
A Silicon Complex of 1,4,7,10-Tetraazacyclododecane (Cyclen) with Unusual Coordination Geometry


Abstract
1. Introduction
2. Experimental Section
2.1. General
2.2. Synthesis of (1-Hydrogen-1,4,7,10-tetraazacyclododecane)silicon(IV) Chloride (2)
2.3. Single-Crystal Structure Determination
2.4. Quantum Chemical Calculations
3. Results and Discussion
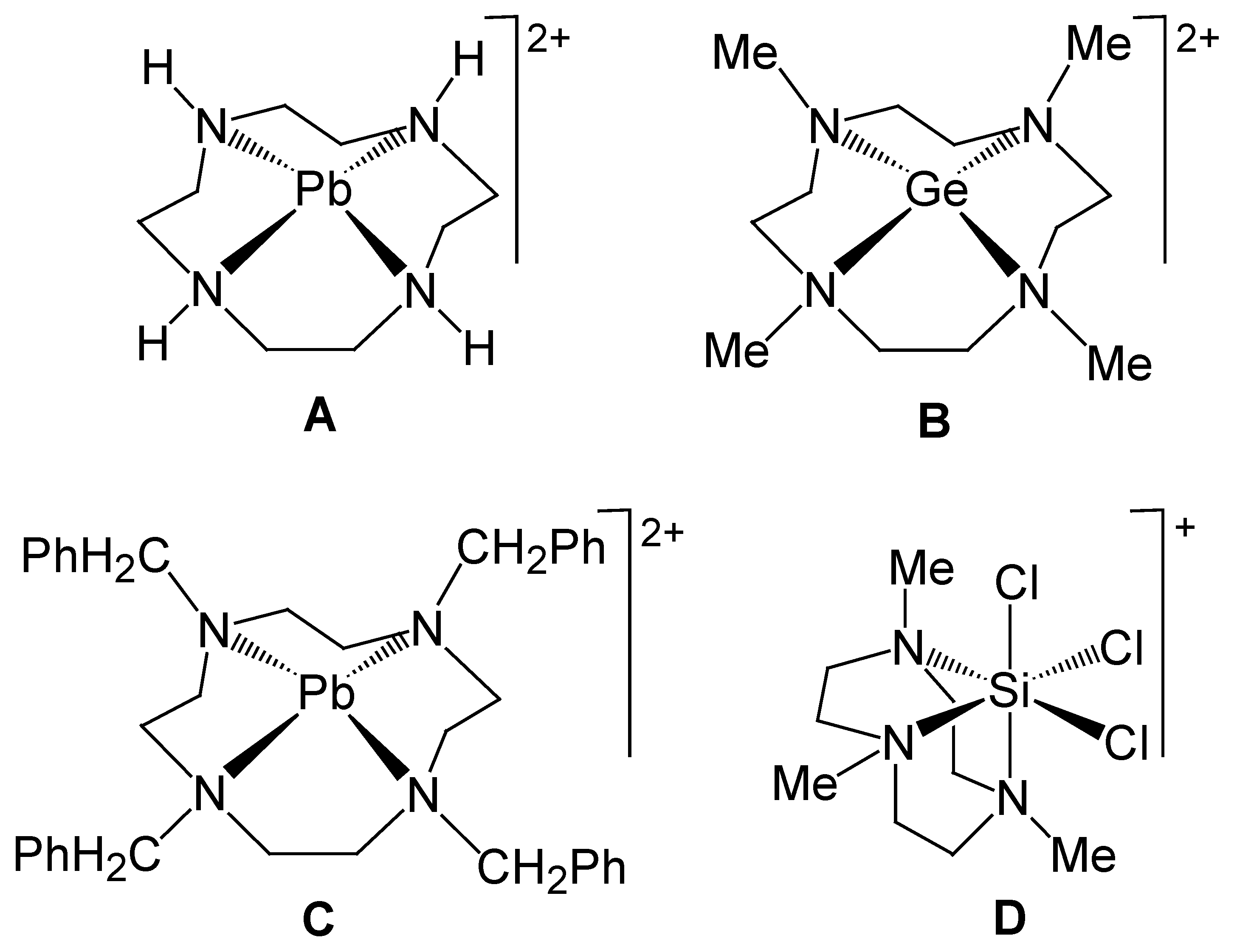
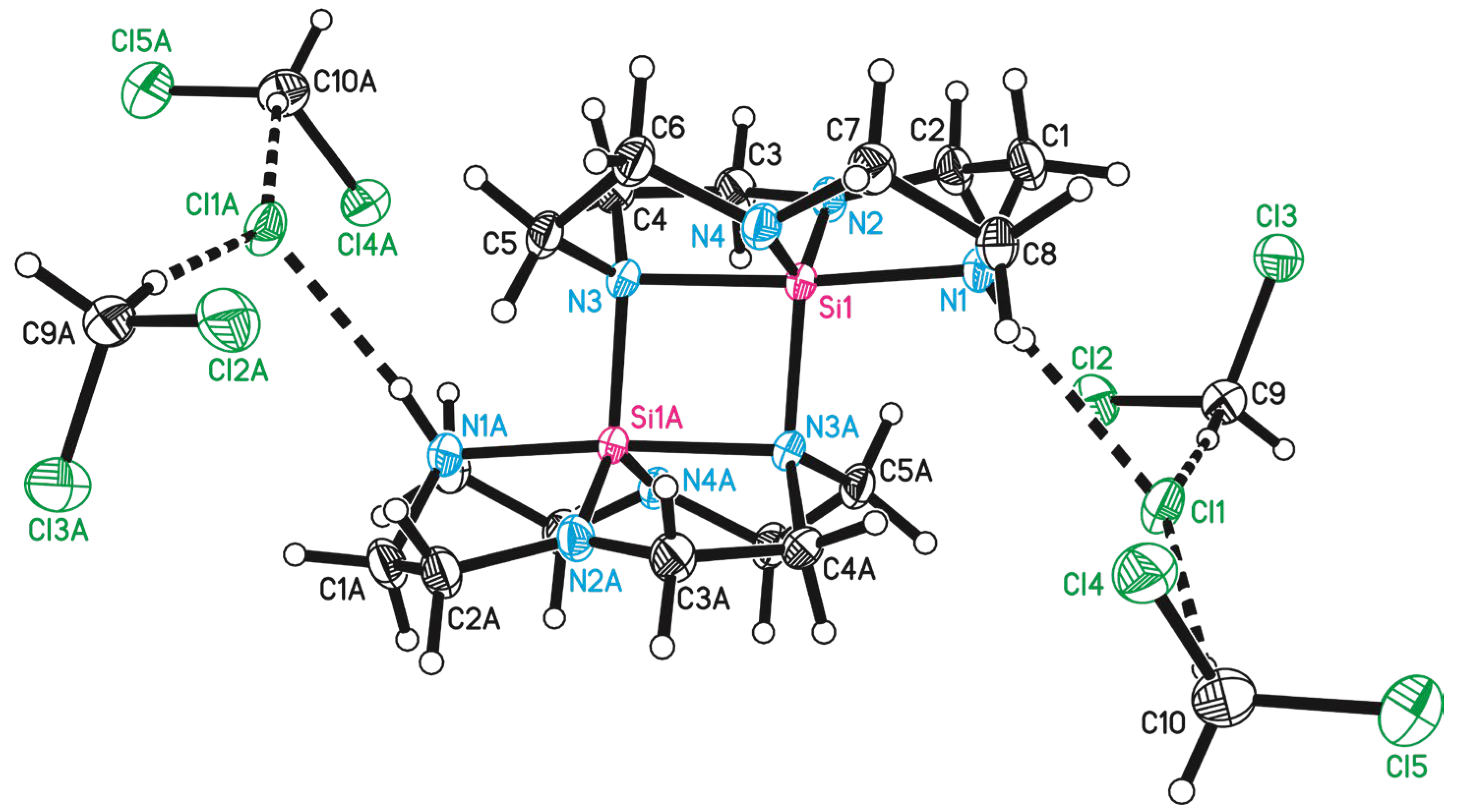
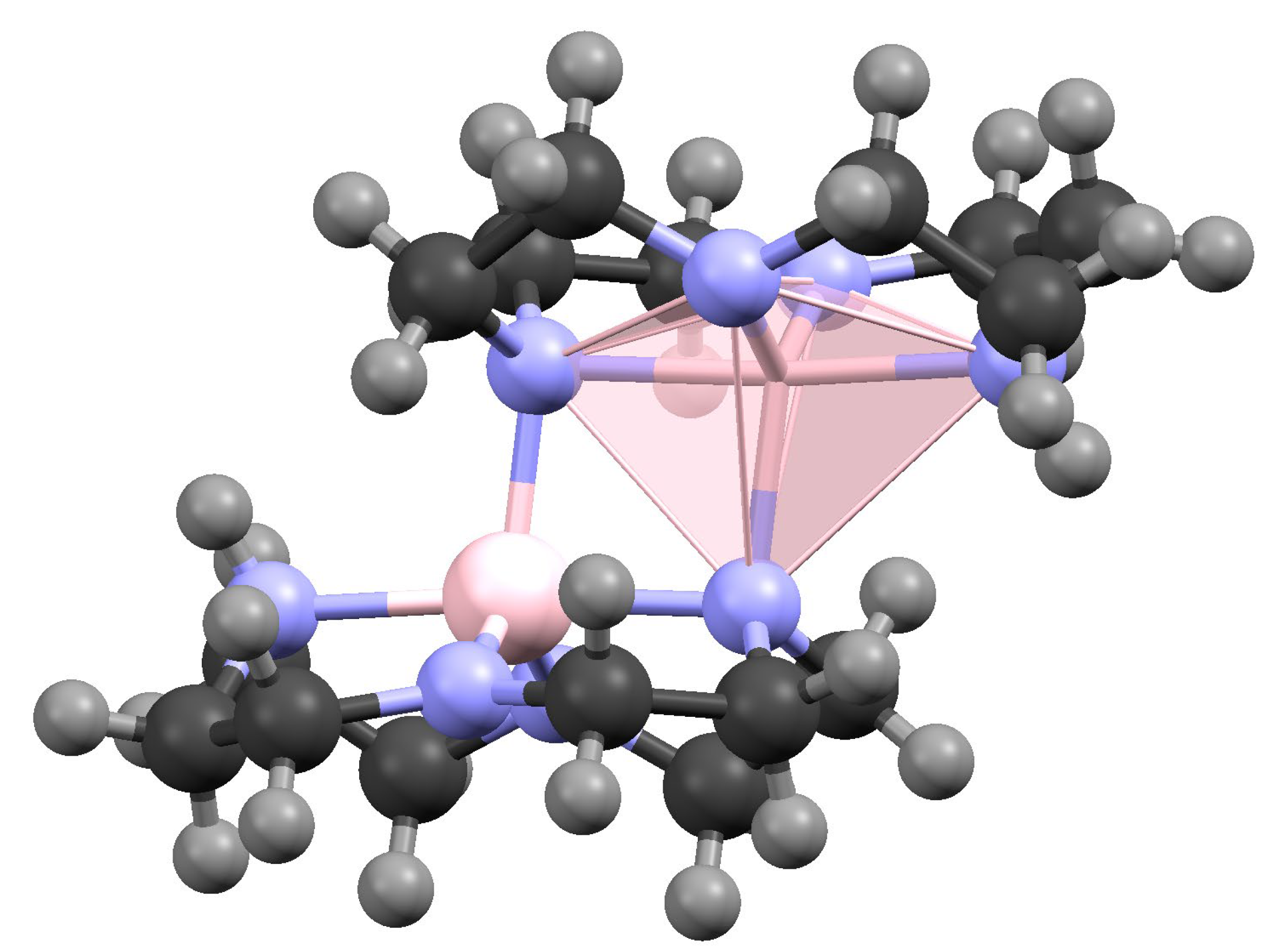
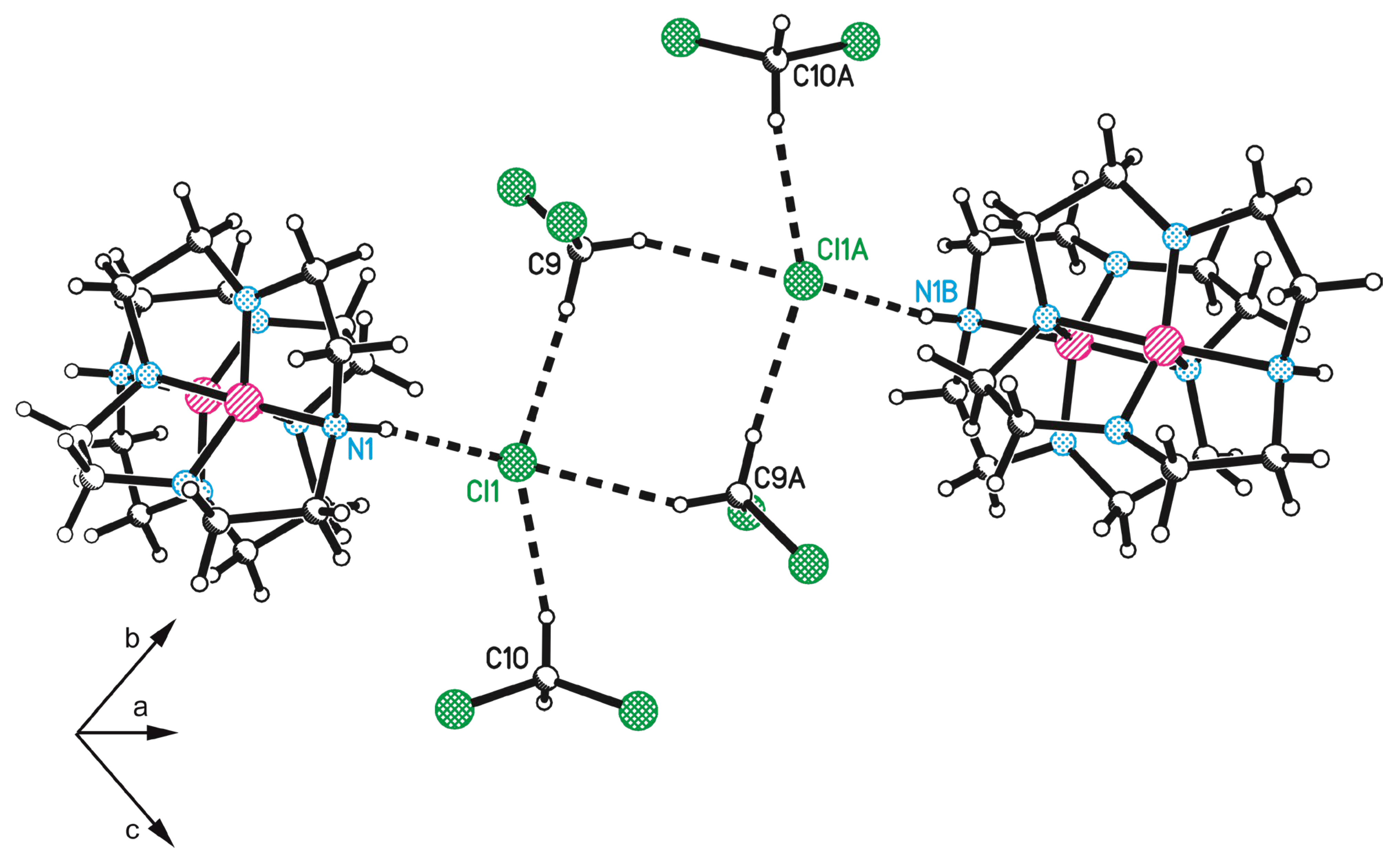
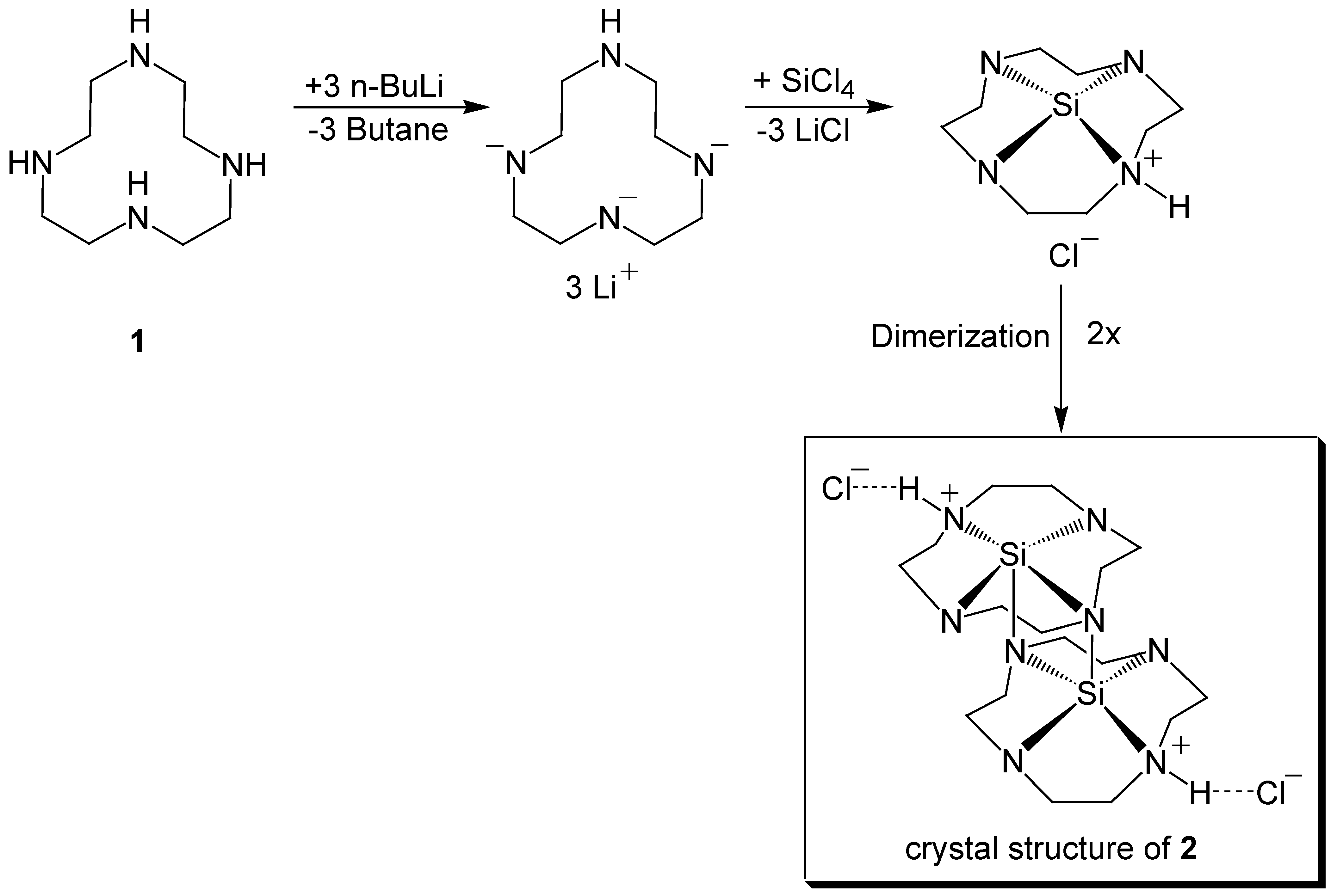
3.1. Preparation and Crystal Structure Analysis
3.2. Discussion of the Outcome of the Reaction
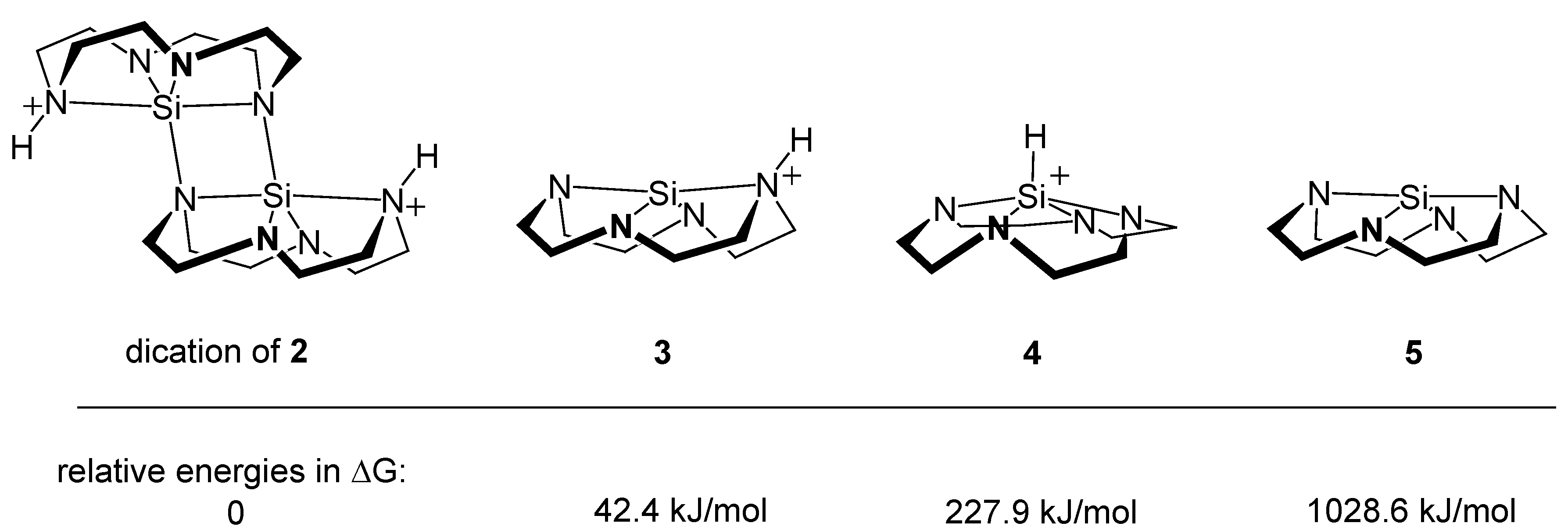
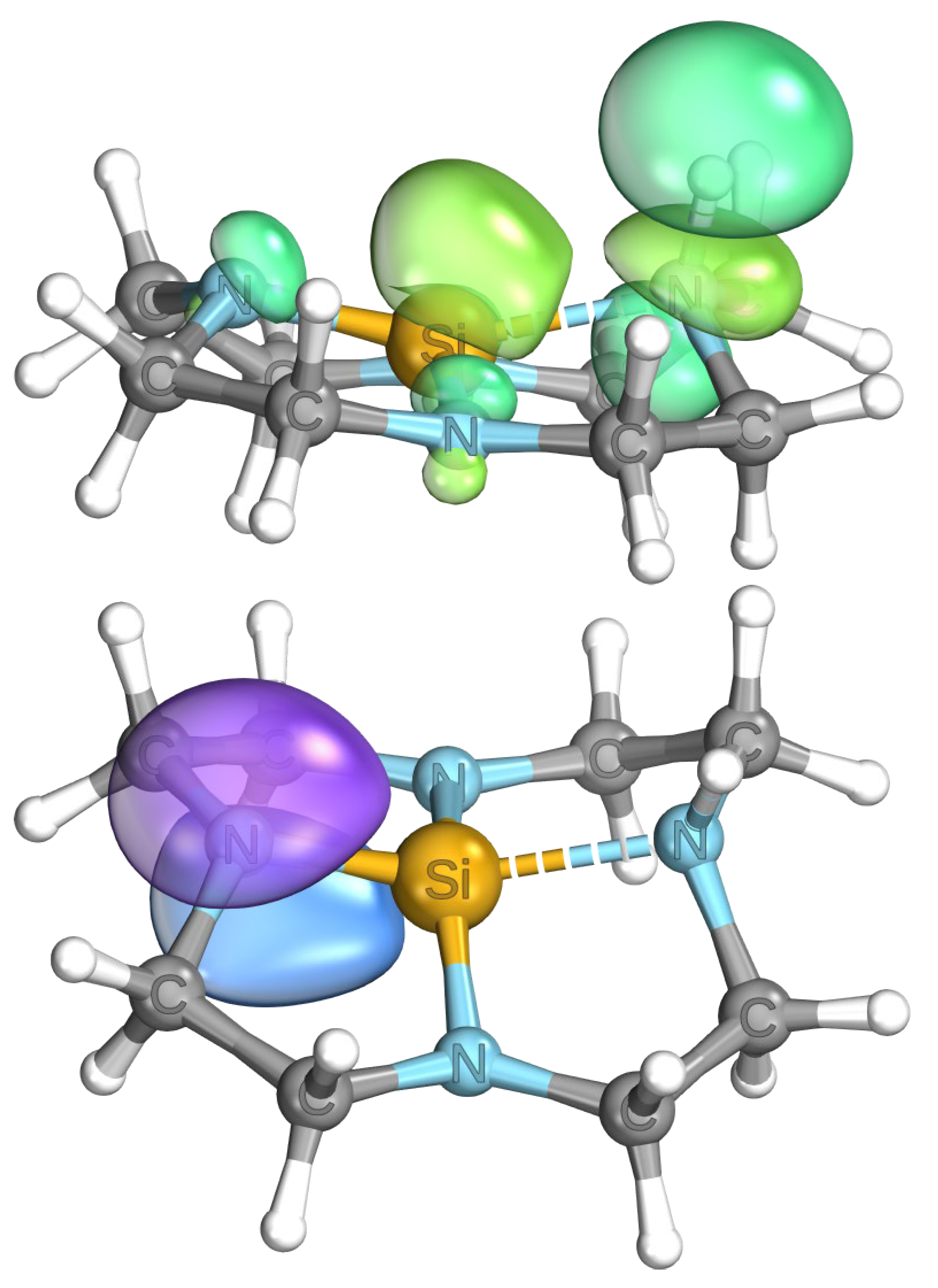
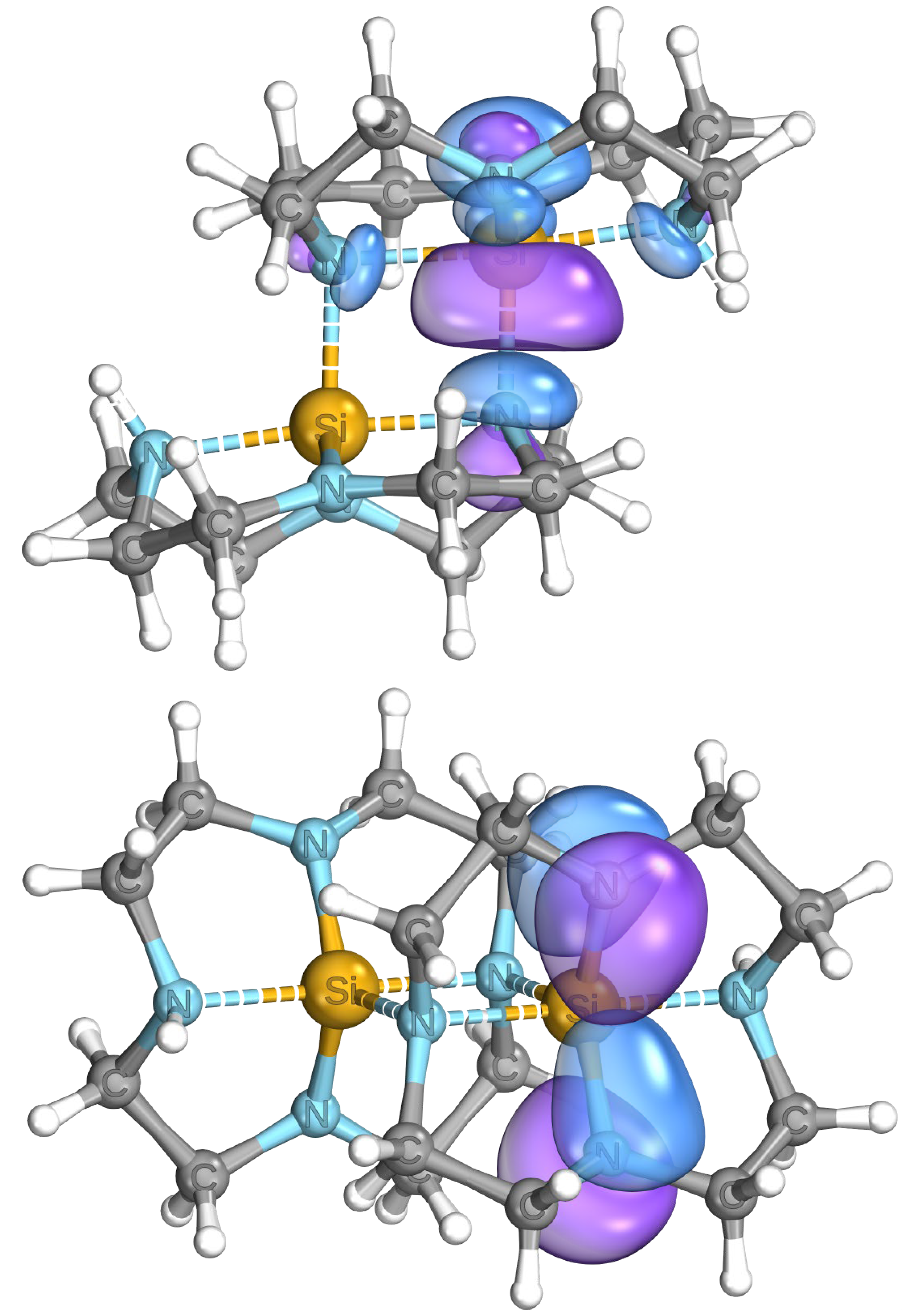
3.3. Quantum Chemical Calculations and Perspectives in Reactivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Curtis, F.N. Macrocyclic Ligands. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 1, pp. 447–473. [Google Scholar]
- Wang, Z.-F.; Zhou, X.-F.; Wei, Q.-C.; Qin, Q.-P.; Li, J.-X.; Tan, M.-X.; Zhang, S.-H. Novel bifluorescent Zn(II)-cryptolepine-cyclen complexes trigger apoptosis induced by nuclear and mitochondrial DNA damage in cisplatin-resistant lung tumor cells. Eur. J. Med. Chem. 2022, 238, 114418. [Google Scholar] [CrossRef] [PubMed]
