
Computational Modeling of Cation Diffusion in Isolated Nanocrystals of Mixed Uranium, Plutonium and Thorium Dioxides

 , ,
, , 
Abstract
1. Introduction
2. Modeling Methodology
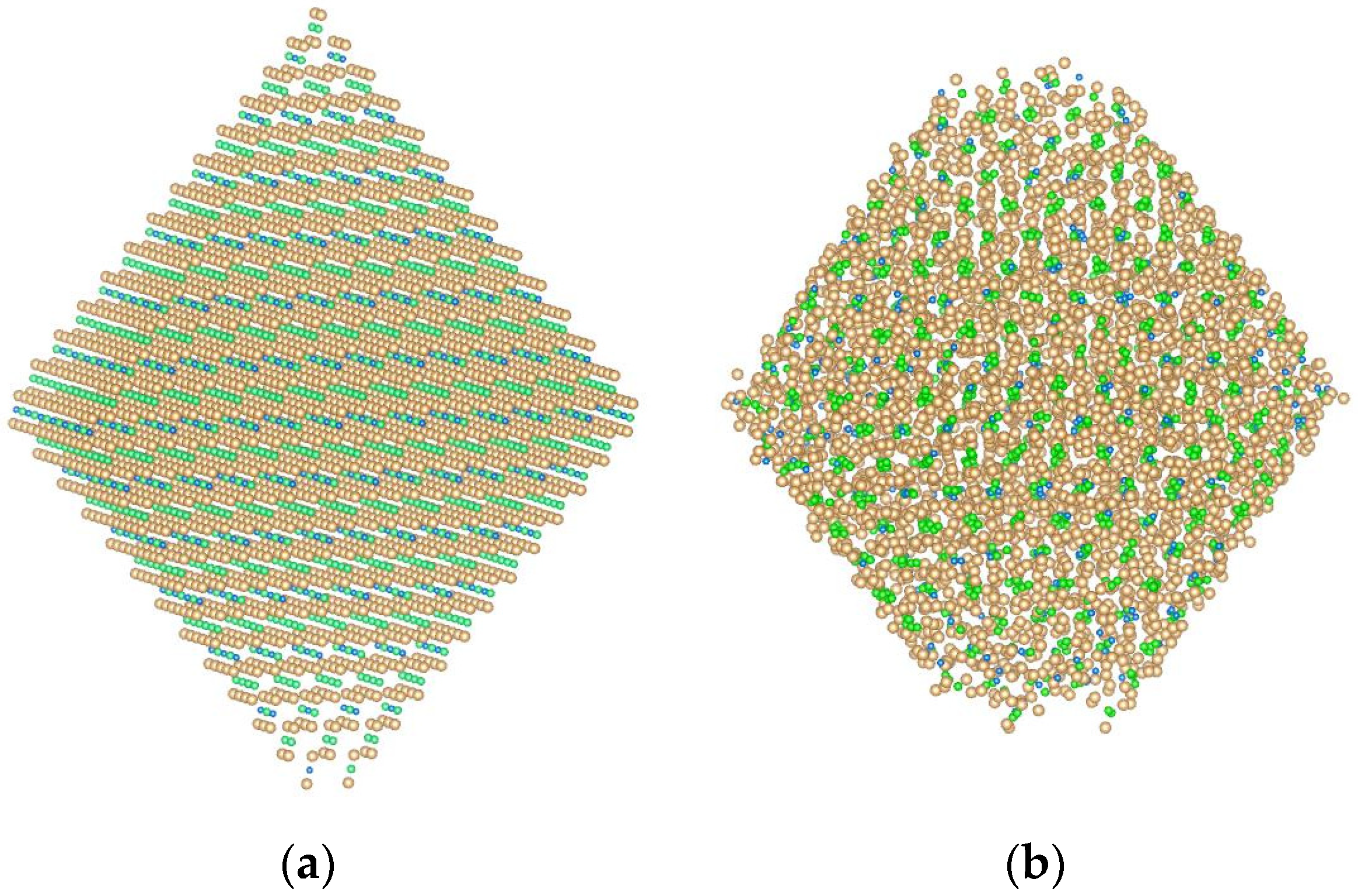
2.1. Model System and Interatomic Potentials
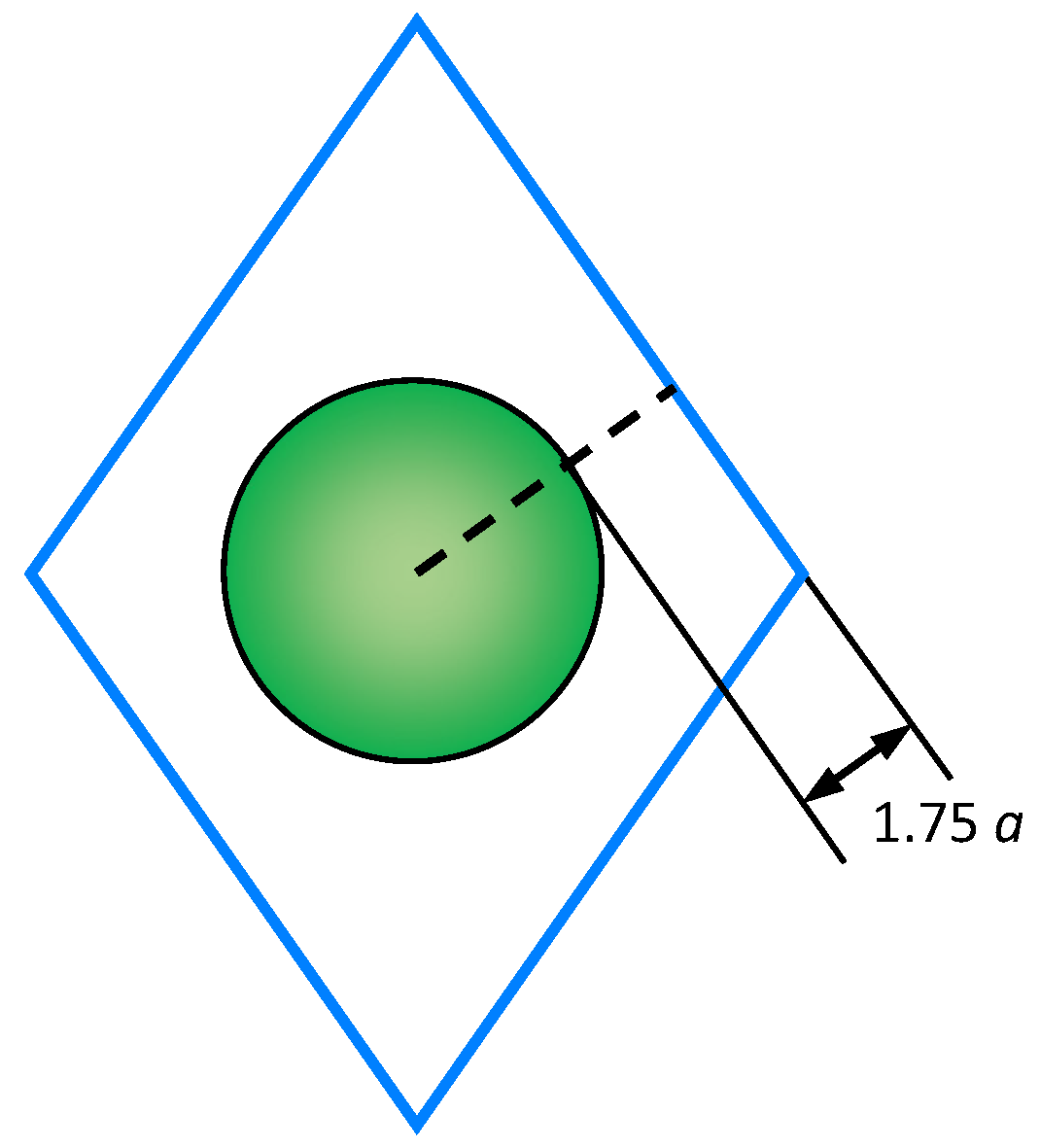
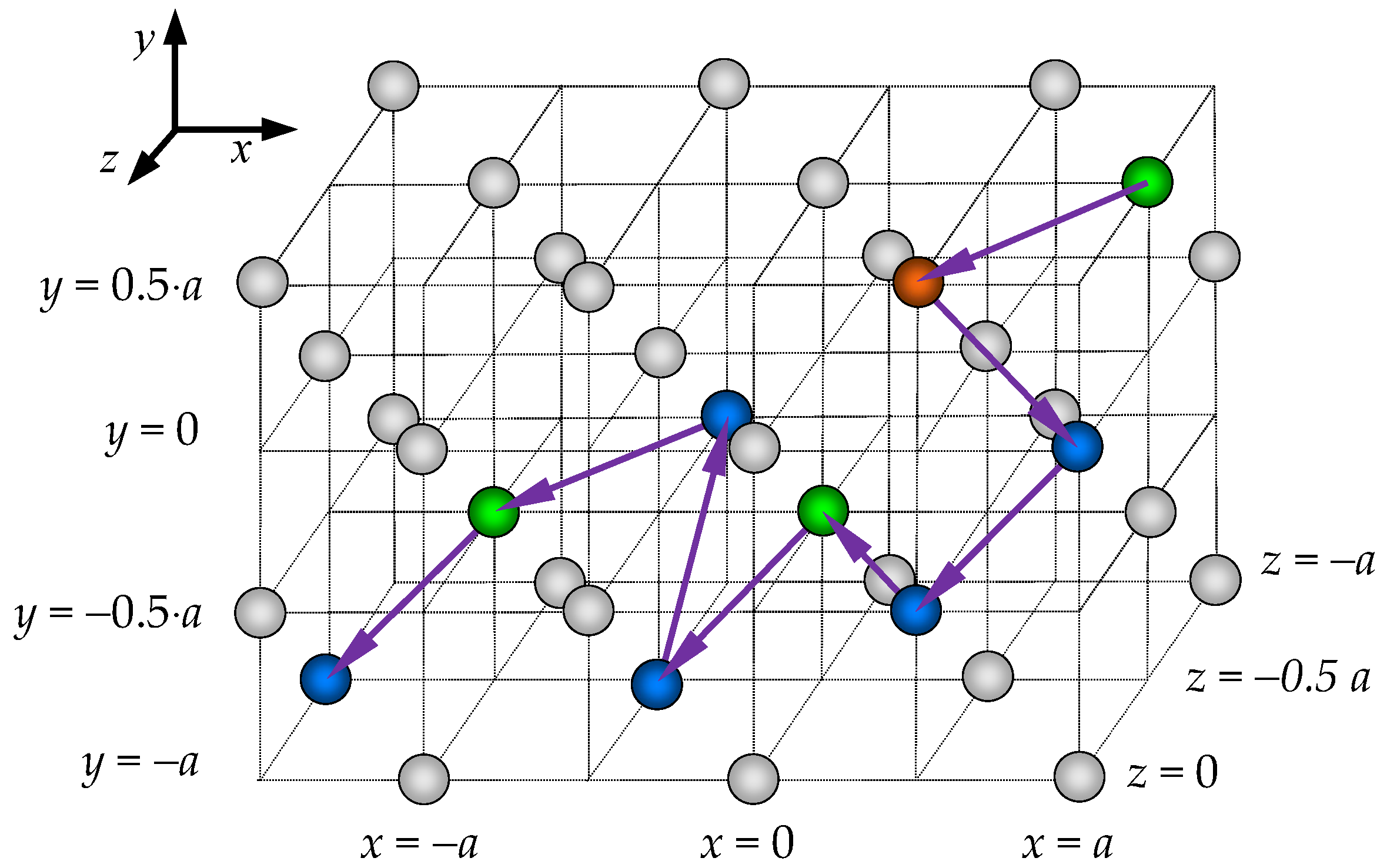
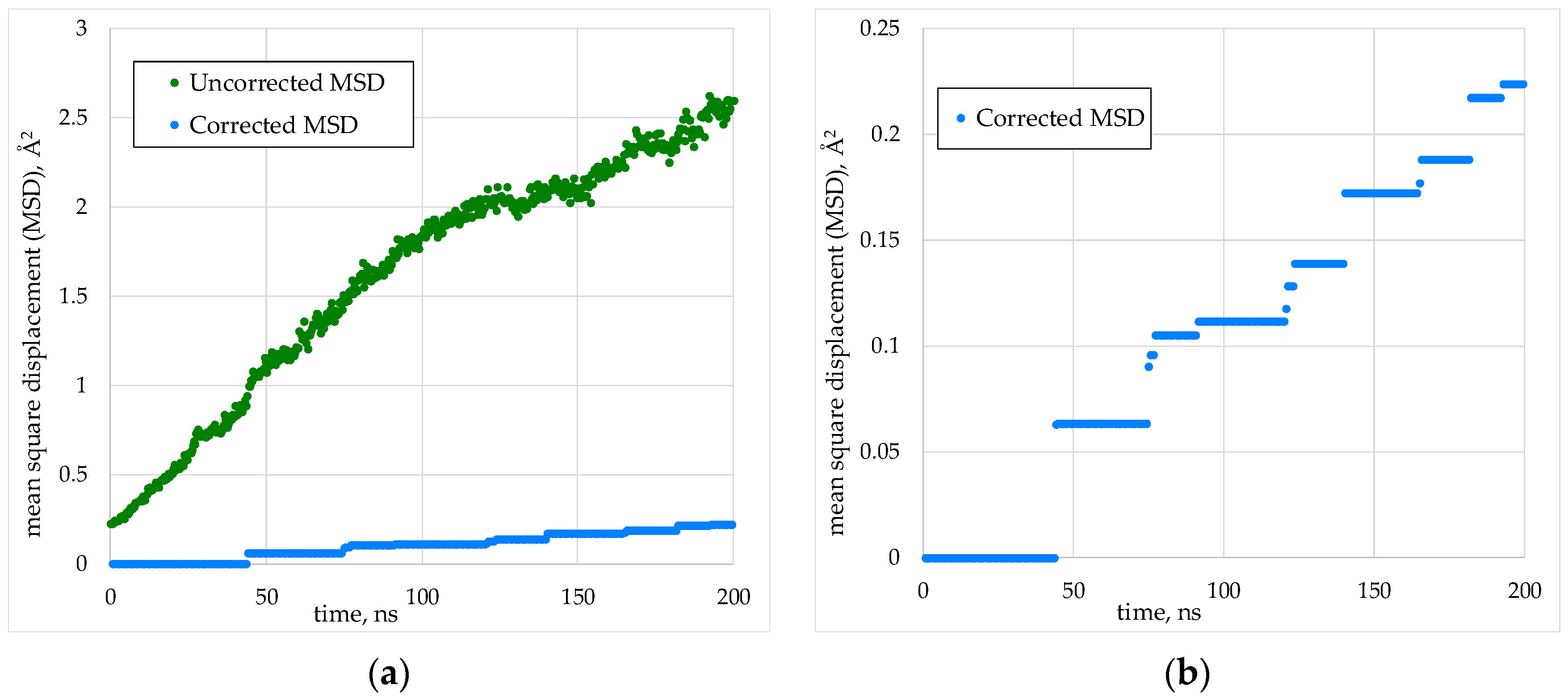
2.2. Calculation of the Cation Diffusion Coefficients
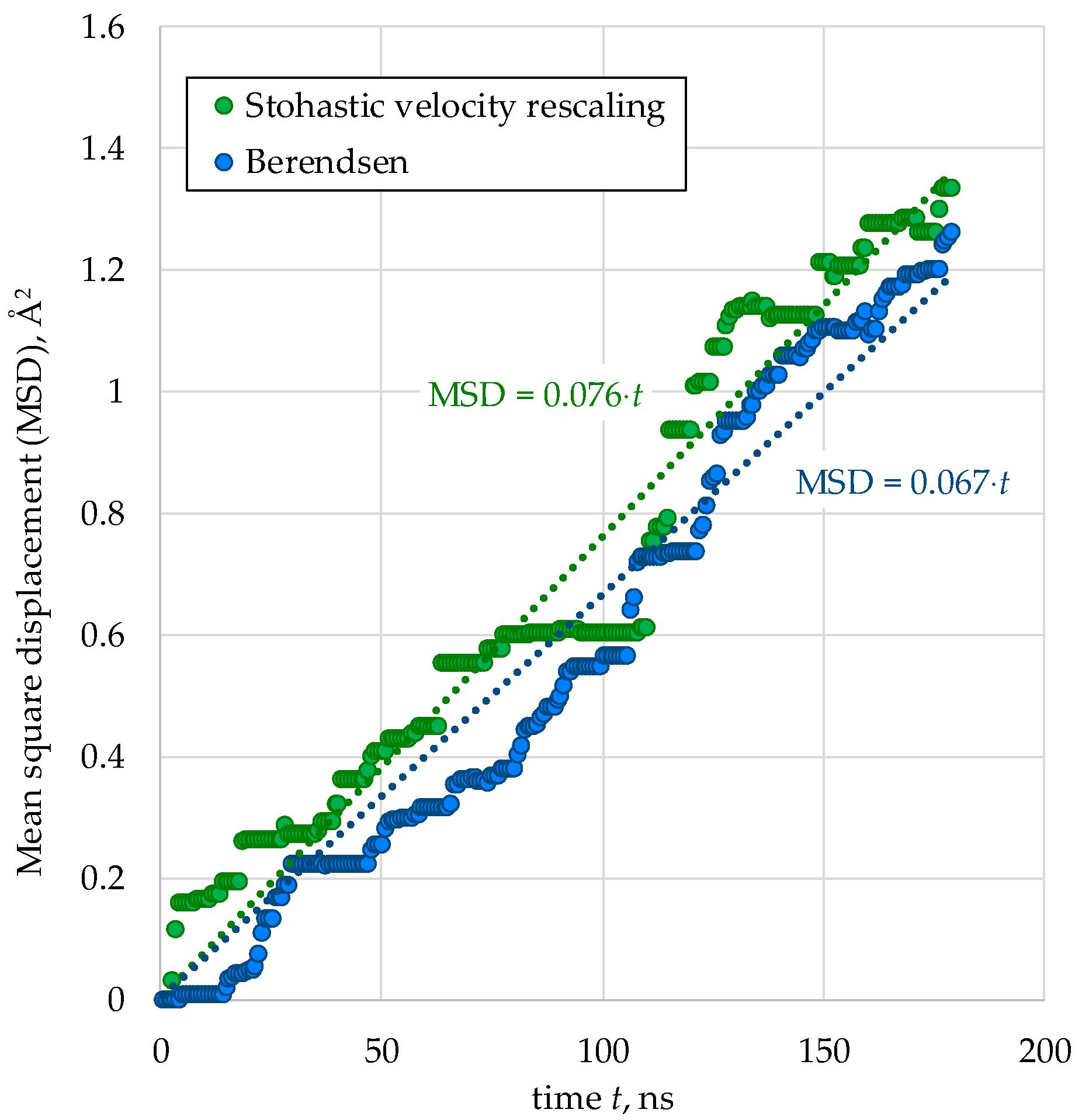
2.3. Thermostating of the Model Systems
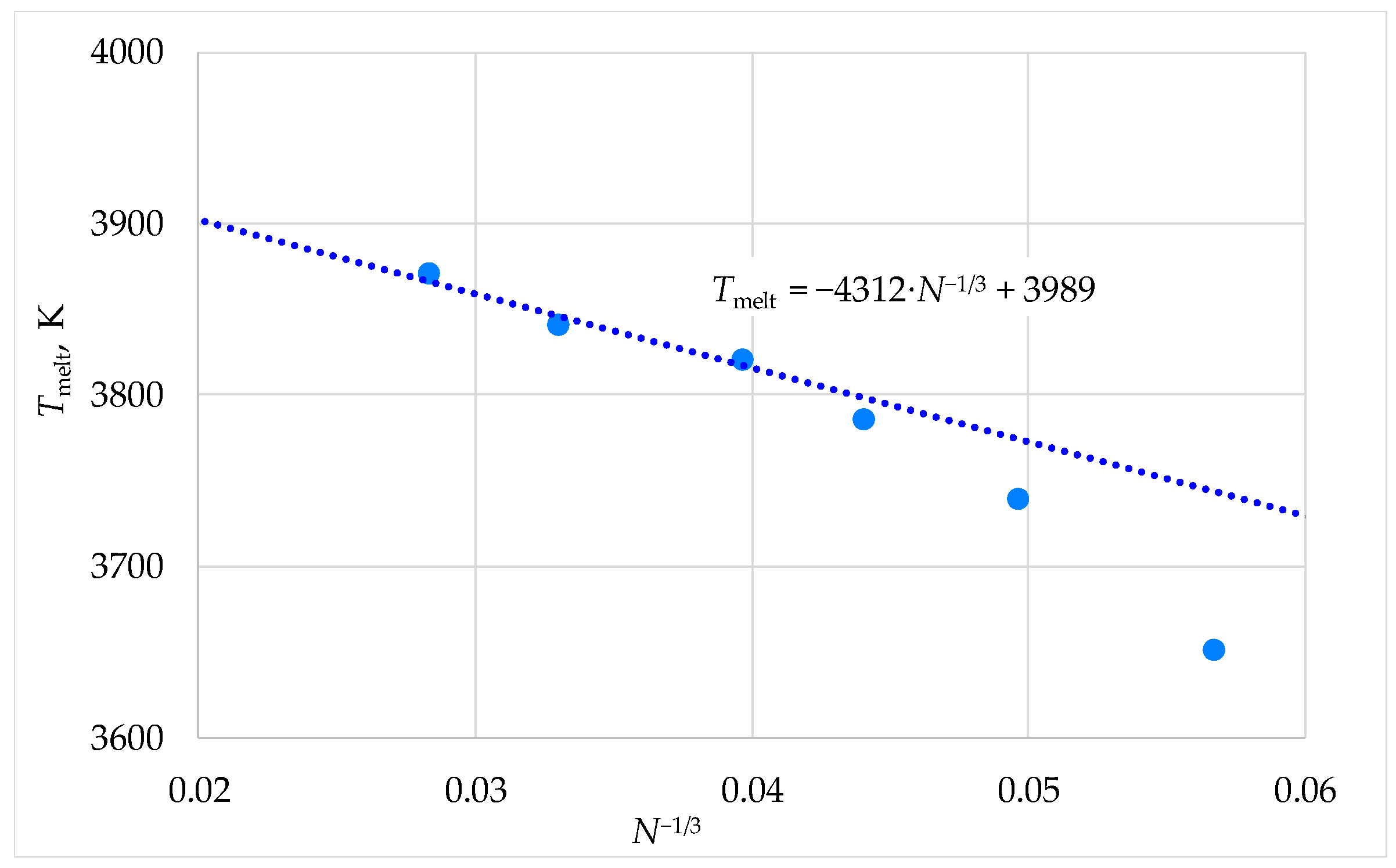
2.4. Calculation of Melting Temperatures
3. Results and Discussion
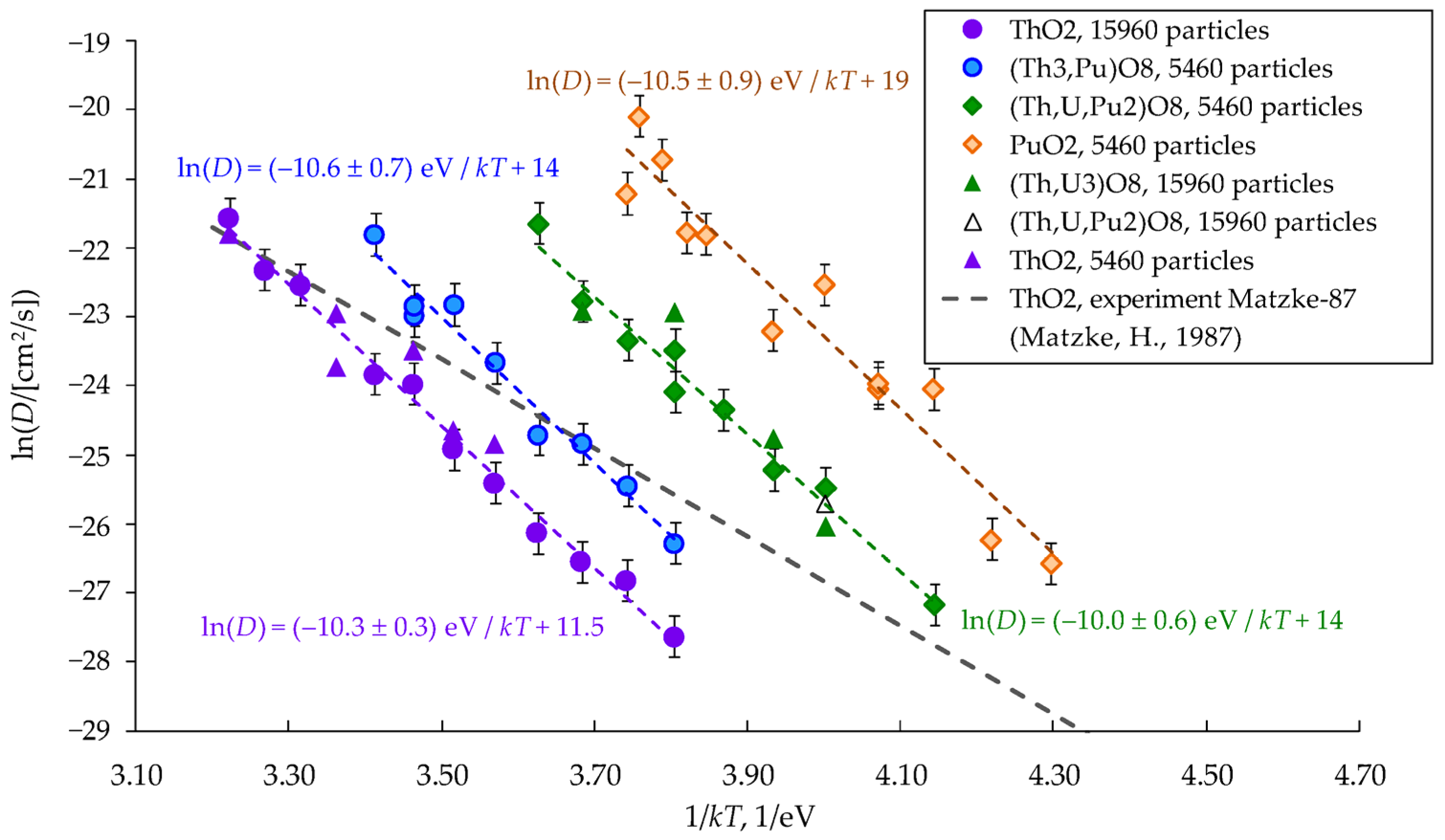
3.1. Cation Diffusion Coefficients
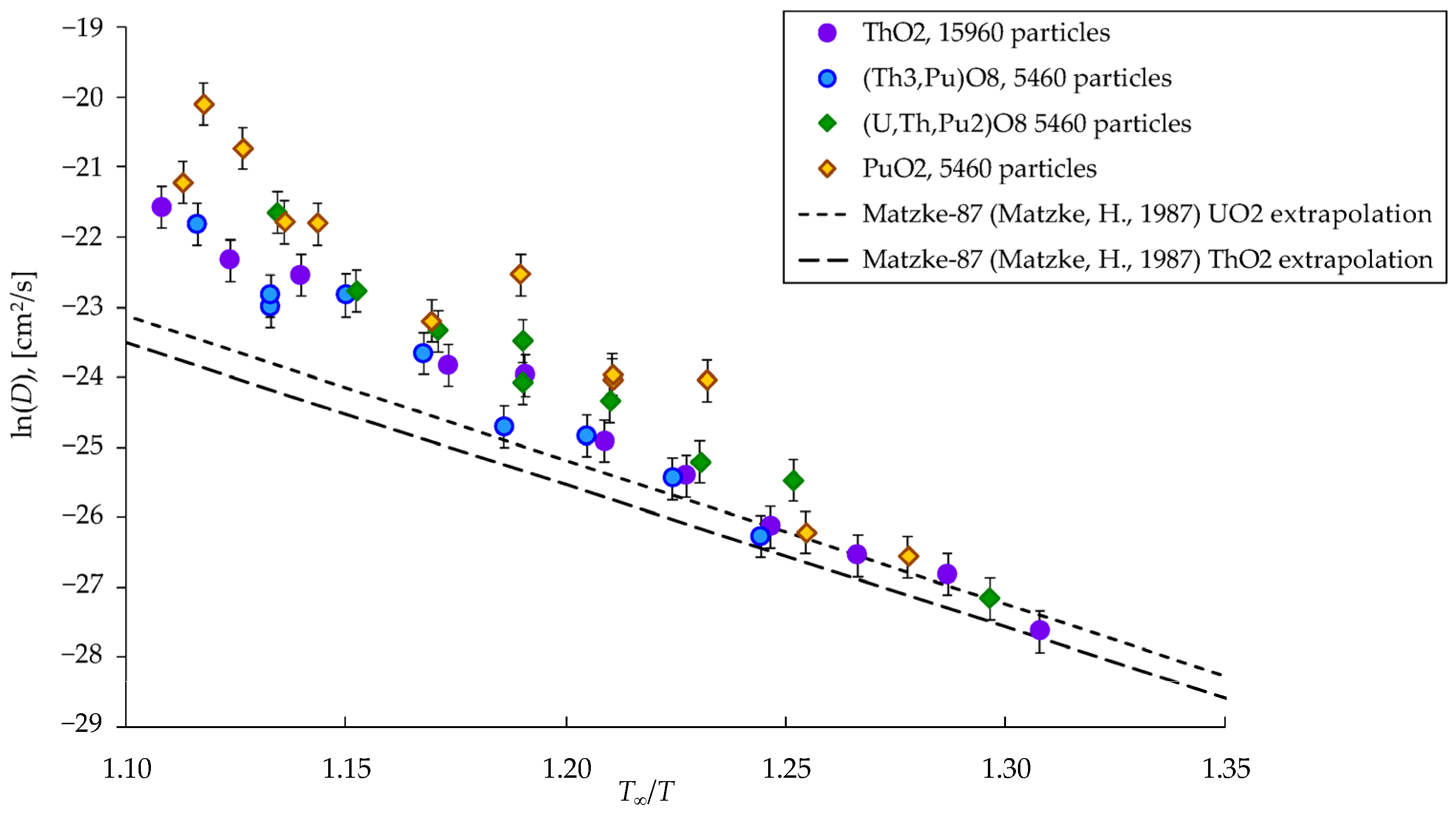
3.2. Activation Energies of Cation Diffusion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Carbajo, J.J.; Yoder, G.L.; Popov, S.G.; Ivanov, V.K. A Review of the Thermophysical Properties of MOX and UO2 Fuels. J. Nucl. Mater. 2001, 299, 181–198. [Google Scholar] [CrossRef]
- Parrish, R.; Aitkaliyeva, A. A Review of Microstructural Features in Fast Reactor Mixed Oxide Fuels. J. Nucl. Mater. 2018, 510, 644–660. [Google Scholar] [CrossRef]
- Ghosh, P.S.; Kuganathan, N.; Galvin, C.O.T.; Arya, A.; Dey, G.K.; Dutta, B.K.; Grimes, R.W. Melting Behavior of (Th,U)O2 and (Th,Pu)O2 Mixed Oxides. J. Nucl. Mater. 2016, 479, 112–122. [Google Scholar] [CrossRef]
- Meyer, M.K.; Fielding, R.; Gan, J. Fuel development for gas-cooled fast reactors. J. Nucl. Mater. 2007, 371, 281–287. [Google Scholar] [CrossRef]
- Crawford, D.C.; Porter, D.L.; Hayes, S.L. Fuels for Sodium-Cooled Fast Reactors: US Perspective. J. Nucl. Mater. 2007, 371, 202–231. [Google Scholar] [CrossRef]
- Matthews, R.B.; Chidester, K.M.; Hoth, C.W.; Mason, R.E.; Petty, R.L. Fabrication and Testing of Uranium Nitride Fuel for Space Power Reactors. J. Nucl. Mater. 1988, 151, 345–351. [Google Scholar] [CrossRef]
- Matzke, H. Science of Advanced LMFBR Fuels; North-Holland: Amsterdam, The Netherlands, 1986. [Google Scholar]
- Godinho, J.R.A.; Piazolo, S.; Stennett, M.C.; Hyatt, N.C. Sintering of CaF2 Pellets as Nuclear Fuel Analog for Surface Stability Experiments. J. Nucl. Mater. 2011, 419, 46–51. [Google Scholar] [CrossRef]
