
Synthesis and High-Pressure Stability Study of Energetic Molecular Perovskite DAI-X1

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Experimental Material
2.2. Sample Preparation
2.3. Experimental Characterization
2.4. High Pressure Experiment of DAI-X1
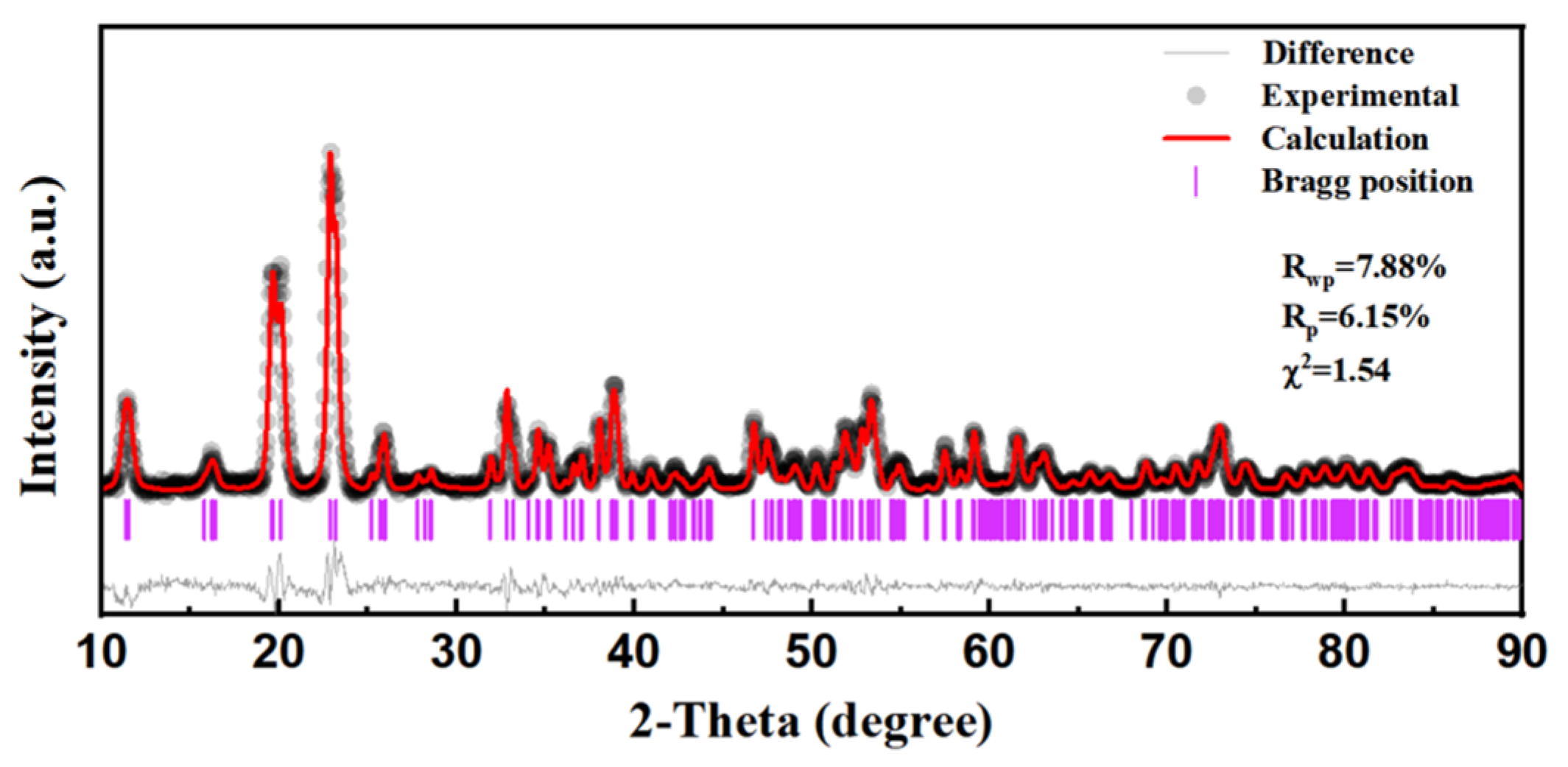
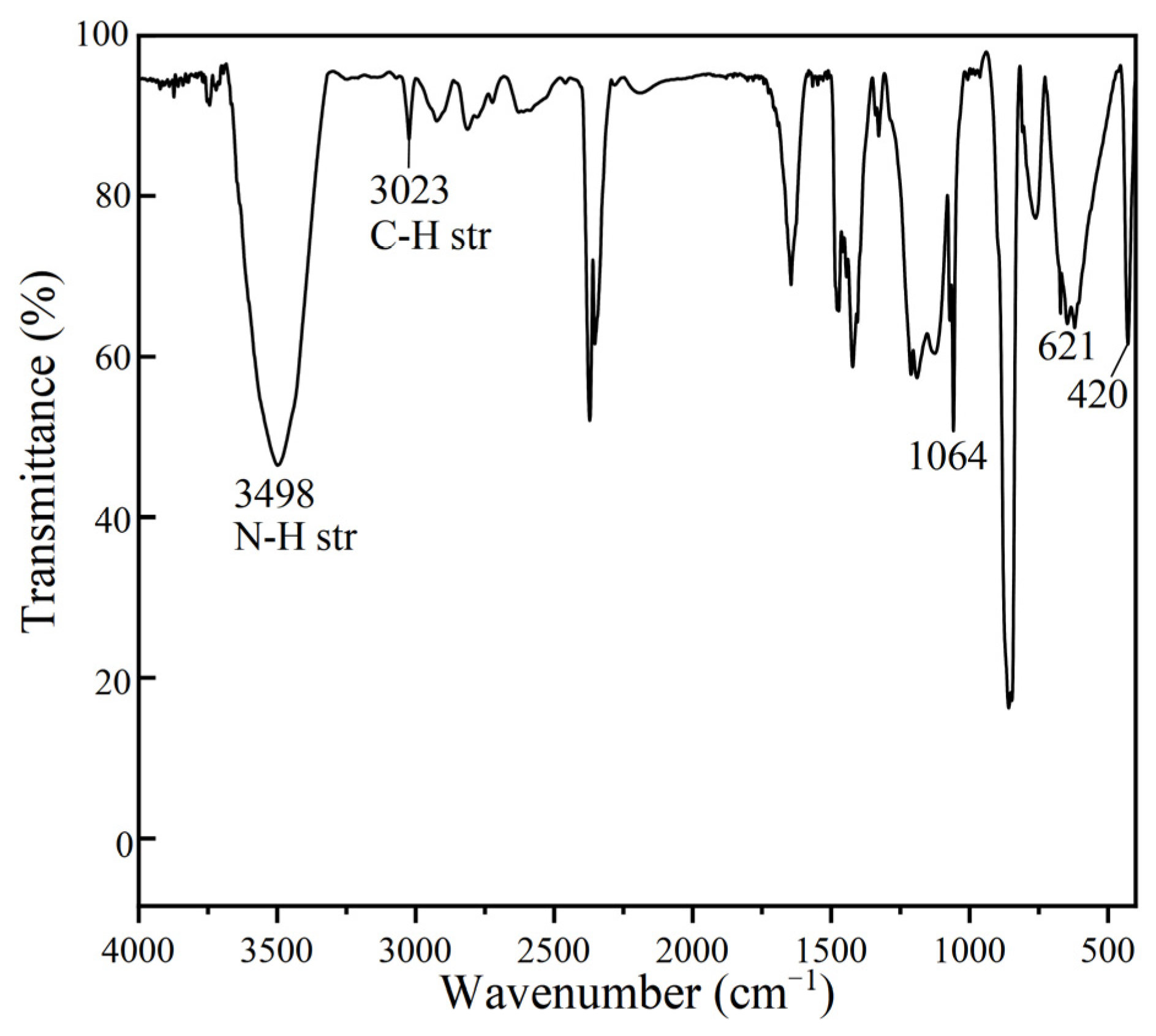
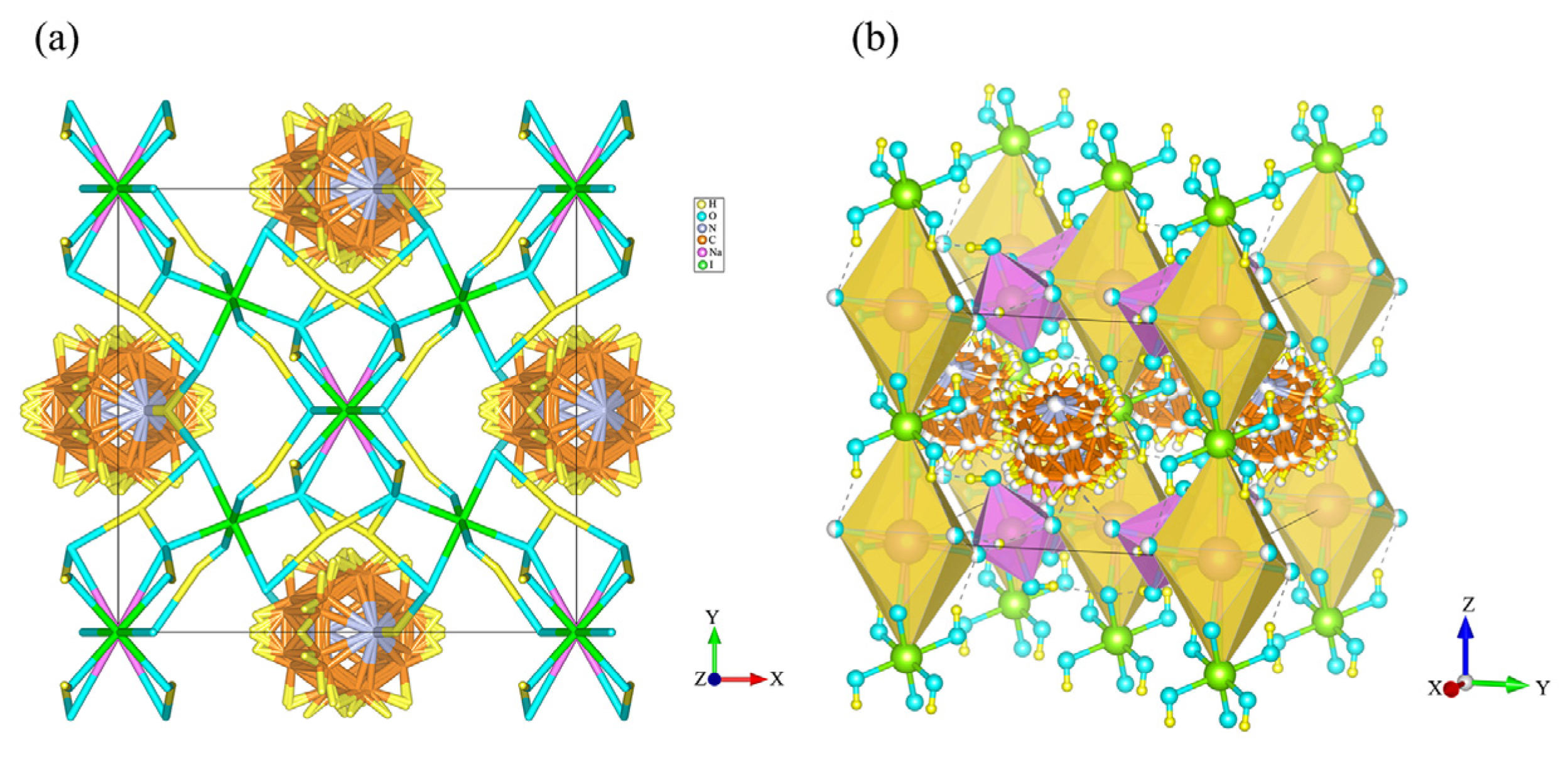
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gao, H.; Zhang, Q.; Jean’ne, M.S. Fused heterocycle-based energetic materials (2012–2019). J. Mater. Chem. A 2020, 8, 4193–4216. [Google Scholar] [CrossRef]
- Shang, Y.; Huang, R.-K.; Chen, S.-L.; He, C.-T.; Yu, Z.-H.; Ye, Z.-M.; Zhang, W.-X.; Chen, X.-M. Metal-free molecular perovskite high-energetic materials. Cryst. Growth Des. 2020, 20, 1891–1897. [Google Scholar] [CrossRef]
- Zlotin, S.G.; Churakov, A.M.; Egorov, M.P.; Fershtat, L.L.; Klenov, M.S.; Kuchurov, I.V.; Makhova, N.N.; Smirnov, G.A.; Tomilov, Y.V.; Tartakovsky, V.A. Advanced energetic materials: Novel strategies and versatile applications. Mendeleev Commun. 2021, 31, 731–749. [Google Scholar] [CrossRef]
- Muravyev, N.V.; Wozniak, D.R.; Piercey, D.G. Progress and performance of energetic materials: Open dataset, tool, and implications for synthesis. J. Mater. Chem. A 2022, 10, 11054–11073. [Google Scholar] [CrossRef]
- Pang, W.-Q.; Yetter, R.A.; DeLuca, L.T.; Zarko, V.; Gany, A.; Zhang, X.-H. Boron-based composite energetic materials (B-CEMs): Preparation, combustion and applications. Prog. Energy Combust. Sci. 2022, 93, 101038. [Google Scholar] [CrossRef]
