Magnetic and Electric Properties of Organic Conductors Probed by 13C-NMR Using Selective-Site Substituted Molecules

Abstract
:1. Introduction

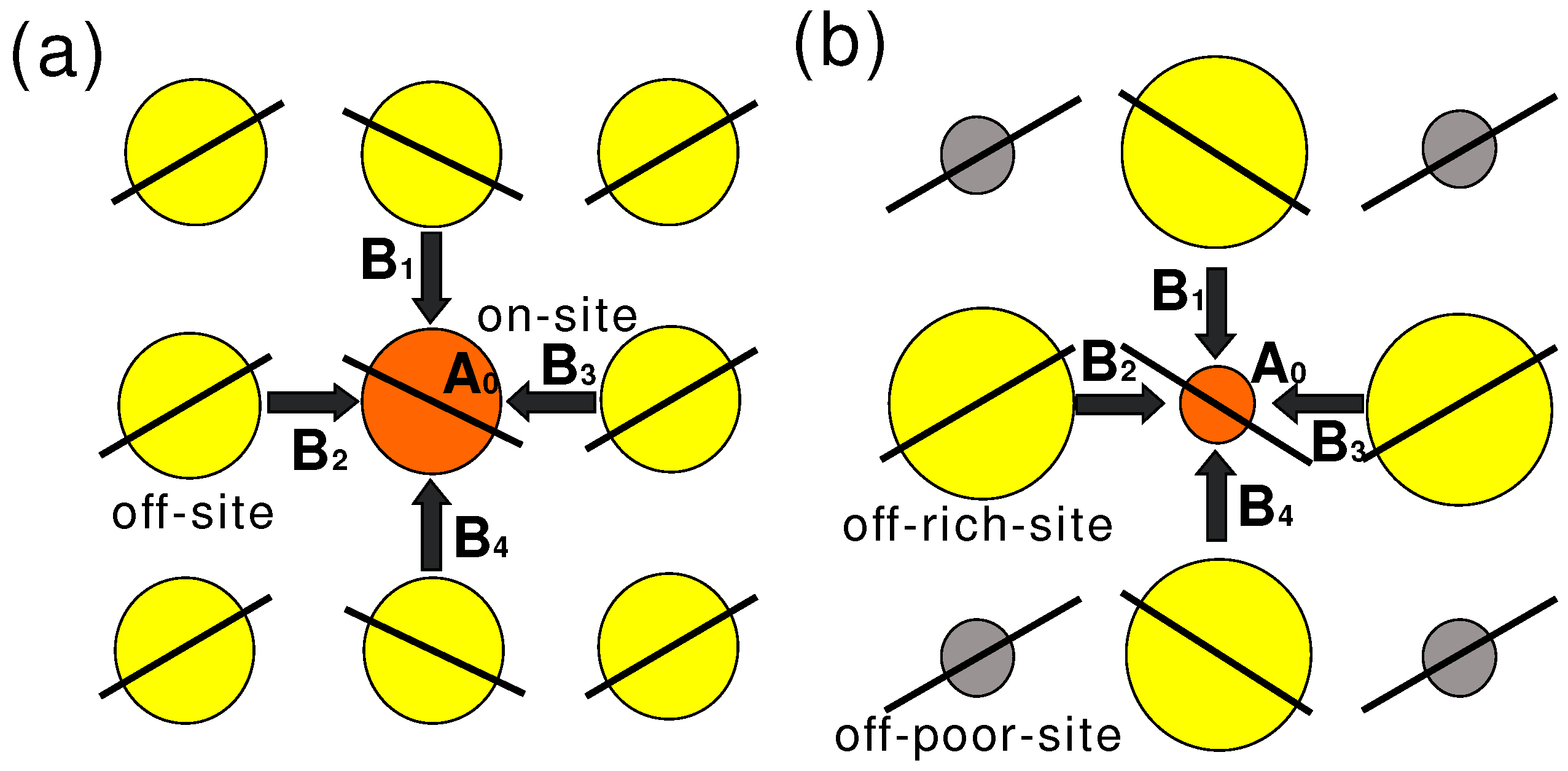
2. Problems of Pake Doublet
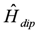 , is added to Zeeman interaction,
, is added to Zeeman interaction, 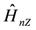 . These interactions are described as,
. These interactions are described as,

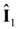 ,
, 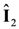 ,
,  ,
,  ,
, 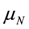 ,
, 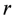 are the spin operators of each nuclear site, the g factor of nuclei, the magnitude of the external magnetic field, the nuclear magneton, and the vector between two nuclei. This dipole–dipole interaction,
are the spin operators of each nuclear site, the g factor of nuclei, the magnitude of the external magnetic field, the nuclear magneton, and the vector between two nuclei. This dipole–dipole interaction, 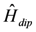 , can be expressed as, using
, can be expressed as, using 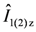 and the creation-annihilation operators,
and the creation-annihilation operators, 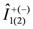 ,
,





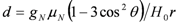 . As a result, the two transitions of
. As a result, the two transitions of 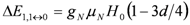 and
and 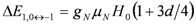 between closest levels in the triplet state are allowed with same intensities. This doublet structure is the so called Pake doublet.
between closest levels in the triplet state are allowed with same intensities. This doublet structure is the so called Pake doublet.  and
and 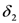 , the nuclear Zeeman interaction,
, the nuclear Zeeman interaction, 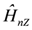 , is modified as,
, is modified as, 

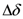 and
and 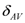 are the difference and the average of NMR shift between two local sites, respectively. Because of the off-diagonal elements in the energy matrix, the hybridization between the singlet and triplet states occurs and the eigenstates are expressed as follow,
are the difference and the average of NMR shift between two local sites, respectively. Because of the off-diagonal elements in the energy matrix, the hybridization between the singlet and triplet states occurs and the eigenstates are expressed as follow, 



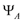 and
and 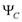 are allowed and we can observed four transitions,
are allowed and we can observed four transitions, 



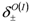 , and intensities,
, and intensities, 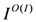 , are expressed as,
, are expressed as, 

 also depends on the angle of θ, the angular dependence of does not show a simple curve but a complicated one.
also depends on the angle of θ, the angular dependence of does not show a simple curve but a complicated one.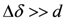 , the mean values of two higher (lower) shift in quartet represent NMR shift at two local sites,
, the mean values of two higher (lower) shift in quartet represent NMR shift at two local sites, 

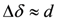 . Since the spin susceptibility is connected to NMR shift at local nucleus via hyperfine coupling constant, NMR shift does not represent the local spin susceptibility. Since the magnitude of d isproportional to H0−1, the problems actually surface in the low field experiments, such as that under superconductivity.
. Since the spin susceptibility is connected to NMR shift at local nucleus via hyperfine coupling constant, NMR shift does not represent the local spin susceptibility. Since the magnitude of d isproportional to H0−1, the problems actually surface in the low field experiments, such as that under superconductivity.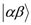 and
and  depending on the parameter of also have an influence on the spin-lattice relaxation time, T1, which intrinsically depends on the magnetic fluctuation at each local nucleus. Therefore, the relaxation to the mixing states is driven by the magnetic fluctuation on both sites, and the problems of Pake doublet causes great loss of the advantage of the local magnetic probe. , which cannot be canceled by the spin echo, π/2-π pulse, sequence. Therefore, the spectrum by the fast Fourier transformation of this modulated spin echo signal contains the spurious information such as the negative intensity [37]. This modulation could be canceled by the solid echo pulse sequence [38]. There are a few experiments using solid echo pulse sequence [39]. However, this sequence requires the fast recovery RF system and restricts the advantage of the conventional spin echo. This modulation also inhibits the measurement of the spin-spin relaxation time, T2. There are some estimations of T2 deduced from the envelope of the modulated spin echo intensity, so called J-modulation [40,41]. However, this method estimates T2 for overall spectrum in the coupled spin system and is not conventional.
depending on the parameter of also have an influence on the spin-lattice relaxation time, T1, which intrinsically depends on the magnetic fluctuation at each local nucleus. Therefore, the relaxation to the mixing states is driven by the magnetic fluctuation on both sites, and the problems of Pake doublet causes great loss of the advantage of the local magnetic probe. , which cannot be canceled by the spin echo, π/2-π pulse, sequence. Therefore, the spectrum by the fast Fourier transformation of this modulated spin echo signal contains the spurious information such as the negative intensity [37]. This modulation could be canceled by the solid echo pulse sequence [38]. There are a few experiments using solid echo pulse sequence [39]. However, this sequence requires the fast recovery RF system and restricts the advantage of the conventional spin echo. This modulation also inhibits the measurement of the spin-spin relaxation time, T2. There are some estimations of T2 deduced from the envelope of the modulated spin echo intensity, so called J-modulation [40,41]. However, this method estimates T2 for overall spectrum in the coupled spin system and is not conventional.  ) is one of alternative methods to avoid Pake doublet problem [8,42]. However, the setting restricts the direction of the external field. Therefore, the single-site substituted TMTCF and BEDT-TTF are desired.