- Ichimaru, Y.; Kato, K.; Nakatani, R.; Sugiura, K.; Mizutani, H.; Kinoshita-Kikuta, E.; Kurosaki, H. Characterization of zinc(II) complex of 1,4,7,10-tetrazacyclododecane and deprotonated 5-fluorouracil (FU) in crystalline/solution states and evaluation of anticancer activity: Approach for improving the anticancer activity of FU. Inorg. Chem. Commun. 2023, 147, 110221. [Google Scholar] [CrossRef]
- Du, M.-M.; Sun, B.-C.; Wang, B.-B.; Luo, Y.; Zhang, L.-L.; Chu, G.-W.; Chen, J.-F. Kinetic study on the reaction between CO2 and tertiary amine catalyzed by zinc(II) aza-macrocyclic complexes. AIChE J. 2024, 70, e18366. [Google Scholar] [CrossRef]
- Ma, X.; Li, Y.; Meng, L.; Li, L.; Li, Y.; Zhang, H.; Hua, J. An 1,4,7,10-tetraazacyclododecane-nickel complex functionalized polymolybdate acting as artificial oxidase in removing methylene blue. J. Mol. Struct. 2024, 1312, 138502. [Google Scholar] [CrossRef]
- Kovács, A. Favorable Symmetric Structures of Radiopharmaceutically Important Neutral Cyclen-Based Ligands. Symmetry 2024, 16, 1668. [Google Scholar] [CrossRef]
- Burcevs, A.; Jonusauskas, G.; Novosjolova, I.; Turks, M. Synthesis of Purine-1,4,7,10-Tetraazacyclododecane Conjugate and Its Complexation Modes with Copper(II). Molecules 2025, 30, 1612. [Google Scholar] [CrossRef]
- Berchel, M.; Malouch, D.; Beyler, M.; Fauchon, M.; Toueix, Y.; Hellio, C.; Jaffrès, P.-A. Antifouling Properties of N,N′-Dialkylated Tetraazamacrocyclic Polyamines and Their Metal Complexes. Molecules 2025, 30, 2368. [Google Scholar] [CrossRef]