- Idriss, H. Surface Reactions of Uranium Oxide Powder, Thin Films and Single Crystals. Surf. Sci. Rep. 2010, 65, 67–109. [Google Scholar] [CrossRef]
- Potashnikov, S.I.; Boyarchenkov, A.S.; Nekrasov, K.A.; Kupryazhkin, A.Y. High-Precision Molecular Dynamics Simulation of UO2–PuO2: Pair Potentials Comparison in UO2. J. Nucl. Mater. 2011, 419, 217–225. [Google Scholar] [CrossRef]
- Murch, G.E.; Catlow, C.R.A. Oxygen Diffusion in UO2, ThO2 and PuO2: A Review. J. Chem. Soc. Faraday Trans. 1987, 83, 1157–1169. [Google Scholar] [CrossRef]
- Potashnikov, S.I.; Boyarchenkov, A.S.; Nekrasov, K.A.; Kupryazhkin, A.Y. High-Precision Molecular Dynamics Simulation of UO2–PuO2: Anion Self-Diffusion in UO2. J. Nucl. Mater. 2013, 433, 215–224. [Google Scholar] [CrossRef]
- Matzke, H. Lattice Disorder and Metal Self-Diffusion in Non-Stoichiometric UO2 and (U, Pu)O2. J. Phys. Colloq. 1973, 34, 317–325. [Google Scholar] [CrossRef]
- Matzke, H. Atomic Transport Properties in UO2 and Mixed Oxides (U, Pu)O2. J. Chem. Soc. Faraday Trans. 2 1987, 83, 1121–1142. [Google Scholar] [CrossRef]
- Ronchi, C.; Sheindlin, M.; Staicu, D.; Kinoshita, M. Effect of Burn-Up on the Thermal Conductivity of Uranium Dioxide up to 100,000 MWdt−1. J. Nucl. Mater. 2004, 327, 58–76. [Google Scholar] [CrossRef]
- Govers, K.; Lemehov, S.E.; Hou, M.; Verwerft, M. Comparison of Interatomic Potentials for UO2. Part I: Static Calculations. J. Nucl. Mater. 2007, 366, 161–177. [Google Scholar] [CrossRef]
- Govers, K.; Lemehov, S.E.; Hou, M.; Verwerft, M. Comparison of Interatomic Potentials for UO2: Part II: Molecular Dynamics Simulations. J. Nucl. Mater. 2008, 376, 66–77. [Google Scholar] [CrossRef]
- Balboa, H.; Van Brutzel, L.; Chartier, A.; Le Bouar, Y. Assessment of Empirical Potential for MOX Nuclear Fuels and Thermomechanical Properties. J. Nucl. Mater. 2017, 495, 67–79. [Google Scholar] [CrossRef]
- Shi, H.; Chu, M.; Zhang, P. Optical Properties of UO2 and PuO2. J. Nucl. Mater. 2010, 400, 151–156. [Google Scholar] [CrossRef]
- Peng-Fei, S.; Zhen-Hong, D.; Xiao-Ling, Z.; Yin-Chang, Z. Electronic Structure and Optical Properties in Uranium Dioxide: The First Principle Calculations. Chin. Phys. Lett. 2015, 32, 077101. [Google Scholar] [CrossRef]
