.; Auban-Senzier, P.; Fourmigué, M. Charge-Assisted Halogen Bonding: Donor-Acceptor Complexes with Variable Ionicity. Chem. Eur. J. 2013, 19, 14804–14813. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Baillargeon, P.; Dory, Y.L. Supramolecular Walls from Cyclic Peptides: Modulating Nature and Strength of Weak Interactions. Cryst. Growth Des. 2009, 9, 3638–3645. [Google Scholar] [CrossRef]
- Baillargeon, P.; Lussier, T.; Dory, Y.L. Hydrogen Bonds between Acidic Protons from Alkynes (C–H···O) and Amides (N–H···O) and Carbonyl Oxygen Atoms as Acceptor Partners. J. Crystallogr. 2014, 2014, 1–5. [Google Scholar] [CrossRef]
- SeethaLekshmi, S.; Varughese, S.; Girl, L.; Pedireddi, V.R. Molecular Complexes of 4-Halophenylboronic Acids: A Systematic Exploration of Isostructurality and Structural Landscape. Cryst. Growth Des. 2014, 14, 4143–4154. [Google Scholar] [CrossRef]
- Dechambenoit, P.; Ferlay, S.; Kyritsakas, N.; Hosseini, M.W. Playing with isostructurality: From tectons to molecular alloys and composite crystals. Chem. Commun. 2009, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Schultheiss, N.C.; Rajbanshi, A.; Desper, J.; Moore, C. Supramolecular Synthesis Based on a Combination of Hydrogen and Halogen Bonds. Cryst. Growth Des. 2009, 9, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, S.; Muthiah, P.T.; Butcher, R.J. Design of a Series of Isostructural Co-Crystals with Aminopyrimidines: Isostructurality through Chloro/Methyl Exchange and Studies on Supramolecular Architectures. Cryst. Growth Des. 2011, 11, 3579–3592. [Google Scholar] [CrossRef]
- Ekkebus, R.; van Kasteren, S.I.; Kulathu, Y.; Scholten, A.; Berlin, I.; Geurink, P.P.; de Jong, A.; Goerdayal, S.; Neefjes, J.; Heck, A.J.R.; et al. On Terminal Alkynes that Can React with Active-Site Cysteine Nucleophiles in Proteases. J. Am. Chem. Soc. 2013, 135, 2867–2870. [Google Scholar] [CrossRef] [PubMed]
- Sommer, S.; Weikart, N.D.; Linne, U.; Mootz, H.D. Covalent inhibition of SUMO and ubiquitin-specific cysteine proteases by an in situ thiol-alkyne addition. Bioorg. Med. Chem. 2013, 21, 2511–2517. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Jiang, H. Haloalkynes: A Powerful and Versatile Building Block in Organic Synthesis. Acc. Chem. Res. 2014, 47, 2483–2504. [Google Scholar] [CrossRef] [PubMed]
- Mevers, E.; Liu, W.-T.; Engene, N.; Mohimani, H.; Byrum, T.; Pevzner, P.A.; Dorrestein, P.C.; Spadafora, C.; Gerwick, W.H. Cytotoxic Veraguamides, Alkynyl Bromide-Containing Cyclic Depsipeptides from the Marine Cyanobacterium cf. Oscillatoria margaritifera. J. Nat. Prod. 2011, 74, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Biggs, J.S.; Paul, V.J.; Luesch, H. Veraguamides A-G, Cyclic Hexadepsipeptides from a Dolastatin 16-Producing Cyanobacterium Symploca cf. hydnoides from Guam. J. Nat. Prod. 2011, 74, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.S. Oxidative Coupling of Acetylenes II. J. Org. Chem. 1962, 27, 3320–3321. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Non-conventional hydrogen bonds. Chem. Soc. Rev. 1998, 27, 163–170. [Google Scholar] [CrossRef]