- Osei, M.K.; Lucht, B.; Xu, H.-L.; Valles, A.; Espinoza Castro, V.M.; La, N.; Hernández Sánchez, R. Cyclen-Based Octaamine Ligand Supporting the Formation of Dinuclear Metal Compounds. Inorg. Chem. 2025, 64, 6408–6413. [Google Scholar] [CrossRef]
- Hierlmeier, I.; Randhawa, P.; McNeil, B.L.; Oliveri, E.; Maus, S.; Radchenko, V.; Bartholomä, M.D. Radiochemistry of cyclen-derived chelators comprising five-membered azaheterocyclic arms with 212Pb2+, 213Bi3+, and 225Ac3+. Nucl. Med. Biol. 2025, 146–147, 109034. [Google Scholar] [CrossRef]
- Everett, M.; Jolleys, A.; Levason, W.; Light, M.E.; Pugh, D.; Reid, G. Cationic aza-macrocyclic complexes of germanium(II) and silicon(IV). Dalton Trans. 2015, 44, 20898–20905. [Google Scholar] [CrossRef]
- McNeil, B.L.; Kadassery, K.J.; McDonagh, A.W.; Zhou, W.; Schaffer, P.; Wilson, J.J.; Ramogida, C.F. Evaluation of the Effect of Macrocyclic Ring Size on [203Pb]Pb(II) Complex Stability in Pyridyl-Containing Chelators. Inorg. Chem. 2022, 61, 9638–9649. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.B.; Brown, T.R.; Reibenspies, J.H.; Lee, H.-S.; Hancock, R.D. Exciplex formation as an approach to selective Copper(II) fluorescent sensors. Inorg. Chim. Acta 2020, 506, 119544. [Google Scholar] [CrossRef]
- Ebner, F.; Greb, L. An isolable, crystalline complex of square-planar silicon(IV). Chem 2021, 7, 2151–2159. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.; Jöst, C.; Mackenroth, A.; Greb, L. Synthesis of Electron-Rich, Planarized Silicon(IV) Species and a Theoretical Analysis of Dimerizing Aminosilanes. Chem. Eur. J. 2017, 23, 17764–17774. [Google Scholar] [CrossRef]