- Boudjemline, A.; Louail, L.; Islam, M.M.; Diawara, B. Dependence of Pressure on Elastic, Electronic and Optical Properties of CeO2 and ThO2: A First Principles Study. Comput. Mater. Sci. 2011, 50, 2280–2286. [Google Scholar] [CrossRef]
- Li, Y. A Universal COMB Potential for the Whole Composition Range of the Uranium-Oxygen System. J. Nucl. Mater. 2019, 513, 102–111. [Google Scholar] [CrossRef]
- Phillpot, S.R.; Antony, A.C.; Shi, L.; Fullarton, M.L.; Liang, T.; Sinnott, S.B.; Zhang, Y.; Biner, S.B. Charge Optimized Many Body (COMB) Potentials for Simulation of Nuclear Fuel and Clad. Comput. Mater. Sci. 2018, 148, 231–241. [Google Scholar] [CrossRef]
- Li, Y.; Liang, T.; Sinnott, S.B.; Phillpot, S.R. A Charge-Optimized Many-Body Potential for the U–UO2–O2 System. J. Phys. Condens. Matter 2013, 25, 505401. [Google Scholar] [CrossRef]
- Ryzhkov, M.V.; Kupryazhkin, A.Y. First-Principles Study of Electronic Structure and Insulating Properties of Uranium and Plutonium Dioxides. J. Nucl. Mater. 2009, 384, 226–232. [Google Scholar] [CrossRef]
- Ryzhkov, M.V.; Kovalenko, M.A.; Kupryazhkin, A.Y.; Gupta, S.K. Transformation of Electron Density Distribution Induced by the Cation Point Defects in Uranium Dioxide. J. Radioanal. Nucl. Chem. 2020, 325, 253–262. [Google Scholar] [CrossRef]
- Boyarchenkov, A.S.; Nekrasov, K.A.; Kupryazhkin, A.Y.; Gupta, S.K. A Novel Empirical Potential for High-Temperature Molecular Dynamics Simulation of ThO2 and MOX Nuclear Fuel Crystals. AIP Conf. Proc. 2020, 2313, 030064. [Google Scholar] [CrossRef]
- Brutzel, L.V.; Rarivomanantsoa, M.; Ghaleb, D. Displacement Cascade Initiated with the Realistic Energy of the Recoil Nucleus in UO2 Matrix by Molecular Dynamics Simulation. J. Nucl. Mater. 2006, 354, 28–35. [Google Scholar] [CrossRef]
- Martin, G.; Garcia, P.; Brutzel, L.V.; Dorado, B.; Maillard, S. Effect of the Cascade Energy on Defect Production in Uranium Dioxide. Nucl. Instrum. Methods Phys. Res. B 2011, 269, 1727–1731. [Google Scholar] [CrossRef]
- Balboa, H.; Van Brutzel, L.; Chartier, A.; Le Bouar, Y. Damage Characterization of (U,Pu)O2 Under Irradiation by Molecular Dynamics Simulations. J. Nucl. Mater. 2018, 512, 440–451. [Google Scholar] [CrossRef]