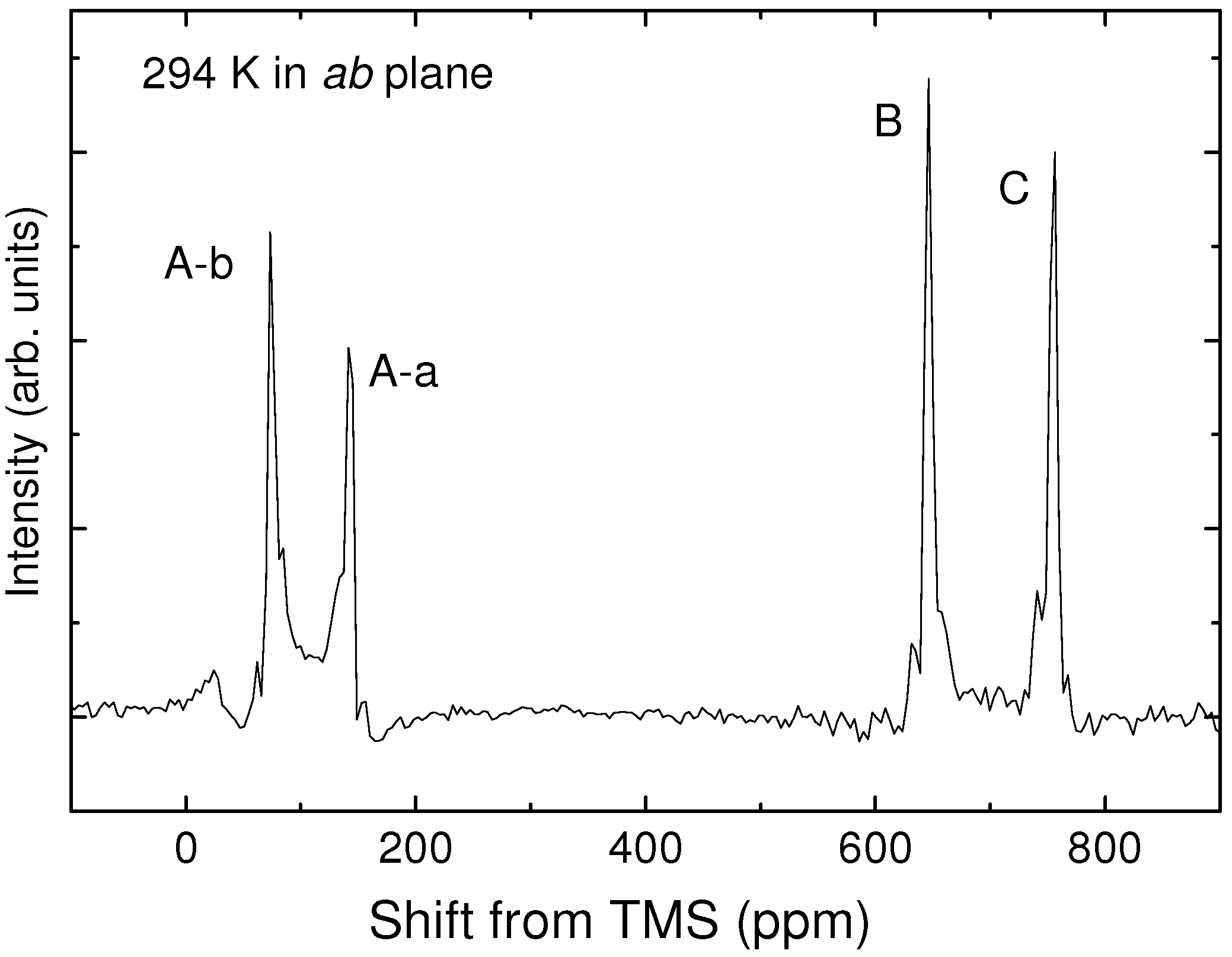
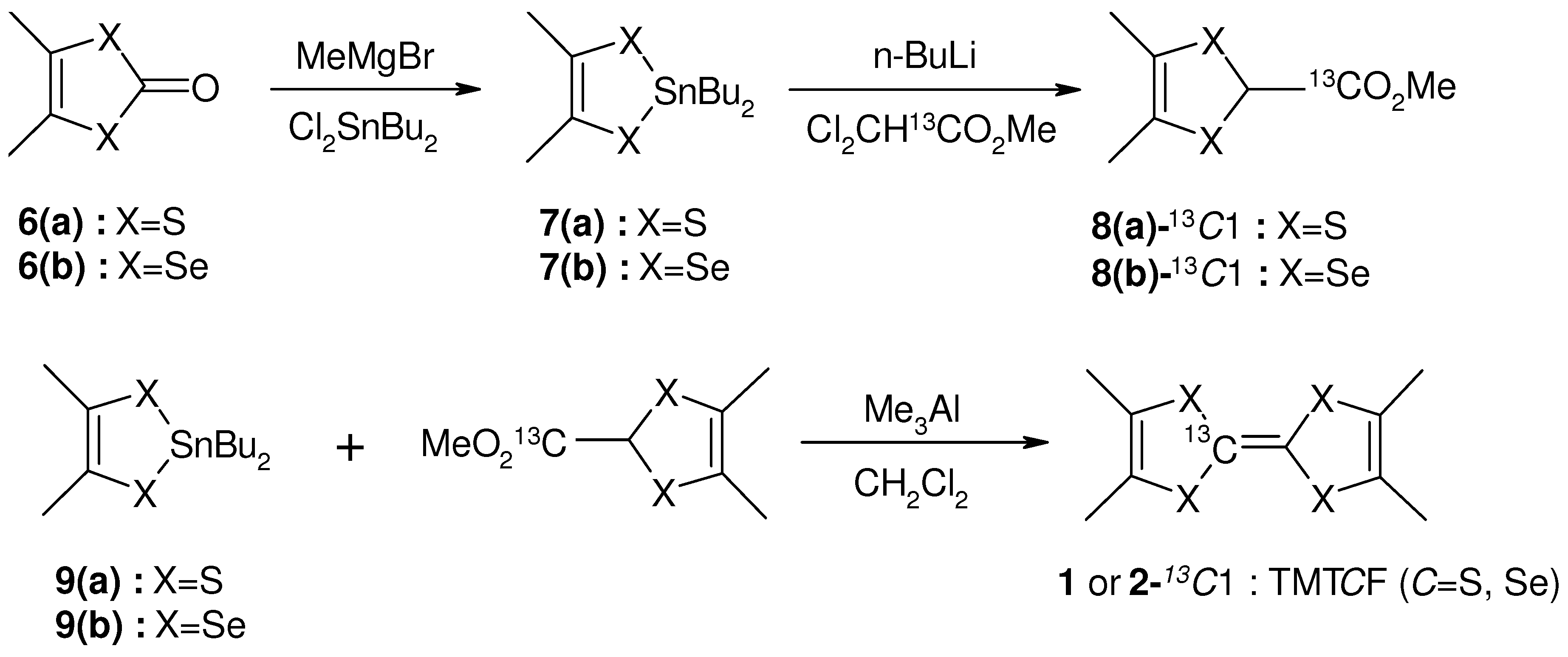
) is one of alternative methods to avoid Pake doublet problem [8,42]. However, the setting restricts the direction of the external field. Therefore, the single-site substituted TMTCF and BEDT-TTF are desired. 3. Synthetic Method of Single-Site 13C- Substituted Molecule
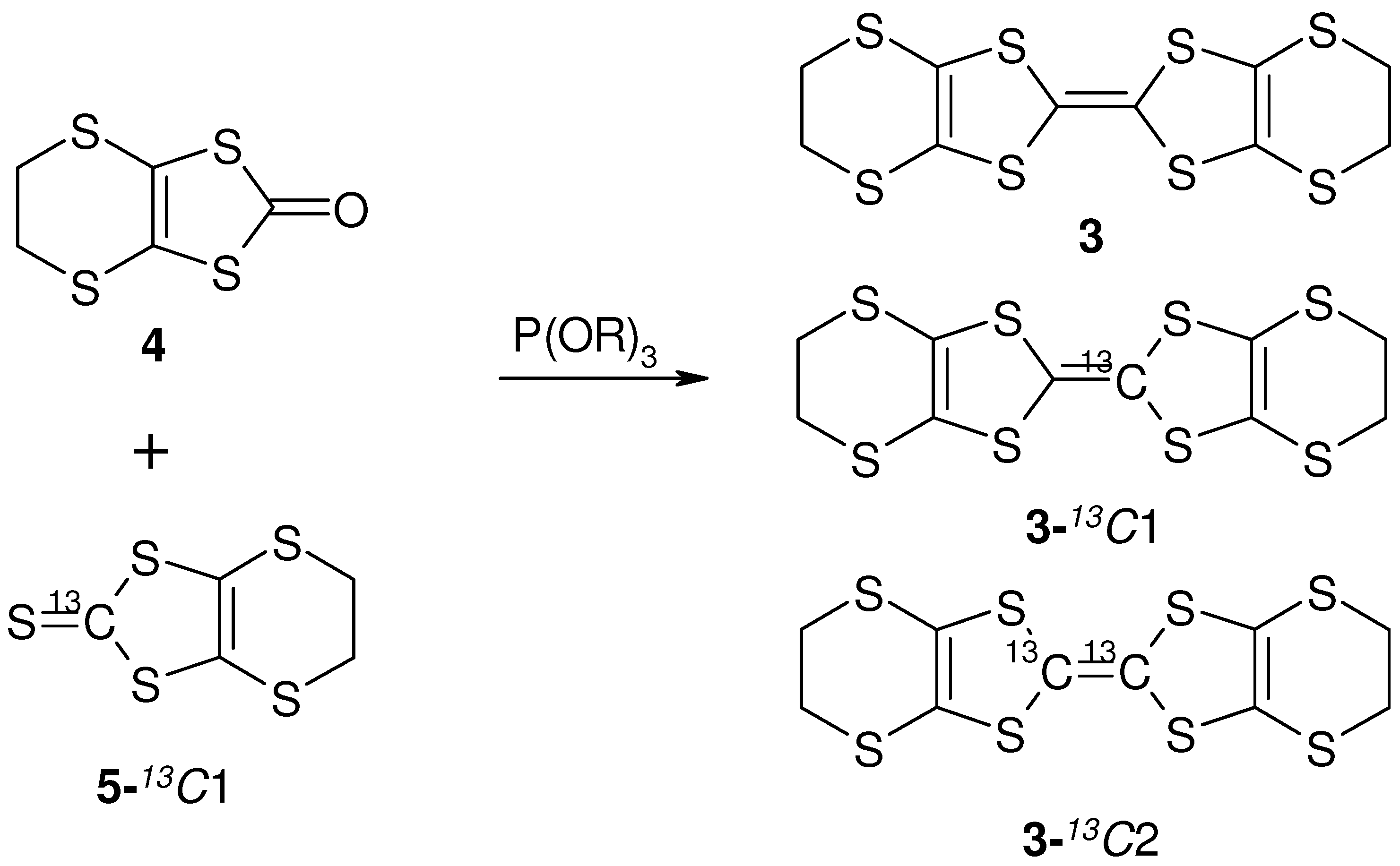
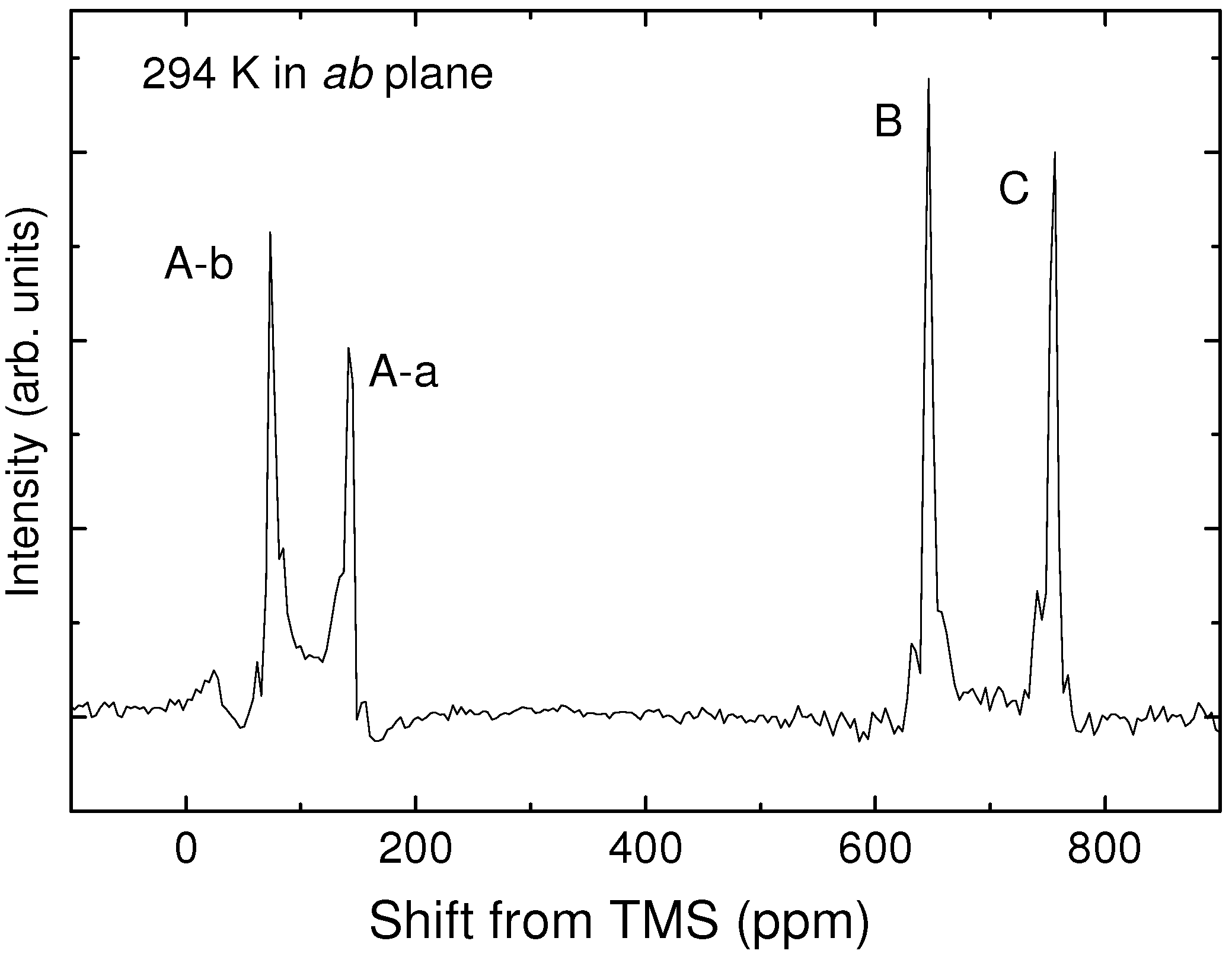

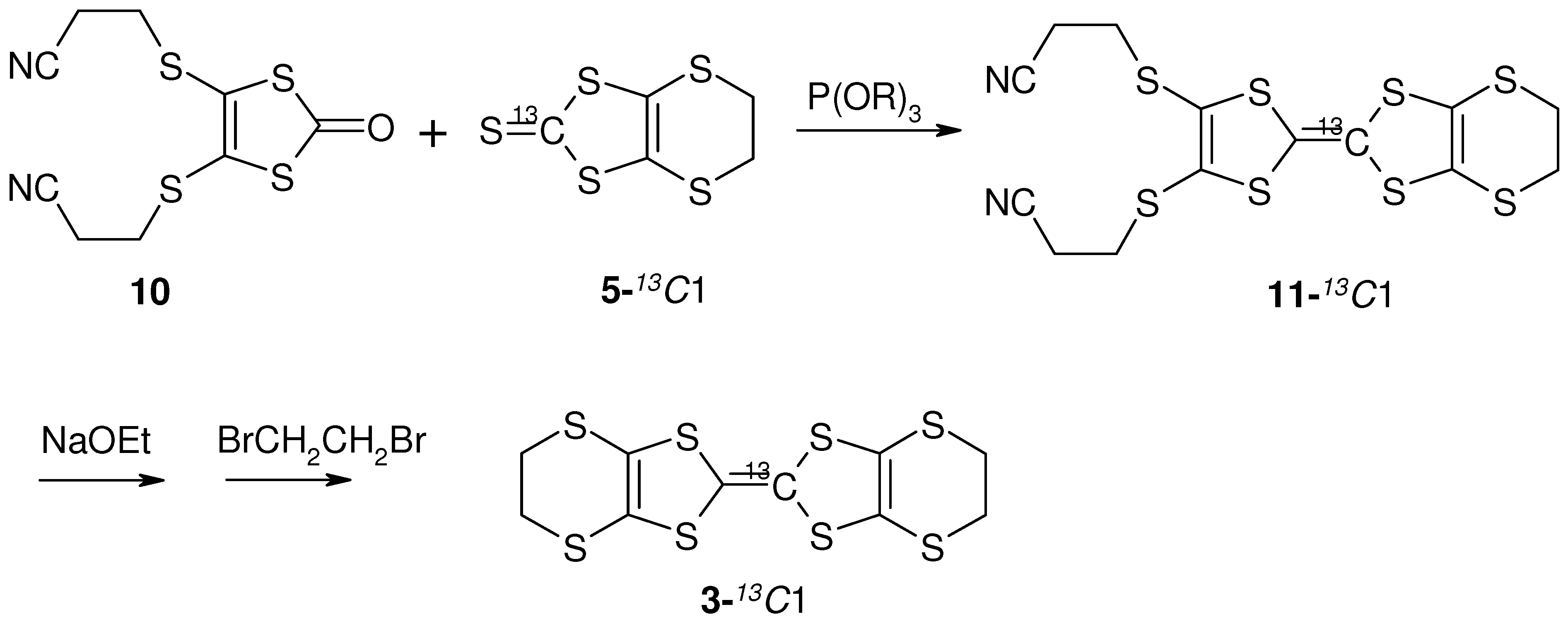
4. NMR Investigation Utilizing Hyperfine Coupling and Chemical Shift Tensor