- Power, P.P. Main-group elements as transition metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef]
- Herzog, S.; Dehnert, J. Eine rationelle anaerobe Arbeitsmethode. Z. Chem. 1964, 4, 1–11. [Google Scholar] [CrossRef]
- Böhme, U. Inertgastechnik; Walter deGruyter: Berlin, Germany, 2020. [Google Scholar] [CrossRef]
- Stoe & Cie. X-RED (Version 2.3.1) and X-AREA (Version 2.3), Stoe & Cie: Darmstadt, Germany, 2009.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian, version 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Knizia, G. Intrinsic Atomic Orbitals: An Unbiased Bridge between Quantum Theory and Chemical Concepts. J. Chem. Theory Comput. 2013, 9, 4834–4843. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef]
- Weigend, F.; Furche, F.; Ahlrichs, R. Gaussian basis sets of quadruple zeta valence quality for atoms H-Kr. J. Chem. Phys. 2003, 119, 12753–12762. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua [1,7-bis (N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]-copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1984, 1349–1356. [Google Scholar] [CrossRef]
- Kaftory, M.; Kapon, M.; Botoshansky, M. The structural chemistry of organosilicon compounds. In The Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; Jonh Wiley & Sons: Hoboken, NJ, USA, 1998; Volume 2, pp. 181–265. [Google Scholar]