- Yu, Q.; Yin, P.; Zhang, J.; He, C.; Imler, G.H.; Parrish, D.A.; Shreeve, J.n.M. Pushing the limits of oxygen balance in 1,3,4-oxadiazoles. J. Am. Chem. Soc. 2017, 139, 8816–8819. [Google Scholar] [CrossRef]
- Anniyappan, M.; Talawar, M.B.; Sinha, R.K.; Murthy, K.P. Review on advanced energetic materials for insensitive munition formulations. Combust. Explos. Shock Waves 2020, 56, 495–519. [Google Scholar] [CrossRef]
- Banik, S.; Yadav, A.K.; Kumar, P.; Ghule, V.D.; Dharavath, S. Unfolding the chemistry of FOX-7: Unique energetic material and precursor with numerous possibilities. Chem. Eng. J. 2022, 431, 133378. [Google Scholar] [CrossRef]
- Herweyer, D.; Brusso, J.L.; Murugesu, M. Modern trends in “Green” primary energetic materials. New J. Chem. 2021, 45, 10150–10159. [Google Scholar] [CrossRef]
- Bu, R.; Xiong, Y.; Zhang, C. π–π stacking contributing to the low or reduced impact sensitivity of energetic materials. Cryst. Growth Des. 2020, 20, 2824–2841. [Google Scholar] [CrossRef]
- Li, G.; Zhang, C. Review of the molecular and crystal correlations on sensitivities of energetic materials. J. Hazard. Mater. 2020, 398, 122910. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Zhang, Y.; Ma, Z.; Guo, M.; Xiao, Z.; Zhang, J.; Dong, M.; Fan, J.; Guo, Z.; Liu, C. Energy characteristics and mechanical properties of cyclotrimethylenetrinitramine (RDX)-based insensitive high-energy propellant. J. Mater. Res. Technol. 2020, 9, 15313–15323. [Google Scholar] [CrossRef]