- Devanathan, R. Molecular Dynamics Simulation of Fission Fragment Damage in Nuclear Fuel and Surrogate Material. MRS Adv. 2017, 2, 1225–1236. [Google Scholar] [CrossRef]
- Yablinsky, C.A.; Devanathan, R.; Pakarinen, J.; Gan, J.; Severin, D.; Trautmann, C.; Allen, T.R. Characterization of Swift Heavy Ion Irradiation Damage in Ceria. J. Mater. Res. 2015, 30, 1473–1484. [Google Scholar] [CrossRef]
- Kovalenko, M.A.; Kupryazhkin, A.Y. Mechanisms of Exchange and Anion Frenkel Diffusion in Uranium Dioxide: Molecular Dynamics Study. J. Nucl. Mater. 2019, 522, 255–264. [Google Scholar] [CrossRef]
- Boyarchenkov, A.S.; Potashnikov, S.I.; Nekrasov, K.A.; Kupryazhkin, A.Y. Molecular Dynamics Simulation of UO2 Nanocrystals Melting Under Isolated and Periodic Boundary Conditions. J. Nucl. Mater. 2012, 427, 311–322. [Google Scholar] [CrossRef]
- Singh, S.; Sonvane, Y.; Nekrasov, K.A.; Boyarchenkov, A.S.; Kupryazhkin, A.Y.; Gajjar, P.N.; Gupta, S.K. Ab-Initio Investigation of Crystal Structure and Pressure Induced Phase Transition in ThO2 and PuO2. Mater. Today Commun. 2021, 28, 102579. [Google Scholar] [CrossRef]
- Kim, K.C.; Olander, D.R. Oxygen Diffusion in UO2−x. J. Nucl. Mater. 1981, 102, 192–199. [Google Scholar] [CrossRef]
- Basak, C.; Sengupta, A.; Kamath, H. Classical Molecular Dynamics Simulation of UO2 to Predict Thermophysical Properties. J. Alloys Compd. 2003, 360, 210–216. [Google Scholar] [CrossRef]
- Morelon, N.-D.; Ghaleb, D.; Delaye, J.-M.; Brutzel, L.V. A New Empirical Potential for Simulating the Formation of Defects and Their Mobility in Uranium Dioxide. Philos. Mag. 2003, 83, 1533–1555. [Google Scholar] [CrossRef]
- Yakub, E.; Ronchi, C.; Staicu, D. Computer Simulation of Defects Formation and Equilibrium in Non-Stoichiometric Uranium Dioxide. J. Nucl. Mater. 2009, 389, 119–129. [Google Scholar] [CrossRef]
- Moxon, S.; Skelton, J.; Joshua, S.T.; Flitcroft, J.; Togo, A.; Cooke, D.J.; Da Silva, E.L.; Harker, R.M.; Storr, M.T.; Parker, S.C. Structural Dynamics of Schottky and Frenkel Defects in ThO2: A Density-Functional Theory Study. J. Mater. Chem. A 2022, 10, 1861–1875. [Google Scholar] [CrossRef]
- Dorado, B.; Andersson, D.A.; Stanek, C.R.; Bertolus, M.; Uberuaga, B.P.; Martin, G.; Freyss, M.; Garcia, P. First-Principles Calculations of Uranium Diffusion in Uranium Dioxide. Phys. Rev. B 2012, 86, 035110. [Google Scholar] [CrossRef]