- Chuit, C.; Corriu, R.J.P.; Catherine, R.; Young, J.C. Reactivity of penta- and hexacoordinate silicon compounds and their role as reaction intermediates. Chem. Rev. 1993, 93, 1371–1448. [Google Scholar] [CrossRef]
- Kost, D.; Kalikhman, I. Hypervalent Silicon Compounds. In The Chemistry of Organic Silicon Compounds; PATAI’S Chemistry of Functional Groups; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 1–107. [Google Scholar] [CrossRef]
- Böhme, U.; Wiesner, S.; Günther, B. Easy access to chiral penta- and hexacoordinate silicon compounds. Inorg. Chem. Commun. 2006, 9, 806–809. [Google Scholar] [CrossRef]
- Böhme, U.; Fels, S. A new type of chiral pentacoordinated silicon compounds with azomethine ligands made from acetylacetone and amino acids. Inorg. Chim. Acta 2013, 406, 251–255. [Google Scholar] [CrossRef]
- Paul, L.E.H.; Foehn, I.C.; Schwarzer, A.; Brendler, E.; Böhme, U. Salicylaldehyde-(2-hydroxyethyl)imine—A flexible ligand for group 13 and 14 elements. Inorg. Chim. Acta 2014, 423, 268–280. [Google Scholar] [CrossRef]
- Schwarzer, S.; Böhme, U.; Fels, S.; Günther, B.; Brendler, E. (S)-N-[(2-hydroxynaphthalen-1-yl)methylidene]valine—A valuable ligand for the preparation of chiral complexes. Inorg. Chim. Acta 2018, 483, 136–147. [Google Scholar] [CrossRef]
- Driess, M.; Muresan, N.; Merz, K. A Novel Type of Pentacoordinate Silicon Complexes and Unusual Ligand Coupling by Intramolecular Electron Transfer. Angew. Chem. Int. Ed. 2005, 44, 6738–6741. [Google Scholar] [CrossRef]