- Kou, Y.; Song, X.; Guo, K.; Cheng, Z.; Wang, Y. Characterization, Thermolysis, and Energetic Properties of an MTNP/PETN Eutectic Prepared via the Solvent/Anti-Solvent Method. Propellants Explos. Pyrotech. 2021, 46, 299–308. [Google Scholar] [CrossRef]
- Singh, J.; Staples, R.J.; Jean’ne, M.S. Coordination-driven safer and sustainable energetic materials. J. Mater. Chem. A 2025, 13, 11475–11485. [Google Scholar] [CrossRef]
- Chen, Q.; Wu, J.; Ou, X.; Huang, B.; Almutlaq, J.; Zhumekenov, A.A.; Guan, X.; Han, S.; Liang, L.; Yi, Z. All-inorganic perovskite nanocrystal scintillators. Nature 2018, 561, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-X.; Chen, S.-L.; Shang, Y.; Yu, Z.-H.; Chen, X.-M. Molecular perovskites as a new platform for designing advanced multi-component energetic crystals. Energet. Mater. Front. 2020, 1, 123–135. [Google Scholar] [CrossRef]
- Chen, S.-L.; Yang, Z.-R.; Wang, B.-J.; Shang, Y.; Sun, L.-Y.; He, C.-T.; Zhou, H.-L.; Zhang, W.-X.; Chen, X.-M. Molecular perovskite high-energetic materials. Sci. China Mater. 2018, 61, 1123–1128. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, J.; Cao, W.; Zhang, J.; Shreeve, J.N.M. A promising perovskite primary explosive. Nat. Commun. 2023, 14, 7765. [Google Scholar] [CrossRef]
- Chen, S.-l.; Shang, Y.; Jiang, J.; Huang, M.; Ren, J.-T.; Guo, T.; Yu, C.-X.; Zhang, W.-X.; Chen, X.-M. A new nitrate-based energetic molecular perovskite as a modern edition of black powder. Energet. Mater. Front. 2022, 3, 122–127. [Google Scholar] [CrossRef]
- Jia, Q.; Bai, X.; Zhu, S.; Cao, X.; Deng, P.; Hu, L. Fabrication and characterization of nano (H2dabco)[K(ClO4)3] molecular perovskite by ball milling. J. Energet. Mater. 2020, 38, 377–385. [Google Scholar] [CrossRef]