- Hassel, O. Structural Aspects of Interatomic Charge-Transfer Bonding. Science 1970, 170, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Bolton, O.; Lee, K.; Kim, H.-J.; Lin, K.Y.; Kim, J. Activating efficient phosphorescence from purely organic materials by crystal design. Nat. Chem. 2011, 3, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G. Halogen Bonding: A Paradigm in Supramolecular Chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Restani, R. The Halogen Bond in the Design of Functional Supramolecular Materials: Recent Advances. Acc. Chem. Res. 2013, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Cody, V.; Murray-Rust, P. Iodine···X(O, N, S) intermolecular contacts: Models of thyroid hormone protein binding interactions using information from the cambridge crystallographic data files. J. Mol. Struct. 1984, 112, 189–199. [Google Scholar] [CrossRef]
- Ouvrard, C.; le Questel, J.-Y.; Berthelot, M.; Laurence, C. Halogen-bond geometry: A crystallographic data-base investigation of dihalogen complexes. Acta Crystallogr. B 2003, 59, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Lommerse, J.P.M.; Stone, A.J.; Taylor, R.; Allen, F.H. The Nature and Geometry of Intermolecular Interactions between Halogens and Oxygen or Nitrogen. J. Am. Chem. Soc. 1996, 118, 3108–3116. [Google Scholar] [CrossRef]
- Hoheisel, T.N.; Schrettl, S.; Marty, R.; Todorova, T.K.; Corminboeuf, C.; Sienkiewicz, A. A multistep single-crystal-to-single-crystal bromodiacetylene dimerization. Nat. Chem. 2013, 5, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Curtis, S.M.; Le, N.; Nguyen, T.; Ouyang, X.; Tran, T.; Fowler, F.W.; Lauher, J.W. What have We Learned about Topochemical Diacetylene Polymerizations? Supramol. Chem. 2005, 17, 31–36. [Google Scholar] [CrossRef]
- Okuno, T.; Yamane, K.; Sandman, D.J. Solid State Polymerization of Diacetylenes with Amide Groups. Mol. Cryst. Liq. Cryst. 2006, 456, 45–53. [Google Scholar] [CrossRef]
- Luo, L.; Wilhelm, C.; Sun, A.; Grey, C.P.; Lauher, J.W.; Goroff, N.S. Poly(diiododiacetylene): Preparation, isolation, and full characterization of a very simple poly(diacetylene). J. Am. Chem. Soc. 2008, 130, 7702–7709. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Fowler, F.W.; Lauher, J.W. Weak Interactions Dominating the Supramolecular Self-Assembly in a Salt: A Designed Single-Crystal-to-Single-Crystal Topochemical Polymerization of a Terminal Aryldiacetylene. J. Am. Chem. Soc. 2009, 131, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Plonka, A.M.; Parise, J.B.; Goroff, N.S. Pressure induced topochemical polymerization of diiodobutadiyne: A single-crystal-to-single-crystal transformation. CrystEngComm 2013, 15, 3106–3110. [Google Scholar] [CrossRef]
- Haridas, V.; Sadanandan, S.; Collart-Dutilleul, P.-Y.; Gronthos, S.; Voelcker, N.H. Lysine-Appended Polydiacetylene Scaffolds for Human Mesenchymal Stem Cells. Biomacromolecules 2014, 15, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.L.; Smith, M.D.; Krause, J.A.; Greytak, A.B.; Ma, S.; Read, C.M.; Shimizu, L.S. Single Crystal to Single Crystal Polymerization of a Self-Assembled Diacetylene Macrocycle Affords Columnar Polydiacetylenes. Cryst. Growth Des. 2014, 14, 993–1002. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Liu, H.; Li, J.; Li, T.; Wu, Y.; Okada, S.; Nakanishi, H. Topochemical polymerization of unsymmetrical aryldiacetylene supramolecules with nitrophenyl substituents utilizing C–H···π interactions. Org. Biomol. Chem. 2015, 13, 5467–5474. [Google Scholar] [CrossRef] [PubMed]