- Frey, G.D.; Lavallo, V.; Donnadieu, B.; Schoeller, W.W.; Bertrand, G. Facile Splitting of Hydrogen and Ammonia by Nucleophilic Activation at a Single Carbon Center. Science 2007, 316, 439–441. [Google Scholar] [CrossRef]
- von Grotthuss, E.; Diefenbach, M.; Bolte, M.; Lerner, H.-W.; Holthausen, M.C.; Wagner, M. Reversible Dihydrogen Activation by Reduced Aryl Boranes as Main-Group Ambiphiles. Angew. Chem. Int. Ed. 2016, 55, 14067–14071. [Google Scholar] [CrossRef]
- Lichtenberg, C. Well-Defined, Mononuclear BiI and BiII Compounds: Towards Transition-Metal-Like Behavior. Angew. Chem. Int. Ed. 2016, 55, 484–486. [Google Scholar] [CrossRef]
- Knerr, S.; Böhme, U.; Herbig, M. Coordinative Unsaturation in an Antimony(III)-Complex with the 2-Salicylidenaminophenolato Ligand: Synthesis, Crystal Structure, Spectroscopic Analysis, and DFT Studies. Crystals 2023, 13, 1300. [Google Scholar] [CrossRef]
- Gerwig, M.; Böhme, U.; Friebel, M. Challenges in the Synthesis and Processing of Hydrosilanes as Precursors for Silicon Deposition. Chem. Eur. J. 2024, 30, e202400013. [Google Scholar] [CrossRef]
- Böhme, U.; Riedel, S. Trichlorosilyl Anions of p-Block Elements—New Applications for Simple Molecules. Eur. J. Inorg. Chem. 2022, 2022, e202200571. [Google Scholar] [CrossRef]