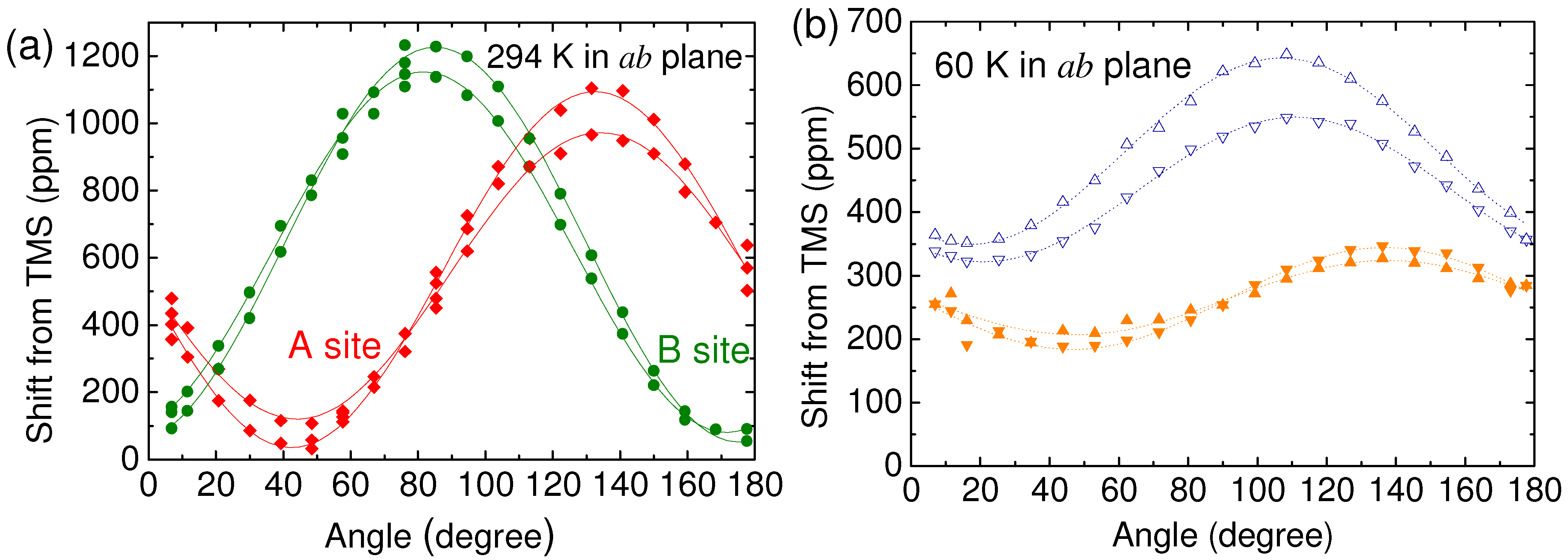
4.1. NMR Shift on Molecular Conductors










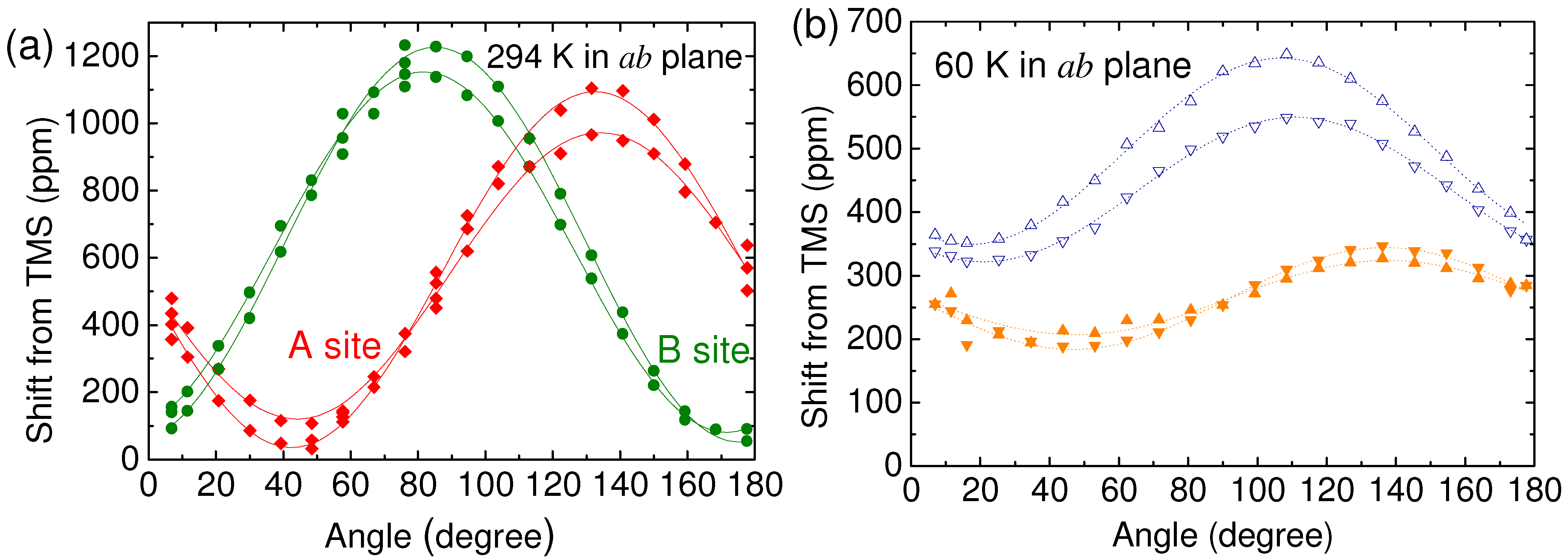
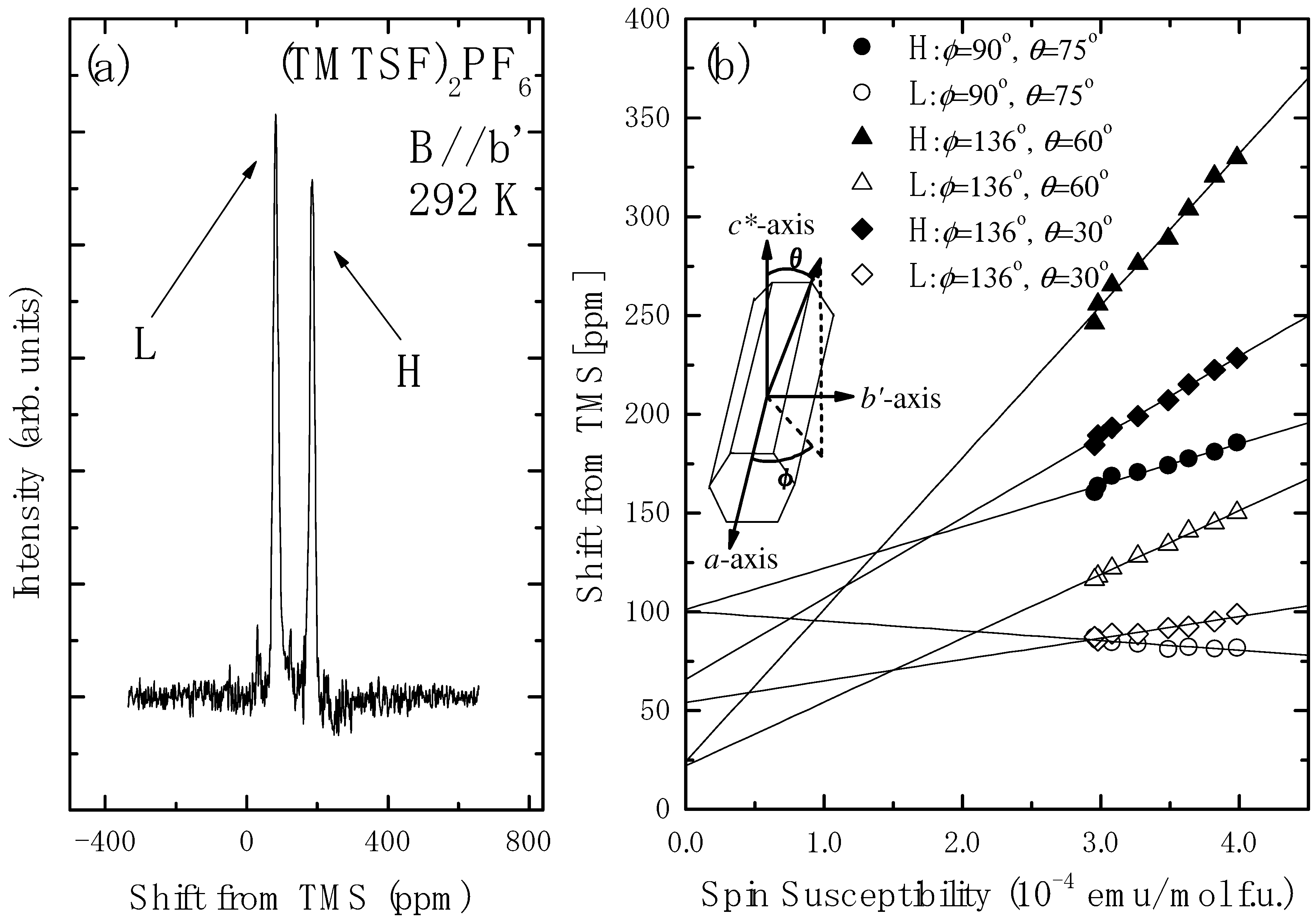
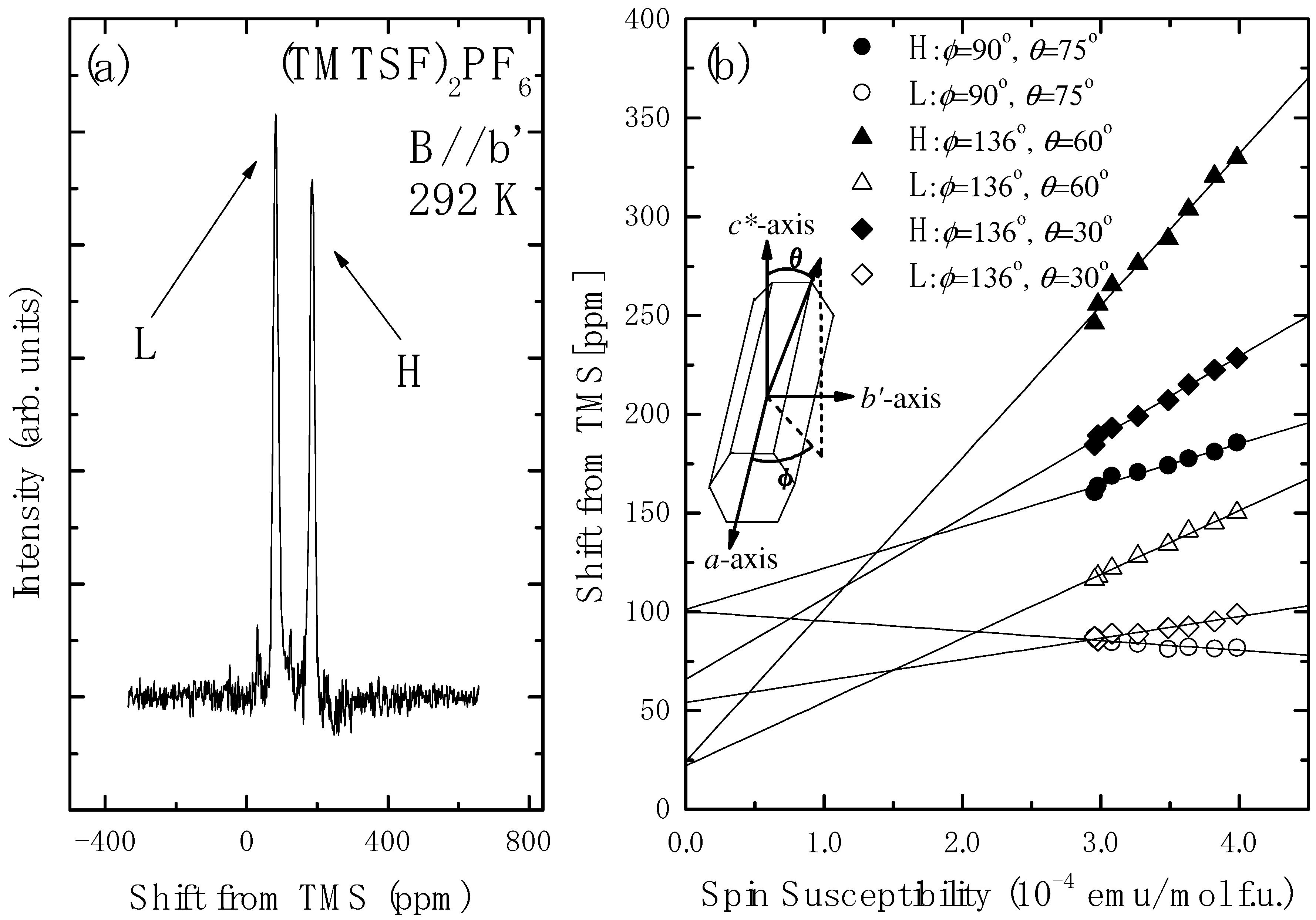
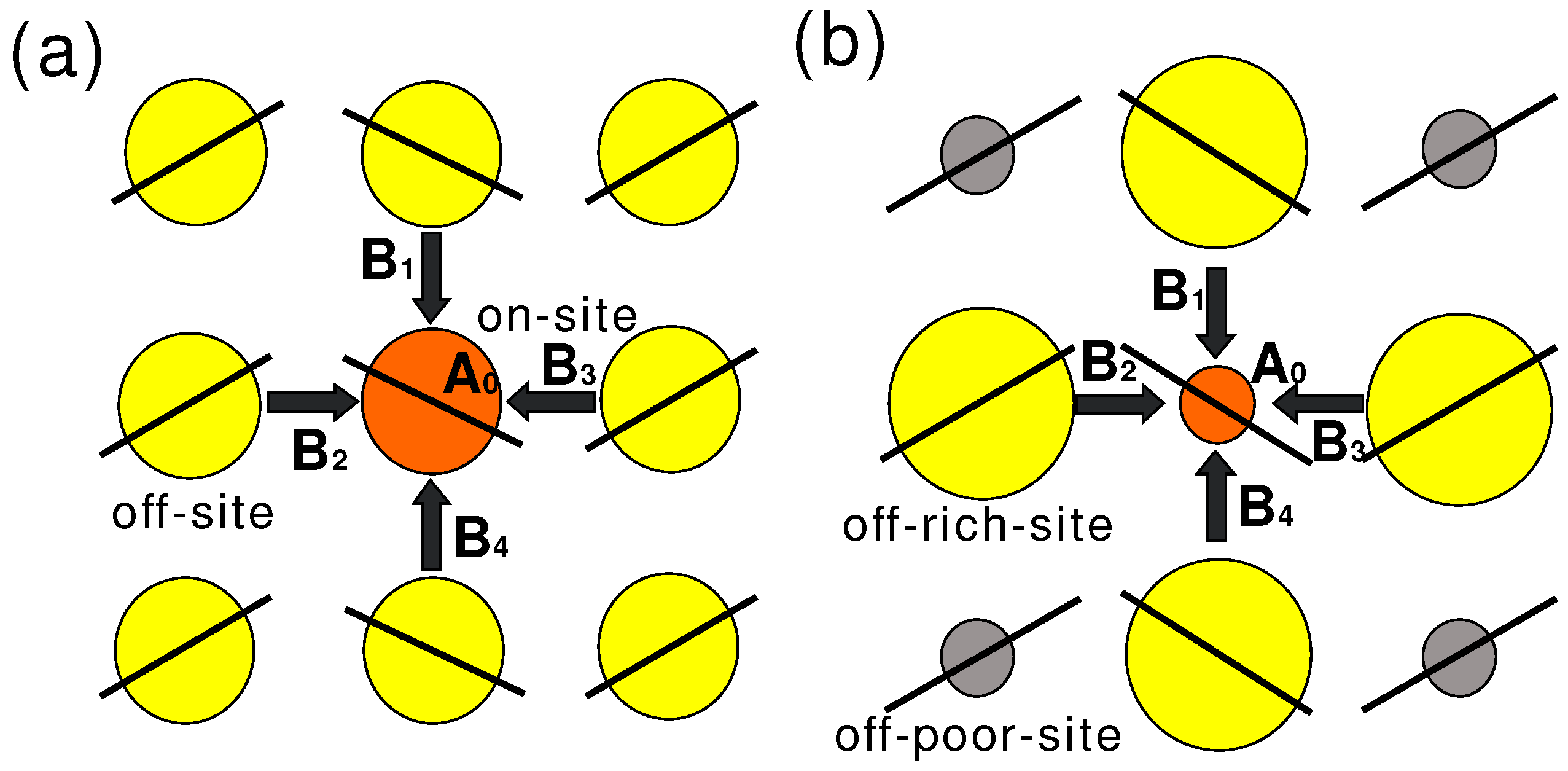
4.2. Determination of the Hyperfine Coupling and Chemical Shift Tensor








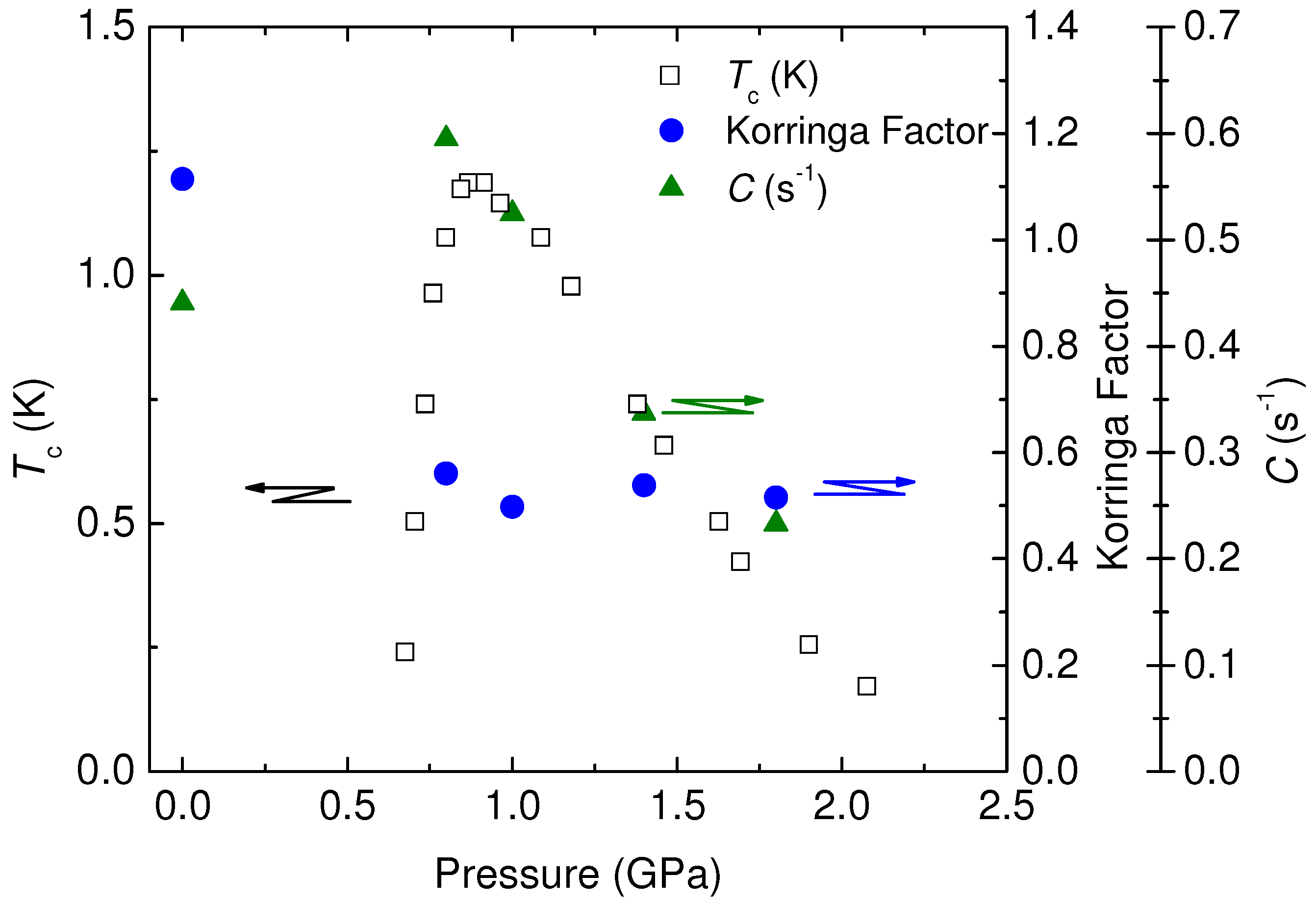
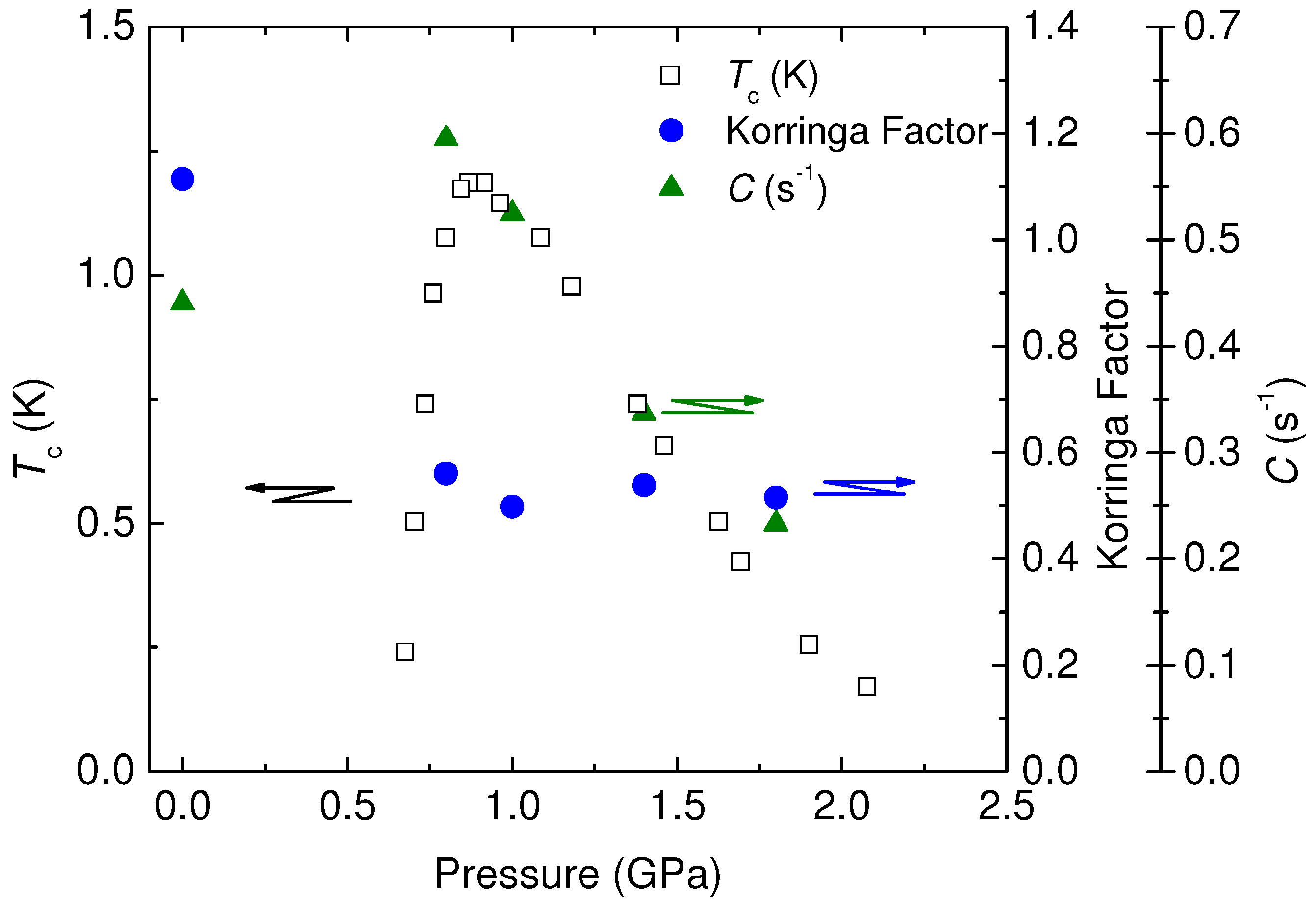
4.3. Korringa Enhancement Factor in (TMTTF)2PF6
 , is important, which is related by,