- Dubois, E.T.; Tranchida, J.; Bouchet, J.; Maillet, J.B. Atomistic Simulations of Nuclear Fuel UO2 with Machine Learning Interatomic Potentials. Phys. Rev. Mater. 2024, 8, 025402. [Google Scholar] [CrossRef]
- Stippell, E.; Alzate-Vargas, L.; Subedi, K.N.; Tutchton, R.M.; Cooper, M.W.D.; Tretiak, S.; Gibson, T.; Messerly, R.A. Building a DFT+U Machine Learning Interatomic Potential for Uranium Dioxide. Artif. Intell. Chem. 2024, 2, 100042. [Google Scholar] [CrossRef]
- Konashi, K.; Kato, N.; Mori, K.; Kurosaki, K. Neural Network Potential for Molecular Dynamics Calculation of UO2. J. Nucl. Mater. 2025, 607, 155660. [Google Scholar] [CrossRef]
- Vincent-Aublant, E.; Delaye, J.-M.; Van Brutzel, L. Self-Diffusion Near Symmetrical Tilt Grain Boundaries in UO2 Matrix: A Molecular Dynamics Simulation Study. J. Nucl. Mater. 2009, 392, 114–122. [Google Scholar] [CrossRef]
- Arima, T.; Yoshida, K.; Idemitsu, K.; Inagaki, Y.; Sato, I. Molecular Dynamics Analysis of Diffusion of Uranium and Oxygen Ions in Uranium Dioxide. IOP Conf. Ser. Mater. Sci. Eng. 2010, 9, 012003. [Google Scholar] [CrossRef]
- Desai, T.G.; Millett, P.; Tonks, M.; Wolf, D. Atomistic Simulations of Void Migration Under Thermal Gradient in UO2. Acta Mater. 2010, 58, 330–339. [Google Scholar] [CrossRef]
- Boyarchenkov, A.S.; Potashnikov, S.I.; Nekrasov, K.A.; Kupryazhkin, A.Y. Investigation of Cation Self-Diffusion Mechanisms in UO2±x Using Molecular Dynamics. J. Nucl. Mater. 2013, 442, 148–161. [Google Scholar] [CrossRef]
- Boyarchenkov, A.S.; Potashnikov, S.I.; Nekrasov, K.A.; Kupryazhkin, A.Y. Molecular Dynamics Simulation of UO2 Nanocrystals Surface. J. Nucl. Mater. 2012, 421, 1–8. [Google Scholar] [CrossRef]
- Pitskhelaury, S.; Seitov, D.; Nekrasov, K.; Boyarchenkov, A.; Kupryazhkin, A.; Gupta, S.K. Influence of the Superionic Transition on the Diffusion of Cations in ThO2 Nanocrystals: A Molecular Dynamics Simulation. Mater. Today Proc. 2023, in press. [Google Scholar] [CrossRef]
- Seitov, D.D.; Pitskhelaury, S.S.; Nekrasov, K.A.; Boyarchenkov, A.S.; Kupryazhkin, A.Y. A Mechanism of Cation Diffusion in ThO2 Nanocrystal Bulk: A Molecular Dynamic Simulation. AIP Conf. Proc. 2022, 2466, 030040. [Google Scholar] [CrossRef]