- Jia, Q.; Deng, P.; Li, X.; Hu, L.; Cao, X. Insight into the thermal decomposition properties of potassium perchlorate (KClO4)-based molecular perovskite. Vacuum 2020, 175, 109257. [Google Scholar] [CrossRef]
- Deng, P.; Wang, H.; Yang, X.; Ren, H.; Jiao, Q. Thermal decomposition and combustion performance of high-energy ammonium perchlorate-based molecular perovskite. J. Alloys Compd. 2020, 827, 154257. [Google Scholar] [CrossRef]
- Gao, Y.; Shi, E.; Deng, S.; Shiring, S.B.; Snaider, J.M.; Liang, C.; Yuan, B.; Song, R.; Janke, S.M.; Liebman-Peláez, A. Molecular engineering of organic-inorganic hybrid perovskites quantum wells. Nat. Chem. 2019, 11, 1151–1157. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, L.-S.; Gong, S.; Guang, C.; Li, L.; Hu, S.; Deng, P. Study of ammonium perchlorate-based molecular perovskite (H2DABCO)[NH4(ClO4)3]/graphene energetic composite with insensitive performance. Cent. Eur. J. Energ. Mater. 2020, 17, 451–469. [Google Scholar] [CrossRef]
- Deng, P.; Guo, X.-Y.; Fang, H.; Liu, R.; Chen, P.-W. Combustion behavior and mechanism of molecular perovskite energetic material DAP-4-based composites with metal fuel Al. Def. Technol. 2023, 27, 53–63. [Google Scholar] [CrossRef]
- Shang, Y.; Sun, L.-Y.; Ye, Z.-M.; Chen, S.-L.; Zhang, W.-X.; Chen, X.-M. Phase transition and thermal expansion of molecular perovskite energetic crystal (C6N2H14)(NH4)(ClO4)3 (DAP-4). FirePhysChem 2022, 2, 221–225. [Google Scholar] [CrossRef]