, is important, which is related by,  is the parameter depending on the electron correlation and is determined from observations of the spin-lattice relaxation time, T1, and Knight shift, K, and other physical constants without analytical assumptions. This factor gives important information for the electron correlation. However, in case the hyperfine coupling is anisotropic, the Equation (39) is modified by the form factor and is written as,
is the parameter depending on the electron correlation and is determined from observations of the spin-lattice relaxation time, T1, and Knight shift, K, and other physical constants without analytical assumptions. This factor gives important information for the electron correlation. However, in case the hyperfine coupling is anisotropic, the Equation (39) is modified by the form factor and is written as,
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
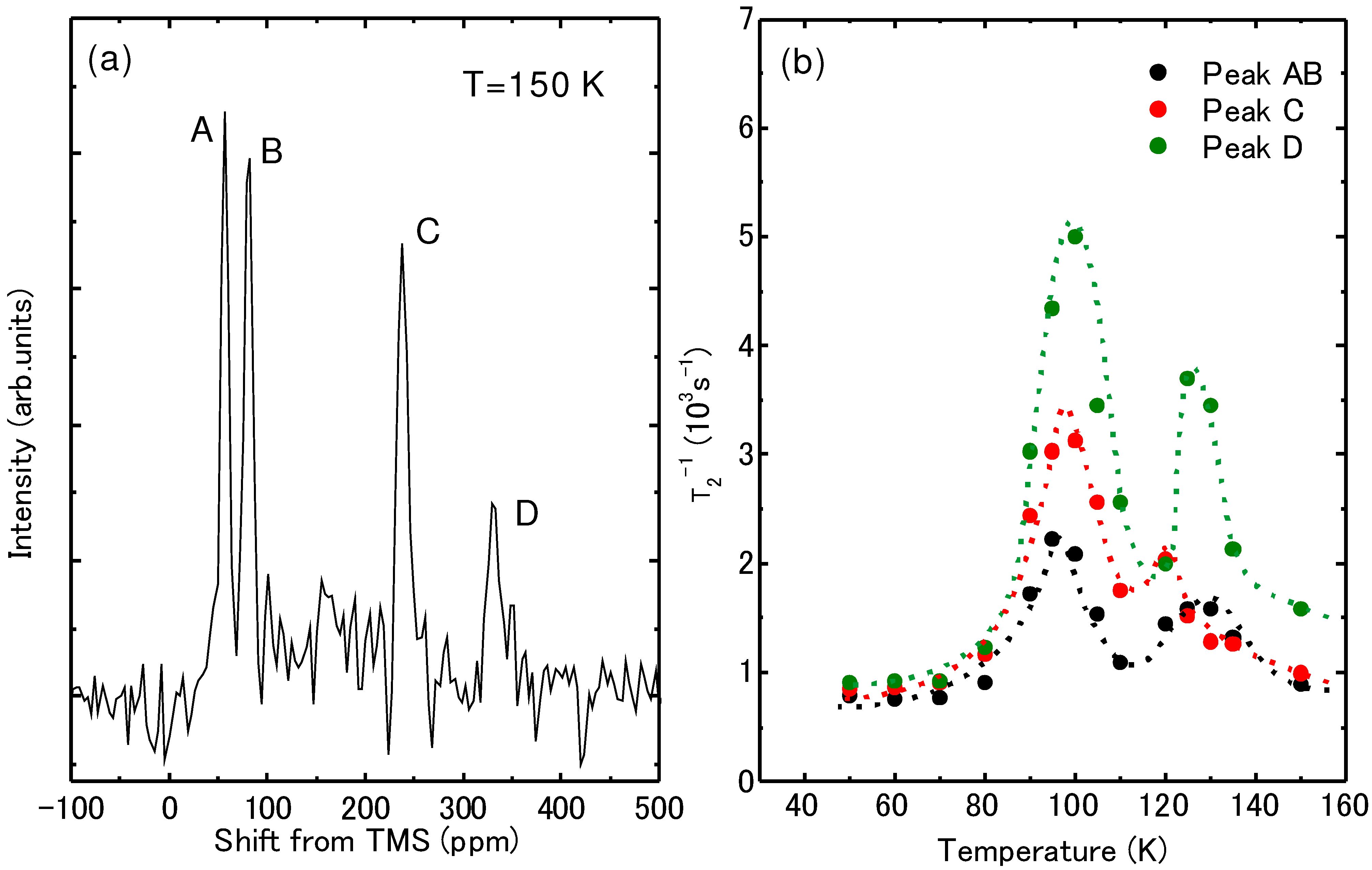
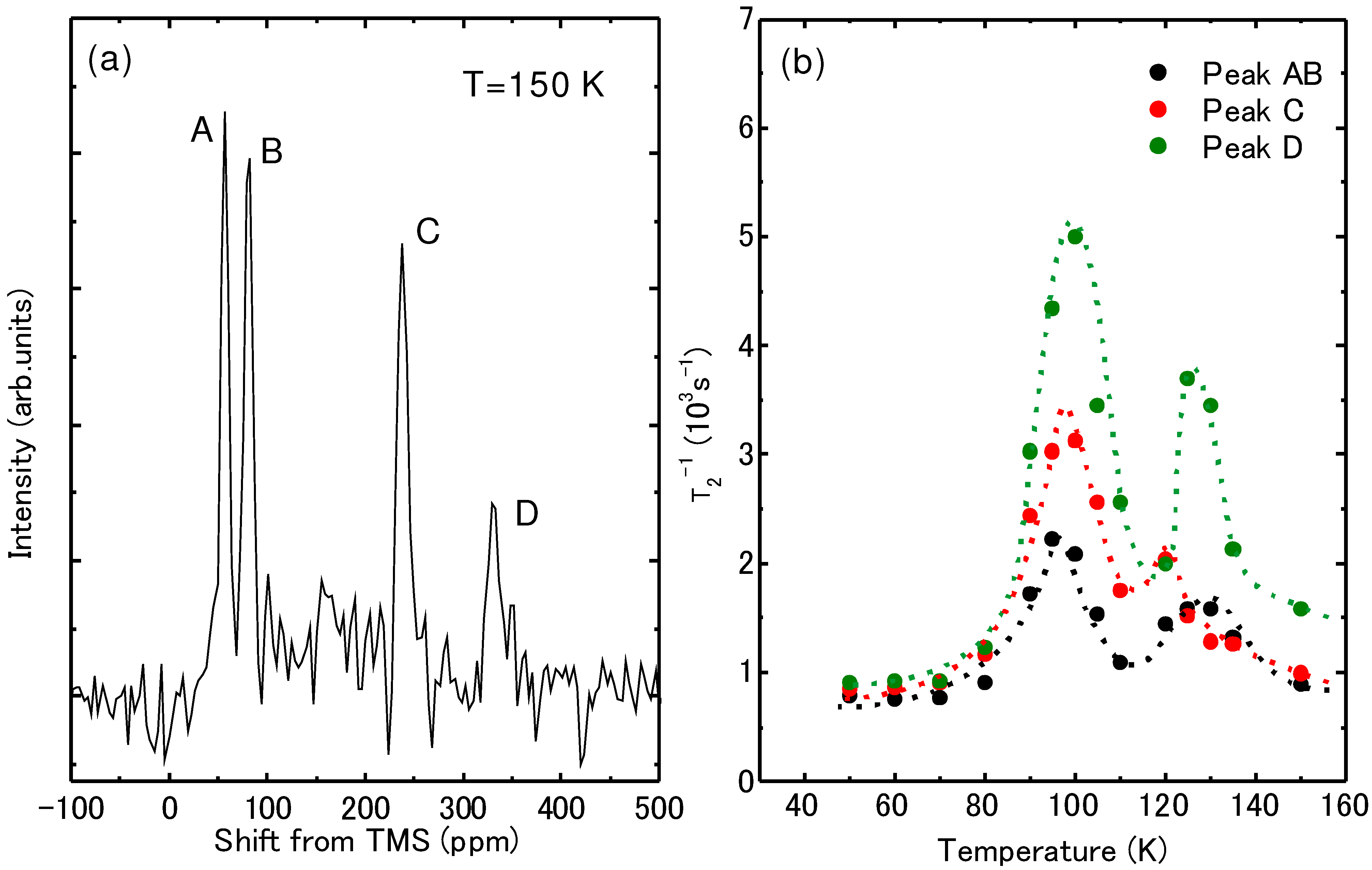{kind=link}
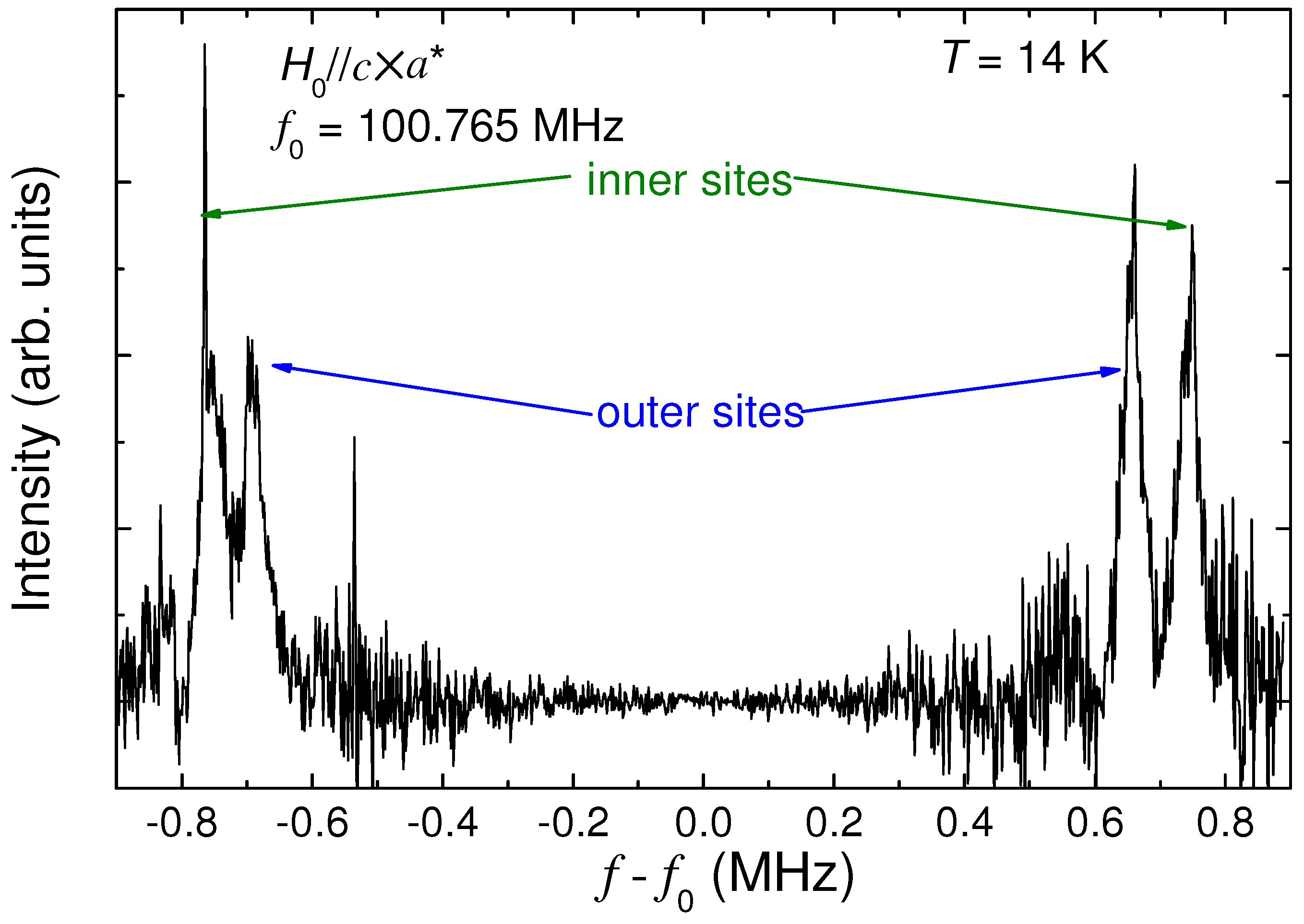
4.4. Determination of the Magnetic Moment in β'-(BEDT-TTF)2ICl2
 [54].