- Cooper, M.W.D.; Rushton, M.J.D.; Grimes, R.W. A Many-Body Potential Approach to Modelling the Thermomechanical Properties of Actinide Oxides. J. Phys. Condens. Matter 2014, 26, 105401. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling Through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Box, G.E.P.; Muller, M.E. A note on the generation of random normal deviates. Ann. Math. Stat. 1958, 29, 610–611. [Google Scholar] [CrossRef]
- Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Nelder, J.A.; Mead, R. A simplex method for function minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
- Nosé, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Nekrasov, K.A.; Boyarchenkov, A.S.; Kupryazhkin, A.Y.; Gupta, S.K. The melting mechanisms of UO2 nanocrystals: A molecular dynamics simulation. AIP Conf. Proc. 2019, 2142, 020001. [Google Scholar] [CrossRef]
- Vanfleet, R.R.; Mochel, J.M. Thermodynamics of Melting and Freezing in Small Particles. Surf. Sci. 1995, 341, 40–50. [Google Scholar] [CrossRef]
- Pavlov, T.R.; Wangle, T.; Wenman, M.R.; Tyrpekl, V.; Vlahovic, L.; Robba, D.; Van Uffelen, P.; Konings, R.J.M.; Grimes, R.W. High Temperature Measurements and Condensed Matter Analysis of the Thermo-Physical Properties of ThO2. Sci. Rep. 2018, 8, 5038. [Google Scholar] [CrossRef]
- De Bruycker, F.; Boboridis, K.; Manara, D.; Pöml, P.; Rini, M.; Konings, R.J.M. Reassessing the Melting Temperature of PuO2. Mater. Today 2010, 13, 52–55. [Google Scholar] [CrossRef]
- Epstein, L.F. Ideal Solution Behavior and Heats of Fusion from the UO2-PuO2 Phase Diagram. J. Nucl. Mater. 1967, 22, 340–348. [Google Scholar] [CrossRef]
- Fink, J.K.; Sofu, T.; Ley, H. International Nuclear Safety Center Database on Thermophysical Properties of Reactor Materials. Int. J. Thermophys. 1999, 20, 279–287. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, Y.; Zhang, P. Thermodynamic Properties and Structural Stability of Thorium Dioxide. J. Phys. Condens. Matter 2012, 24, 225801. [Google Scholar] [CrossRef]
- Murphy, S.T.; Cooper, M.W.D.; Grimes, R.W. Point Defects and Non-Stoichiometry in Thoria. Solid State Ionics 2014, 267, 80–87. [Google Scholar] [CrossRef]