- Ebner, F.; Greb, L. Calix[4]pyrrole Hydridosilicate: The Elusive Planar Tetracoordinate Silicon Imparts Striking Stability to Its Anionic Silicon Hydride. J. Amer. Chem. Soc. 2018, 140, 17409–17412. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Dong, S.; Yao, S.; Zhu, J.; Driess, M. Synthesis and Reactivity of an Anti-van’t Hoff/Le Bel Compound with a Planar Tetracoordinate Silicon(II) Atom. J. Amer. Chem. Soc. 2023, 145, 7084–7089. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Formula | C20H42Cl10N8Si2 |
| Mr | 805.29 |
| T (K) | 153 K |
| λ (Å) | 0.71073 |
| Crystal system | Triclinic |
| Space group | P 1 |
| a (Å) | 9.0804(6) |
| b (Å) | 9.7215(7) |
| c (Å) | 11.0648(7) |
| α (°) | 67.070(5) |
| β (°) | 88.222(6) |
| γ (°) | 75.748(5) |
| V (Å3) | 869.57(11) |
| Z | 1 |
| ρcalc (g.cm−3) | 1.538 |
| μ (mm−1) | 0.898 |
| F(000) | 416 |
| θmax (°) | 27.323 |
| Reflections collected/unique [Rint] | 25,897/3833 [R(int) = 0.0392] |
| Completeness to θ = 25.242° | 98.1% |
| Absorption correction | face-indexing absorption correction |
| Max. and min. transmission | 0.9405 and 0.7674 |
| Data/restraints/parameters | 3833/0/185 |
| GoF on F2 | 1.096 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0322, wR2 = 0.0811 |
| R indices (all data) | R1 = 0.0351, wR2 = 0.0852 |
| Largest peak and hole (e.Å−3) | 0.348 and −0.552 |
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| Si1-N1 | 1.922(1) | Si1-N3A | 1.830(1) |
| Si1-N2 | 1.695(1) | Si1-Si1A | 2.7972(8) |
| Si1-N3 | 1.956(1) | ||
| Si1-N4 | 1.708(1) | ||