[54].
[54].
[54].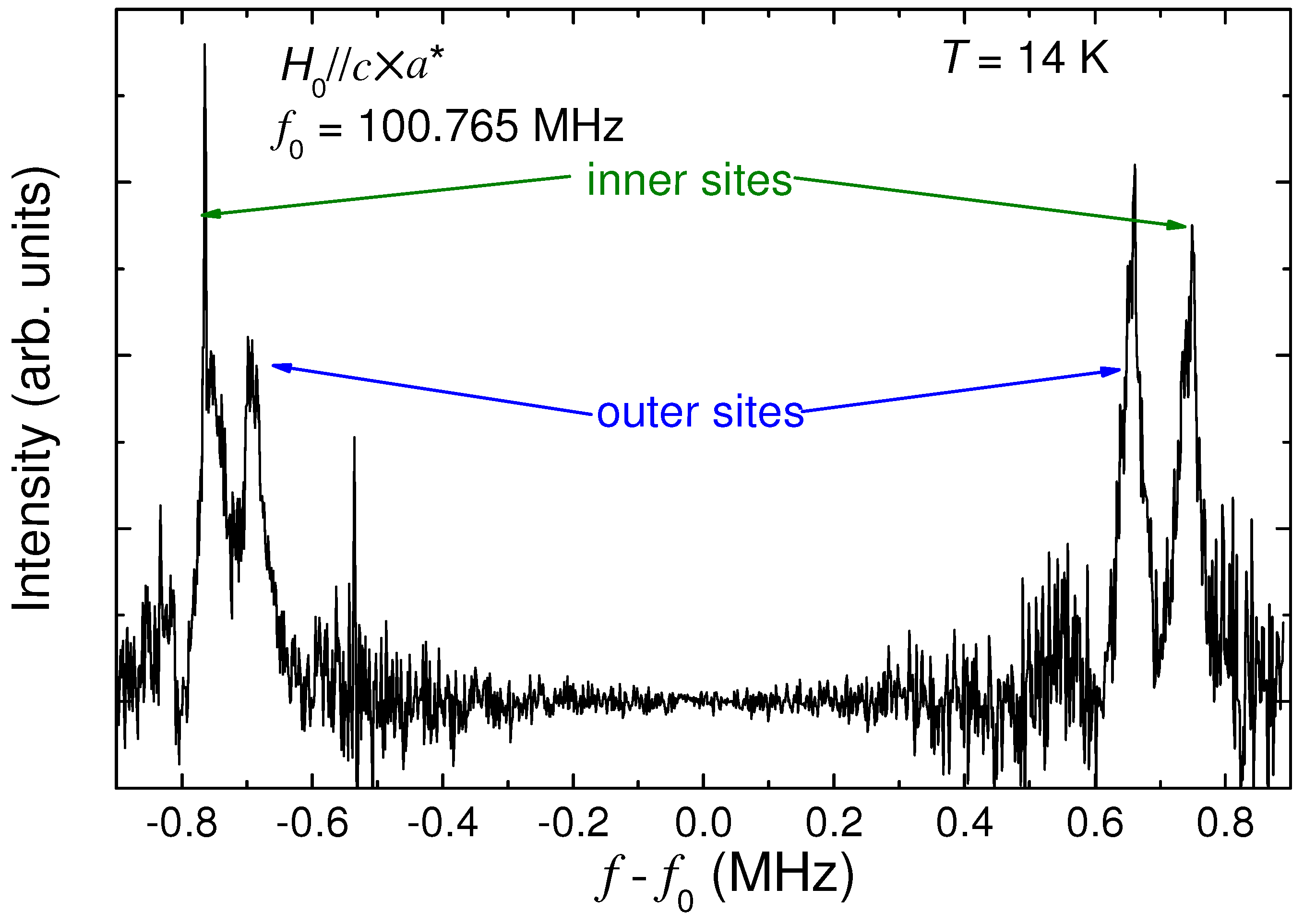
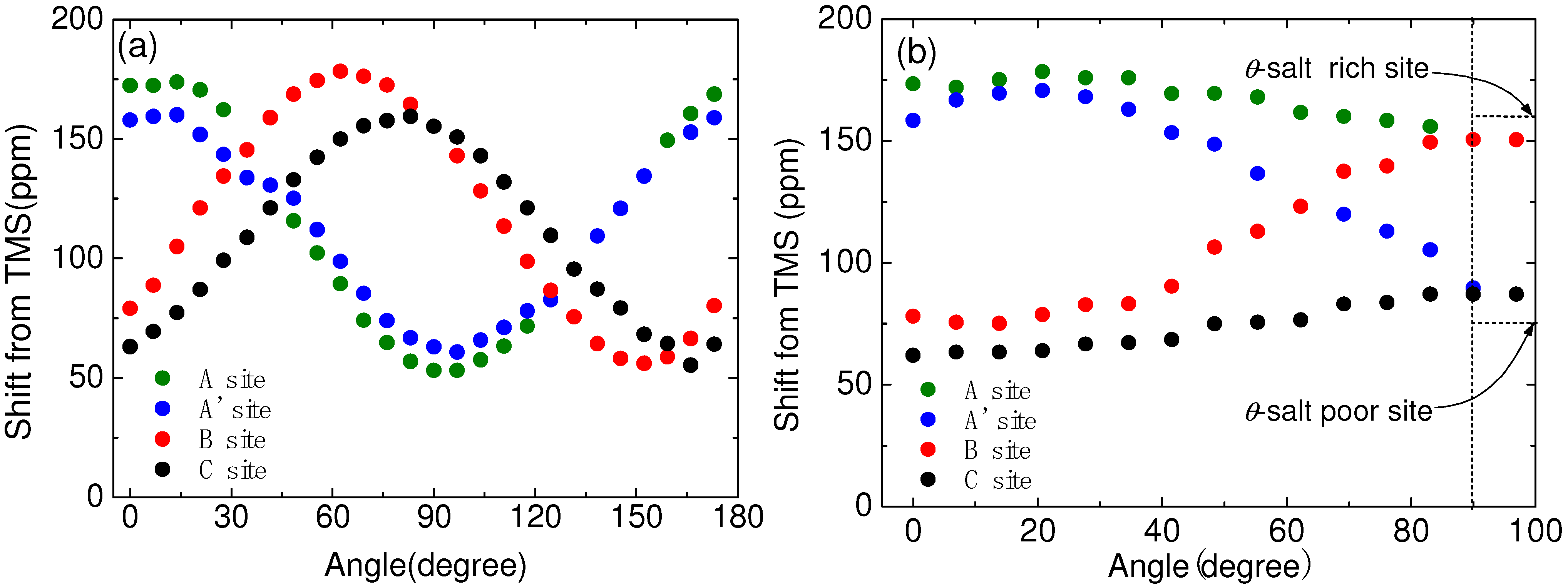
4.5. Site Assignment on Charge Order Phase in α-(BEDT-TTF)2I3




4.6. Slow Dynamics of Ethylene Motions in BEDT-TTF
5. Conclusions
Acknowledgments
References and Notes
- Wang, H.H.; Beno, M.A.; Geiser, U.; Firestone, M.A.; Webb, K.S.; Nunez, L.; Crabtree, G.W.; Carlson, K.D.; Williams, J.M. Ambient-Pressure superconductivity at the highest temperature (5 K) observed in an organic system: β-(BEDT-TTF)2AuI2. Inorg. Chem. 1985, 24, 2465–2466. [Google Scholar]
- Wang, H.H.; Geiser, U.; Williams, J.M.; Mason, J.M.; Perr, J.T.; Heindl, J.E.; Lathrop, M.W.; Love, B.J.; Watkins, D.M.; Yaconi, G.A. Phase selectivity in the simultaneous synthesis of the Tc = 12.8 K (0.3 kbar) organic superconductor κ-(BEDT-TTF)2Cu[N(CN)2]Cl or the semiconductor (BEDT-TTF)Cu[N(CN)2]2. Chem. Mater. 1992, 4, 247–249. [Google Scholar]
- Geiser, U.; Wang, H.H.; Carlson, K.D.; Williams, J.M.; Charlier, H.A.; Heindl, J.E.; Yaconi, G.A.; Love, B.J.; Lathrop, M.W.; Schirber, J.E.; et al. Superconductivity at 2.8 K and 1.5 kbar in κ-(BEDT-TTF)2Cu2(CN)3: The first organic superconductor containing a polymeric copper cyanide anion. Inorg. Chem. 1991, 30, 2586–2588. [Google Scholar]
- Urayama, H.; Yamochi, H.; Saito, G.; Nozawa, K.; Sugano, T.; Kinoshita, M.; Sato, S.; Oshima, K.; Kawamoto, A.; Tanaka, A. A new ambient pressure organic superconductor based on BEDT-TTF with TC higher than 10 K (TC = 10.4 K). J. Chem. Lett. 1988, 17, 55–58. [Google Scholar]
- Jérome, D. The physics of organic superconductors. Science 1991, 252, 1509–1514. [Google Scholar]
- Jérome, D.; Mazaud, A.; Ribault, M.; Bechgaard, K. Superconductivity in a synthetic organic conductor (TMTSF)2PF6. J. Phys. Lett. 1980, 41, 95–98. [Google Scholar]
- Bechgaard, K.; Carneiro, K.; Olsen, M.; Rasmussen, F.B.; Jacobsen, C.S. Zero-Pressure organic superconductor: Di-(tetramethyltetraselenafulvalenium)-perchlorate [(TMTSF)2ClO4]. Phys. Rev. Lett. 1981, 46, 852–855. [Google Scholar]
- Chow, D.S.; Zamborszky, F.; Alavi, B.; Tantillo, D.J.; Baur, A.; Merlic, C.A.; Brown, S.E. Charge ordering in the TMTTF family of molecular conductors. Phys. Rev. Lett. 2000, 85, 1698. [Google Scholar]
- Zamborszky, F.; Yu, W.; Raas, W.; Brown, S.E.; Alavi, B.; Merlic, C.A.; Baur, A. Competition and coexistence of bond and charge orders in (TMTTF)2AsF6. Phys. Rev. B 2002, 66, 1–4. [Google Scholar]
- Yu, W.; Zhang, F.; Zamborsky, F.; Alavi, B.; Baur, A.; Merlic, C.A.; Brown, S.E. Electron-Lattice coupling and broken symmetries of the molecular salt (TMTTF)2SbF6. Phys. Rev. B 2004, 70, 1–4. [Google Scholar]
- Barthel, E.; Quirion, G.; Wzietek, P.; Jérome, D.; Christensen, J.B.; Jørgensen, M.; Bechgaard, K. NMR in commensurate and incommensurate spin density waves. Europhys. Lett. 1993, 21, 87–92. [Google Scholar]
- Klemme, B.J.; Brown, S.E.; Wzietek, P.; Kriza, G.; Batail, P.; Jerome, D.; Fabre, J.M. Commensurate and incommensurate spin-density waves and a modified phase diagram of the Bechgaard salts. Phys. Rev. Lett. 1995, 75, 2408–2411. [Google Scholar]
- Mckenzie, R.H. Similarities between organic and cuprate superconductors. Science 1997, 278, 820–821. [Google Scholar] [CrossRef]
- Lang, M. Quasi-Two dimensional organic superconductors. Supercond. Rev. 1996, 2, 1. [Google Scholar]
- Kanoda, K. Electron correlation, metal-insulator transition and superconductivity in quasi-2D organic systems, (ET)2X. Phys. C 1997, 282–287, 299–302. [Google Scholar] [CrossRef]
- Kawamoto, A.; Miyagawa, K.; Nakazawa, Y.; Kanoda, K. Electron correlation in the κ-phase family of BEDT-TTF compounds studied by 13C-NMR, where BEDT-TTF is bis(ethylenedithio)tetrathiafulvalene. Phys. Rev. B 1995, 52, 15522–15533. [Google Scholar]
- Kondo, H.; Moriya, T. Spin fluctuation-induced superconductivity in organic compounds. J. Phys. Soc. Jpn. 1998, 67, 3695–3698. [Google Scholar] [CrossRef]
- Kino, H.; Kontani, H. Phase diagram of superconductivity on the anisotropic triangular lattice hubbard model: An effective model of κ-(BEDT-TTF) salts. J. Phys. Soc. Jpn. 1998, 67, 3691–3694. [Google Scholar] [CrossRef]
- Schmalian, J. Pairing due to spin fluctuations in layered organic superconductors. Phys. Rev. Lett. 1998, 81, 4232–4235. [Google Scholar] [CrossRef]
- Vojta, M.; Dagotto, E. Indications of unconventional superconductivity in doped and undoped triangular antiferromagnets. Phys. Rev. B 1999, 59, 713–716. [Google Scholar] [CrossRef]
- Schirber, J.E.; Overmyer, D.L.; Carlson, K.D.; William, J.M.; Kini, A.M.; Wang, H.H.; Charlier, H.A.; Love, B.J.; Watkins, D.M.; Yaconi, G.A. Pressure-Temperature phase diagram, inverse isotope effect, and superconductivity in excess of 13 K in κ-(BEDT-TTF)2Cu[N(CN)2]Cl, where BEDT-TTF is bis(ethylenedithio)tetrathiafulvalene. Phys. Rev. B 1991, 44, 4666–4669. [Google Scholar]
- Kakiuchi, T.; Wakabashi, Y.; Sawa, H.; Takahash, T.; Nakamura, T. Charge ordering in α-(BEDT-TTF)2I3 by synchrotron X-ray diffraction. J. Phys. Soc. Jpn. 2007, 76, 1–4. [Google Scholar]
- Moroto, S.; Hiraki, K.-I.; Takano, Y.; Kubo, Y.; Takahashi, T.; Yamamoto, H.M.; Nakamura, T. Charge disproportionation in the metallic state of α-(BEDT-TTF)2I3. J. Phys. IV 2004, 114, 399–340. [Google Scholar]
- Wojciechowski, R.; Yamamoto, K.; Yakushi, K.; Inokuchi, M.; Kawamoto, A. High-Pressure Raman study of the charge ordering in α-(BEDT-TTF)2I3. Phys. Rev. B 2003, 67, 1–11. [Google Scholar]