- Lauher, J.W.; Fowler, F.W. Single-Crystal-to-Single-Crystal Topochemical Polymerizations by Design. Acc. Chem. Res. 2008, 41, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, R.; Ritenberg, M. Polydiacetylenes—Recent molecular advances and applications. RSC Adv. 2013, 3, 21192–21201. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L. Self-Consistent Equations Including Exchange and Correlation Effects. J. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Becke, A.D. Perspective: Fifty years of density-functional theory in chemical physics. J. Phys. Chem. 2014, 140, 18A301. [Google Scholar] [CrossRef] [PubMed]
- Rezác, J.; Hobza, P. Benchmark Calculations of Interaction Energies in Noncovalent Complexes and Their Applications. Chem. Rev. 2016. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016. [Google Scholar] [CrossRef]
- Feldblum, E.S.; Arkin, I.T. Strength of a bifurcated H bond. Proc. Natl. Acad. Sci. 2014, 111, 4085–4090. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsson, H.; Maulitz, A.H.; Bruice, T.C. Calculation of the Potential Energy Surface for Intermolecular Amide Hydrogen Bonds Using Semiempirical and Ab Initio Methods. J. Am. Chem. Soc. 1996, 118, 7689–7693. [Google Scholar] [CrossRef]
- Li, A.; Muddana, H.S.; Gilson, M.K. Quantum Mechanical Calculation of Noncovalent Interactions: A Large-Scale Evaluation of PMx, DFT, and SAPT Approaches. J. Chem. Theory Comput. 2014, 10, 1563–1575. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, B.; Spiteller, M. Binding affinity of terrestrial and aquatic humics toward organic xenobiotics. J. Environ. Chem. Eng. 2016, 4, 498–510. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Shirman, T.; Boterashvili, M.; Orbach, M.; Freeman, D.; Shimon, L.J.W.; Lahav, M.; van der Boom, M.E. Finding the Perfect Match: Halogen vs. Hydrogen Bonding. Cryst. Growth Des. 2015, 15, 4756–4759. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diyne 1 | Diyne 2 | |
|---|---|---|
| formula | C10H13NO2 | C10H12BrNO2 |
| MW/g·mol−1 | 179.21 | 258.12 |
| crystal system | monoclinic | monoclinic |
| space group | P 21/c | P 21/c |
| a/Å | 9.3613(15) | 10.4435(16) |
| b/Å | 9.3135(14) | 9.2090(15) |
| c/Å | 11.8981(19) | 12.2744(19) |
| β/deg | 102.497(5) | 102.599(4) |
| V/Å3 | 1012.8(3) | 1152.1(3) |
| Z | 4 | 4 |
| ρcalc/g·cm−3 | 1.175 | 1.488 |
| meas. reflns | 5204 | 16523 |
| ind. reflns | 1848 | 2175 |
| Rint | 0.0403 | 0.0656 |
| R1 [I > 2σ(I)] | 0.0382 | 0.0644 |
| wR2 [I > 2σ(I)] | 0.0840 | 0.1662 |
| GoF | 1.027 | 1.133 |
| System | Energy (Eh) 1 | |
|---|---|---|
| B3LYP | M06-2X | |
| 8 | −323.59792 | −323.62775 |
| 9 | −192.70191 | −192.73108 |
| 10 | −2765.83708 | −2766.01159 |
| 8-8 | −647.20438 | −647.26691 |
| 8-8-9 | −839.90921 | −840.00619 |
| 8-8-10 | −3413.04394 | −3413.28685 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baillargeon, P.; Caron-Duval, É.; Pellerin, É.; Gagné, S.; Dory, Y.L. Isomorphous Crystals from Diynes and Bromodiynes Involved in Hydrogen and Halogen Bonds. Crystals 2016, 6, 37. https://doi.org/10.3390/cryst6040037
Baillargeon P, Caron-Duval É, Pellerin É, Gagné S, Dory YL. Isomorphous Crystals from Diynes and Bromodiynes Involved in Hydrogen and Halogen Bonds. Crystals. 2016; 6(4):37. https://doi.org/10.3390/cryst6040037
Chicago/Turabian StyleBaillargeon, Pierre, Édouard Caron-Duval, Émilie Pellerin, Simon Gagné, and Yves L. Dory. 2016. "Isomorphous Crystals from Diynes and Bromodiynes Involved in Hydrogen and Halogen Bonds" Crystals 6, no. 4: 37. https://doi.org/10.3390/cryst6040037
APA StyleBaillargeon, P., Caron-Duval, É., Pellerin, É., Gagné, S., & Dory, Y. L. (2016). Isomorphous Crystals from Diynes and Bromodiynes Involved in Hydrogen and Halogen Bonds. Crystals, 6(4), 37. https://doi.org/10.3390/cryst6040037