- Yun, Y.; Oppeneer, P.M.; Kim, H.; Park, K. Defect Energetics and Xe Diffusion in UO2 and ThO2. Acta Mater. 2009, 57, 1655–1659. [Google Scholar] [CrossRef]
- Singh, S.; Sonvane, Y.; Nekrasov, K.A.; Kupryazhkin, A.Y.; Gajjar, P.N.; Gupta, S.K. A First Principles Investigation of Defect Energetics and Diffusion in Actinide Dioxides. J. Nucl. Mater. 2024, 591, 154901. [Google Scholar] [CrossRef]
- Konings, R.J.M.; Beneš, O. The Heat Capacity of NpO2 at High Temperatures: The Effect of Oxygen Frenkel Pair Formation. J. Phys. Chem. Solids 2013, 74, 653–655. [Google Scholar] [CrossRef]
- Gupta, F.; Brillant, G.; Pasturel, A. Correlation Effects and Energetics of Point Defects in Uranium Dioxide: A First Principle Investigation. Philos. Mag. 2007, 87, 2561–2569. [Google Scholar] [CrossRef]
- Nerikar, P.; Watanabe, T.; Tulenko, J.S.; Phillpot, S.R.; Sinnott, S.B. Energetics of Intrinsic Point Defects in Uranium Dioxide from Electronic-Structure Calculations. J. Nucl. Mater. 2009, 384, 61–69. [Google Scholar] [CrossRef]
- Dorado, B.; Jomard, G.; Freyss, M.; Bertolus, M. Stability of Oxygen Point Defects in UO2 by First-Principles DFT+U Calculations: Occupation Matrix Control and Jahn-Teller Distortion. Phys. Rev. B 2010, 82, 035114. [Google Scholar] [CrossRef]
- Freyss, M.; Vergnet, N.; Petit, T. Ab Initio Modeling of the Behavior of Helium and Xenon in Actinide Dioxide Nuclear Fuels. J. Nucl. Mater. 2006, 352, 144–150. [Google Scholar] [CrossRef]
- Hutchings, M.T. High-Temperature Studies of UO2 and ThO2 Using Neutron Scattering Techniques. J. Chem. Soc. Faraday Trans. 2 1987, 83, 1083–1103. [Google Scholar] [CrossRef]
- Wang, L.F.; Sun, B.; Liu, H.F.; Lin, D.Y.; Song, H.F. Thermodynamics and Kinetics of Intrinsic Point Defects in Plutonium Dioxides. J. Nucl. Mater. 2019, 526, 151762. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, Y.; Zhang, P. Charge States of Point Defects in Plutonium Oxide: A First-Principles Study. J. Alloys Compd. 2015, 649, 544–552. [Google Scholar] [CrossRef]
- Yun, Y.; Eriksson, O.; Oppeneer, P.M. First-Principles Study of Helium Behavior in Nuclear Fuel Materials. Preprints 2011. Available online: https://www.researchgate.net/publication/287350512_Firstprinciples_study_of_helium_behavior_in_nuclear_fuel_materials (accessed on 4 May 2025).