- Yue, Y.; Yamamoto, K.; Uruichi, M.; Nakano, C.; Yakushi, K.; Yamada, S.; Hiejima, T.; Kawamoto, A. Nonuniform site-charge distribution and fluctuations of charge order in the metallic state of α-(BEDT-TTF)2I3. Phys. Rev. B 2010, 82, 1–8. [Google Scholar]
- Bender, K.; Hennig, I.; Schweitzer, D.; Dietz, K.; Endres, H.; Keller, H.J. Synthesis, structure and physical properties of a two-dimensional organic metal, di[dis(ethylene-dithiolo)tetrathiofulvalene] triiodide, (BEDT-TTF)+2I−3. Mol. Cryst. Liq. Cryst. 1984, 107, 359–371. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yakushi, K.; Miyagawa, K.; Kanoda, K.; Kawamoto, A. Charge ordering in θ-(BEDT-TTF)2RbZn(SCN)4 studied by vibrational spectroscopy. Phys. Rev. B 2002, 65, 1–8. [Google Scholar]
- Miyagawa, K.; Kawamoto, A.; Kanoda, K. Charge ordering in a quasi-two-dimensional organic conductor. Phys. Rev. B 2000, 62, 7679–7682. [Google Scholar]
- Mori, H.; Tanaka, S.; Mori, T. Systematic study of the electronic state in θ-type BEDT-TTF organic conductors by changing the electronic correlation. Phys. Rev. B 1998, 57, 12023–12029. [Google Scholar]
- Seo, H.; Fukuyama, H. Antiferromagnetic phases of one-dimensional quarter-filled organic conductors. J. Phys. Soc. Jpn. 1997, 66, 1249–1252. [Google Scholar]
- Seo, H. Charge ordering in organic ET compounds. J. Phys. Soc.Jpn. 2000, 69, 805–820. [Google Scholar] [CrossRef]
- Merino, J.; Mckenzie, R.H. Superconductivity mediated by charge fluctuations in layered molecular crystals. Phys. Rev. Lett. 2001, 87, 1–4. [Google Scholar]
- Emery, V.J.; Bruinsma, R.; Barišić, S. Electron-Electron Umklapp scattering in organic superconductors. Phys. Rev. Lett. 1982, 48, 1039–1043. [Google Scholar] [CrossRef]
- Scott, J.C.; Pedersen, H.J.; Bechgaard, K. Magnetic properties of the organic conductor bis-tetramethyltetraselenafulvalene hexafluorophosphate [(TMTSF)2PF6]: A new phase transition. Phys. Rev. Lett. 1980, 45, 2125–2128. [Google Scholar]
- Lasjaunias, J.C.; Biljaković, K.; Nad’, F.; Monceau, P.; Bechgaard, K. Glass transition in the spin-density wave phase of (TMTSF)2PF6. Phys. Rev. Lett. 1994, 72, 1283–1286. [Google Scholar]
- Lee, I.J.; Chow, D.S.; Clark, W.G.; Strouse, M.J.; Naughton, M.J.; Chaikin, P.M.; Brown, S.E. Evidence from 77Se Knight shifts for triplet superconductivity in (TMTSF)2PF6. Phys. Rev. B 2003, 68, 1–4. [Google Scholar]
- Takahashi, T. 13C-NMR studies of charge ordering in organic conductors. Syth. Met. 2003, 133–134, 261–264. [Google Scholar] [CrossRef]
- Powles, J.G.; Manfield, P. Double-Pulse nuclear-resonance transients in solid. Phys. Lett. 1962, 2, 58–59. [Google Scholar] [CrossRef]
- Miyagawa, K.; Kawamoto, A.; Kanoda, K. 13C NMR study of nesting instability in α-(BEDT-TTF)2RbHg(SCN)4. Phys. Rev. B 1997, 56, 8487–8490. [Google Scholar]
- De Soto, S.M.; Slichter, C.P.; Kini, A.M.; Wang, H.H.; Geiser, U.; Williams, J.M. 13C NMR line-shape studies of the organic superconductor κ-(ET)2Cu[N(CN)2]Br. Phys. Rev. B 1996, 54, 16101–16107. [Google Scholar]
- Shimizu, Y.; Miyagawa, K.; Kanoda, K.; Maesato, M.; Saito, G. Emergence of inhomogeneous moments from spin liquid in the triangular-lattice Mott insulator κ-(ET)2Cu2(CN)3. Phys. Rev. B 2006, 73, 140407/1–140407/4. [Google Scholar]
- De Sono, S.M.; Slichter, C.P.; Kini, A.M.; Wang, H.H.; Geiser, U.; Williams, J.M. 13C-NMR studies of the normal and superconducting states of the organic superconductor κ-(ET)2Cu[N(CN)2]Br. Phys. Rev. B 1995, 52, 10364–10368. [Google Scholar]
- Ferraris, J.P.; Poehler, T.O.; Bloch, A.N.; Cowan, D.O. Synthesis of the highly conducting organic salt: Tetramethyltetrathiofulvalenium-tetracyano-p-quinodimethanide. Tetrahedron Lett. 1973, 14, 2553–2556. [Google Scholar]
- Moradpour, A.; Peyrussan, V.; Johansen, I.; Bechgaard, K. High-Yield synthesis of tetramethyltetraselenafulvalene (TMTSF) avoiding the use of gaseous hydrogen selenide. J. Org. Chem. 1983, 48, 388–389. [Google Scholar]
- Bechgaard, K.; Cowan, D.O.; Bloch, A.N. Synthesis of the organic conductor tetramethyltetraselenofulvalenium 7,7,8,8-tetracyano-p-quinodimethanide (TMTSF-TCNQ)[4,4′,5,5′-tetramethyl-Δ2,2′-bis-1,3-diselenolium 3,6-bis-(dicyanomethylene)cyclohexadienide]. J. Chem. Soc. Chem. Commun. 1974, 937–938. [Google Scholar]
- Yamada, J.; Satoki, S.; Mishima, S.; Akashi, N.; Takahashi, K.; Masuda, N.; Nishimoto, Y.; Takasaki, S.; Anzai, H. Synthesis of unsymmetrical tetrathiafulvalene derivatives via Me3Al-promoted reactions of organotin compounds with esters. J. Org. Chem. 1996, 61, 3987–3995. [Google Scholar]
- Tetra-Methyl-Tetra-Thia-Fulvalene (TMTTF, 1). To a solution of 4,5-dimethyl-2,2-dibutyl-2-stanna-1,3-dithiole (4(a); 1600 mg, 4.56 mmol) in anhydrous dichloromethane (8.6 mL) was added at −78 °C a hexane solution of Me3Al (1.08 mol/L × 9.4 mL = 10.15 mmol). And then a solution of Methyl 4,5-dimethyl-1, 3-dithiole- 2-carboxylate (5(a); 720 mg, 3.81 mmol) in anhydrous dichloromethane (8.6 mL) was added. The mixture was warmed up to room temperature, and left stirring overnight. The reaction was quenched by the addition of saturated aqueous NaHCO3 solution at 0 °C. The aqueous layer was extracted with five portions of chloroform. The extracts were combined, dried over MgSO4, and the solvent was evaporated in vacuo. Recrystallization of the product from acetonitrile gave 1 (TMTTF), orange crystals; yield: 360 mg (43.7%). 1H-NMR (CDCl3/TMS): δ = 2.01(s, 12H). MS(FD): m/z (%): 263(22), 262(15), 261(M+, 100).