| N1-Si1-N3 | 175.12(6) | N2-Si1-N3A | 113.77(7) |
| N2-Si1-N4 | 128.60(7) | N4-Si1-N3A | 117.12(6) |
| N2-Si1-N1 | 88.69(6) | N1-Si1-N3A | 99.96(6) |
| N4-Si1-N1 | 89.46(6) | N3-Si1-N3A | 84.81(6) |
| N2-Si1-N3 | 88.42(6) | ||
| N4-Si1-N3 | 89.28(6) |
| D-H⋯A | d(D-H) | d(H⋯A) | d(D⋯A) | <(DHA) |
|---|---|---|---|---|
| C5-H5A⋯N2#1 | 0.99 | 2.55 | 3.176(2) | 121.3 |
| N1-H1C⋯Cl1 | 0.89(2) | 2.28(2) | 3.1556(14) | 166.1(19) |
| C9-H9A⋯Cl1 | 0.99 | 2.60 | 3.578(2) | 170.2 |
| C9-H9B⋯Cl1#2 | 0.99 | 2.74 | 3.5987(19) | 145.8 |
| C10-H10B⋯Cl1 | 0.99 | 2.61 | 3.533(2) | 155.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Böhme, U.; Herbig, M.; Günther, B. A Silicon Complex of 1,4,7,10-Tetraazacyclododecane (Cyclen) with Unusual Coordination Geometry. Crystals 2025, 15, 635. https://doi.org/10.3390/cryst15070635
Böhme U, Herbig M, Günther B. A Silicon Complex of 1,4,7,10-Tetraazacyclododecane (Cyclen) with Unusual Coordination Geometry. Crystals. 2025; 15(7):635. https://doi.org/10.3390/cryst15070635
Chicago/Turabian StyleBöhme, Uwe, Marcus Herbig, and Betty Günther. 2025. "A Silicon Complex of 1,4,7,10-Tetraazacyclododecane (Cyclen) with Unusual Coordination Geometry" Crystals 15, no. 7: 635. https://doi.org/10.3390/cryst15070635
APA StyleBöhme, U., Herbig, M., & Günther, B. (2025). A Silicon Complex of 1,4,7,10-Tetraazacyclododecane (Cyclen) with Unusual Coordination Geometry. Crystals, 15(7), 635. https://doi.org/10.3390/cryst15070635