- Nakamura, H.; Machida, M. A First-Principles Study on Point Defects in Plutonium Dioxide. Prog. Nucl. Sci. Technol. 2018, 5, 132–135. [Google Scholar] [CrossRef]
- Tian, X.; Gao, T.; Lu, C.; Shang, J.; Xiao, H. First Principle Study of the Behavior of Helium in Plutonium Dioxide. Eur. Phys. J. B 2013, 86, 179. [Google Scholar] [CrossRef]
- Matthews, J.R. Technological Problems and the Future of Research on the Basic Properties of Actinide Oxides. J. Chem. Soc. Faraday Trans. 2 1987, 83, 1273–1285. [Google Scholar] [CrossRef]
- Crocombette, J.P.; Jollet, F.; Nga, L.T.; Petit, T. Plane-Wave Pseudopotential Study of Point Defects in Uranium Dioxide. Phys. Rev. B 2001, 64, 104107. [Google Scholar] [CrossRef]
- Freyss, M.; Petit, T.; Crocombette, J.P. Point Defects in Uranium Dioxide: Ab Initio Pseudopotential Approach in the Generalized Gradient Approximation. J. Nucl. Mater. 2005, 347, 44–51. [Google Scholar] [CrossRef]
- Iwasawa, M.; Chen, Y.; Kaneta, Y.; Ohnuma, T.; Geng, H.Y.; Kinoshita, M. First-Principles Calculation of Point Defects in Uranium Dioxide. Mater. Trans. 2006, 47, 2651–2657. [Google Scholar] [CrossRef]
- Dorado, B.; Freyss, M.; Martin, G. GGA+U Study of the Incorporation of Iodine in Uranium Dioxide. Eur. Phys. J. B 2009, 69, 203–209. [Google Scholar] [CrossRef]
- Adamson, M.G.; Aitken, E.A.; Caputi, R.W. Experimental and thermodynamic evaluation of the melting behavior of irradiated oxide fuels. J. Nucl. Mater. 1985, 130, 245–252. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | T∞, K, Present Work | T∞, K, Experiment |
|---|---|---|
| ThO2 | 3990 | 3665 ± 70 K [63] |
| (Th0.75Pu0.25)O2 | 3795 | – |
| (Th0.25U0.25Pu0.5)O2 | 3630 | – |
| PuO2 | 3450 | 3017 ± 28 K [64] |
| Compound | L, kJ/mol | γ, J/m2 |
|---|---|---|
| ThO2 | 37.1 | 4.40 |
| (Th0.75Pu0.25)O2 | 45.3 | 4.33 |
| (Th0.25U0.25Pu0.5)O2 | 65.1 | 3.94 |
| PuO2 | 68.2 | 4.20 |
| Mechanism of Disordering | Formation Energy, eV | ||
|---|---|---|---|
| ThO2 | UO2 | PuO2 | |
| Unbound interstitial anion and anion vacancy (EAF) | 4.5, MOX-07 [50] 6.8, GGA [67] 5.0, GGA [68] 9.8, GGA [69] 9.5, GGA [70] 2.3–4.7, exp [12] 4.42, exp [71] | 4.1, MOX-07 [30] 5.9, CRG [30] 4.0, GGA + U [72] 3.6, GGA [72] 4.5, GGA [69] 3.95, GGA + U [73] 5.8, GGA + U [74] 3.3, GGA + U [41] 3.6, GGA [75] 3.5 ± 0.5, exp [14] 4.6, exp [76] | 3.9, this work 3.9, MOX-07 [30] 5.5, CRG [30] 5.5, CRG [77] 3.48, LDA + U [78] 5.3, GGA [75] 4.4, GGA, [79] 4.6, GGA + U, [80] 4.2, GGA [70] 9.8, GGA + U [81] 2.7–2.9, exp [12] |
| Classic Schottky trio V4− + 2V2+ (ESh) | 12.7, MOX-07 [50] 8.2, GGA [67] 8.05, GGA [68] 20.6, GGA, [69] | 8.9, this work 9.7, MOX-07 [30] 10.9, CRG [30] 7.2, GGA + U [72] 5.2, GGA [72] 7.2, GGA [69] 7.6, GGA + U [73] 6.0, GGA + U [41] 5.6, GGA [75] 6–7, exp [14] | 9.5, MOX-07 [30] 10.0, CRG [30] 10.4, CRG [77] 7.5, LDA + U [78] 9.1, GGA [75] 7.1, GGA [79] 6.09, GGA + U [80] 14.9, GGA + U [81] |
| Bound Schottky trio (V4−·2V2+)111 | 6.9, MOX-07 [50] 5.4, GGA [40] 4.5, GGA [68] 4.6, GGA [77] | 4.8, this work 4.8, MOX-07 [30] 5.0, CRG [30] 3.6, GGA [70] | 5.0, MOX-07 [30] 4.8, CRG [30] 4.8, CRG [77] 3.6, GGA [70] |
| Partially bound Schottky trio V4−·V2 + + V2+ | 9.7, MOX-07 [50] | 6.7, this work | 7.0, this work |
| Migration of a single cation vacancy V4− (height of potential barrier EM) | 5.45, this work 4.5, GGA [69] 5.7, GGA [70] | 3.1, GGA [69] 4.2, GGA + U [82] 3.6, GGA + U [41] 5.4, GGA [70] 2.4, exp [14] | 4.5, this work 3.4, CRG [77] 5.8, GGA [70] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seitov, D.D.; Nekrasov, K.A.; Pitskhelaury, S.S.; Abuova, F.U.; Kabdrakhimova, G.D.; Abuova, A.U.; Gupta, S.K. Computational Modeling of Cation Diffusion in Isolated Nanocrystals of Mixed Uranium, Plutonium and Thorium Dioxides. Crystals 2025, 15, 532. https://doi.org/10.3390/cryst15060532
Seitov DD, Nekrasov KA, Pitskhelaury SS, Abuova FU, Kabdrakhimova GD, Abuova AU, Gupta SK. Computational Modeling of Cation Diffusion in Isolated Nanocrystals of Mixed Uranium, Plutonium and Thorium Dioxides. Crystals. 2025; 15(6):532. https://doi.org/10.3390/cryst15060532
Chicago/Turabian StyleSeitov, Dastan D., Kirill A. Nekrasov, Sergey S. Pitskhelaury, Fatima U. Abuova, Gaukhar D. Kabdrakhimova, Aisulu U. Abuova, and Sanjeev K. Gupta. 2025. "Computational Modeling of Cation Diffusion in Isolated Nanocrystals of Mixed Uranium, Plutonium and Thorium Dioxides" Crystals 15, no. 6: 532. https://doi.org/10.3390/cryst15060532
APA StyleSeitov, D. D., Nekrasov, K. A., Pitskhelaury, S. S., Abuova, F. U., Kabdrakhimova, G. D., Abuova, A. U., & Gupta, S. K. (2025). Computational Modeling of Cation Diffusion in Isolated Nanocrystals of Mixed Uranium, Plutonium and Thorium Dioxides. Crystals, 15(6), 532. https://doi.org/10.3390/cryst15060532