- Tetra-Methyl-Tetra-Selena-Fulvalene (TMTSF, 2). To a solution of 4,5-Dimethyl-2,2-dibutyl-2-stanna-1,3-diselenole (4(b); 1361 mg, 3.06 mmol) in anhydrous dichloromethane (9.8 mL) was added at −78 °C a hexane solution of Me3Al (1.08 mol/L × 5.6 mL = 6.02 mmol). And then a solution of Methyl 4,5-dimethyl-1, 3-diselenole- 2-carboxylate (5(b); 840 mg, 2.93 mmol) in anhydrous dichloromethane (9.8 mL) was added. The mixture was warmed up to room temperature, and left stirring overnight. The reaction was quenched by the addition of saturated aqueous NaHCO3 solution at 0 °C, and the mixture was filtered through Celite. The aqueous layer was extracted with three portions of chloroform. The extracts were combined, dried over MgSO4, and the solvent was evaporated in vacuo. And then the product was chromatographed (silica gel, hexane- chloroform). Recrystallization of the product from acetonitrile gave 2 (TMTSF), purple crystals; yield: 100 mg (7.6%). 1H-NMR (CDCl3/TMS): δ = 1.98(s,12H). MS(FD): m/z (%): 455(35), 454(13), 453(84), 452(29), 451(100), 450(46), 449(M+, 99), 448(50), 447(76), 446(42), 445(45), 444(22), 443(20).
- Svenstrup, N.; Rasmussen, K.M.; Hansen, T.K.; Becher, J. The chemistry of TTFTT; 1: New efficient synthesis and reactions of tetrathiafulvalene-2,3,6,7-tetrathiolate (TTFTT): An important building block in TTF-syntheses. Synthesis 1994, 809–812. [Google Scholar]
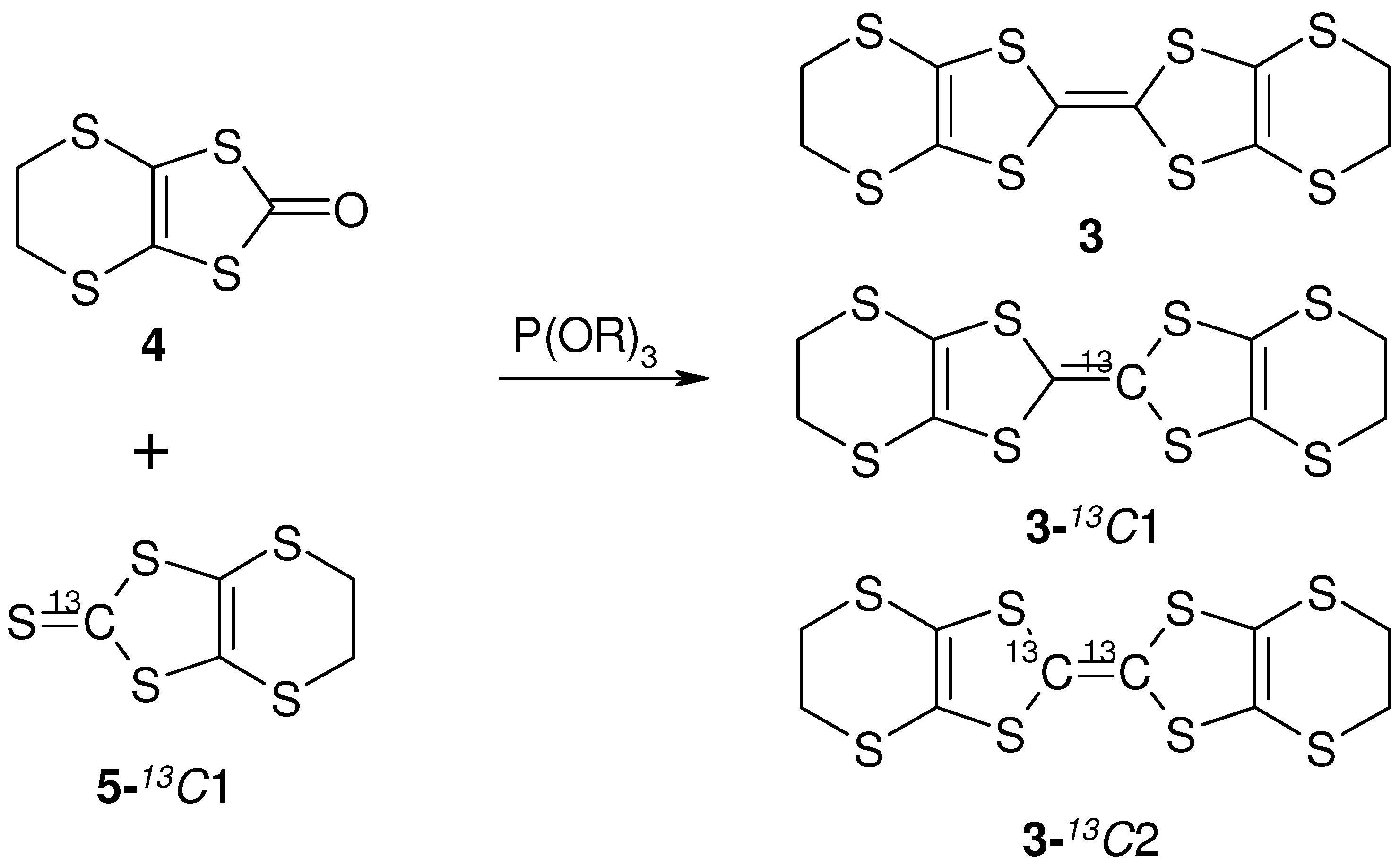
- Bis-(Ethylene-Dithio)-Tetra-Thia-Fulvalene (BEDT-TTF-13C1, 3). Enriched 2,3-Bis(cyanoethylthio)-6,7-ethylenedithiotetrathiafulvalene was prepared from the enriched thioketone form by the literature. To a solution of enriched 2,3-bis(cyanoethylthio)-6,7-ethylenedithiotetrathiafulvalene (12; 200 mg, 0.42 mmol) in degassed EtOH (10 mL) was added Sudium (40mg, 3.5mmol) in 3 mL of degassed EtOH under Ar and the mixture was stirred for 4 h. To the deep red solution was added 1,2-dibromoethane (219 mg, 1.1 mmol) in degassed EtOH (22 mL) and stirred overnight under Ar. The mixture was filtered. The product was purified by chromatography (silica gel, CS2) and recrystallized from chlorobenzene to give 3 (BEDT-TTF-13C1,), bright red crystals; yield: 58 mg (36%). 1H-NMR (CDCl3/TMS): δ = 3.27(s, 8H). MS(FD): m/z (%): 389(6), 388(5), 387(37), 386(18), 385(100), 384(3).
- Kimura, Y.; Misawa, M.; Kawamoto, A. Correlation between non-Fermi-liquid behavior and antiferromagnetic fluctuations in (TMTSF)2PF6 observed using 13C-NMR spectroscopy. Phys. Rev. B 2011, 84, 1–5. [Google Scholar]
- Hirose, S.; Kawamoto, A. 13C NMR study on the charge-ordering salt α'-(BEDT-TTF)2IBr2. Phys. Rev. B 2009, 80, 1–6. [Google Scholar]
- Hiraki, K.; Kanoda, K. Wigner crystal type of charge ordering in an organic conductor with a quarter-filled band: (DI-DCNQI)2Ag. Phys. Rev. Lett. 1998, 80, 4737–4740. [Google Scholar] [CrossRef]
- Eto, Y.; Kawamoto, A. Antiferromagnetic phase in β'-(BEDT-TTF)2ICl2 under pressure as seen via 13C NMR. Phys. Rev. B 2010, 81, 1–4. [Google Scholar]
- Leyraud, N.D.; Senzier, P.A.; Cotret, R.S.; Bourbonnais, C.; J’erome, D.; Bechgaard, K.; Taillefer, L. Correlation between linear resistivity and Tc in the Bechgaard salts and the pnictide superconductor Ba(Fe1−xCox)2As2. Phys. Rev. B 2009, 80, 1–5. [Google Scholar]
- Taniguchi, H.; Miyashita, M.; Uchiyama, K.; Satoh, K.; Mori, N.; Okamoto, H.; Miyagawa, K.; Kanoda, K.; Hedo, M.; Uwatoko, Y. Superconductivity at 14.2 K in layered organics under extreme pressure. J. Phys. Soc. Jpn. 2003, 72, 468–471. [Google Scholar]
- Yoneyama, N.; Miyazaki, A.; Enoki, T.; Saito, G. Magnetic properties of TTF-type charge transfer salts in the Mott insulator regime. Bull. Chem. Soc. Jpn. 1999, 72, 639–651. [Google Scholar] [CrossRef]
- Tokumoto, M.; Anzai, H.; Ishiguro, T.; Saito, G.; Kobayashi, H.; Kato, R.; Kobayashi, A. Electrical and magnetic properties of organic semiconductors, (BEDT-TTF)2X (X = IBr2, IBrCl, and ICl2). Synth. Met. 1987, 19, 215–220. [Google Scholar]
- Yamamoto, T.; Uruichi, M.; Yamamoto, K.; Yakushi, K.; Kawamoto, A.; Taniguchi, H. Examination of the charge-sensitive vibrational modes in bis(ethylenedithio)tetrathiafulvalene. J. Phys. Chem. B 2005, 109, 15226–15235. [Google Scholar]
- Chiba, R.; Hiraki, K.; Takahashi, T.; Yamamoto, H.M.; Nakamura, T. Charge disproportionation and dynamics in θ-(BEDT-TTF)2CsZn(SCN)4. Phys. Rev. B 2008, 77, 1–10. [Google Scholar]
- Kawai, T.; Kawamoto, A. 13C-NMR study of charge ordering state in the organic conductor, α-(BEDT-TTF)2I3. J. Phys. Soc. Jpn. 2009, 78, 1–6. [Google Scholar]
- Klutz, T.; Henning, I.; Haeberlen, U.; Schweitzer, D. Knight shift tensors and p-spin density in the organic metals αt-(BEDT-TTF)2I3 and (BEDT-TTF)2Cu(NCS)2. Appl. Magn. Reson. 1991, 2, 441. [Google Scholar]
- Itaya, M.; Eto, Y.; Kawamoto, A.; Taniguchi, H. Antiferromagnetic fluctuations in the organic superconductor κ-(BEDT-TTF)2Cu(NCS)2 under Pressure. Phys. Rev. Lett. 2009, 102, 1–4. [Google Scholar]
- Strack, C.; Akinci, C.; Paschenko, V.; Wolf, B.; Uhrig, E.; Assmus, W.; Lang, M.; Schreuer, J.; Wiehl, L.; Schlueter, J.A.; et al. Resistivity studies under hydrostatic pressure on a low-resistance variant of the quasi-two-dimensional organic superconductor κ-(BEDT-TTF)2Cu[N(CN)2]Br: Search for intrinsic scattering contributions. J. Phys. Rev. B 2005, 72, 054511. [Google Scholar]
- Müller, J.; Lang, M.; Steglich, F.; Schlueter, J.A.; Kini, A.M.; Sasaki, T. Evidence for structural and electronic instabilities at intermediate temperatures in κ-(BEDT-TTF)2X for X = Cu[N(CN)2]Cl, Cu[N(CN)2]Br and Cu(NCS)2: Implications for the phase diagram of these quasi-two-dimensional organic superconductors. Phys. Rev. B 2002, 65, 1–14. [Google Scholar]
- Kuwata, Y.; Itaya, M.; Kawamoto, A. Low-Frequency dynamics of κ-(BEDT-TTF)2Cu(NCS)2 observed by 13C NMR. Phys. Rev. B 2011, 83, 1–5. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hirose, S.; Misawa, M.; Kawamoto, A. Magnetic and Electric Properties of Organic Conductors Probed by 13C-NMR Using Selective-Site Substituted Molecules. Crystals 2012, 2, 1034-1057. https://doi.org/10.3390/cryst2031034
Hirose S, Misawa M, Kawamoto A. Magnetic and Electric Properties of Organic Conductors Probed by 13C-NMR Using Selective-Site Substituted Molecules. Crystals. 2012; 2(3):1034-1057. https://doi.org/10.3390/cryst2031034
Chicago/Turabian StyleHirose, Shinji, Masaki Misawa, and Atsushi Kawamoto. 2012. "Magnetic and Electric Properties of Organic Conductors Probed by 13C-NMR Using Selective-Site Substituted Molecules" Crystals 2, no. 3: 1034-1057. https://doi.org/10.3390/cryst2031034
APA StyleHirose, S., Misawa, M., & Kawamoto, A. (2012). Magnetic and Electric Properties of Organic Conductors Probed by 13C-NMR Using Selective-Site Substituted Molecules. Crystals, 2(3), 1034-1057. https://doi.org/10.3390/cryst2031034