- Han, K.; Zhang, X.; Deng, P.; Jiao, Q.; Chu, E. Study of the thermal catalysis decomposition of ammonium perchlorate-based molecular perovskite with titanium carbide MXene. Vacuum 2020, 180, 109572. [Google Scholar] [CrossRef]
- Deng, P.; Ren, H.; Jiao, Q. Enhanced the combustion performances of ammonium perchlorate-based energetic molecular perovskite using functionalized graphene. Vacuum 2019, 169, 108882. [Google Scholar] [CrossRef]
- Chang, J.; Zhao, G.; Zhao, X.; He, C.; Pang, S.; Shreeve, J.N.M. New promises from an old friend: Iodine-rich compounds as prospective energetic biocidal agents. Acc. Chem. Res. 2020, 54, 332–343. [Google Scholar] [CrossRef]
- Wei, L.; You, Z.; Meng, X.; Fang, Y.; Li, A.; Li, S.; Wang, K.; Li, Q. High-pressure exciton engineering in two-dimensional metal halide perovskite toward intense deep-blue emission and regulated photocurrent property. Appl. Phys. Lett. 2024, 125, 211901. [Google Scholar] [CrossRef]
- Meng, X.; Wei, L.; Wu, M.; Li, A.; Li, L.; Wang, K.; Li, Q. Resolving multiple emissions of zero-dimensional heterometallic halide hybrid via pressure-triggered antenna effect. Mater. Res. Lett. 2024, 12, 417–424. [Google Scholar] [CrossRef]
- Yan, T.-T.; Xu, Y.-F.; Xi, D.-Y.; Yu, Z.-Q.; Jiang, R.; Zhang, D.-D. High pressure study of hydrogen-bonded energetic material 4-nitropyrazole. Phys. Lett. A 2024, 512, 129567. [Google Scholar] [CrossRef]
- Yan, T.-T.; Jiang, R.; Xi, D.-Y.; Ma, L.; Zhang, D.-D.; Xu, Y.-F. High-pressure behavior of hydrogen-bonded organic crystal trifluoroacetamide. Chem. Phys. Lett. 2024, 850, 141472. [Google Scholar] [CrossRef]
- Ma, Z.; Li, Q.; Luo, J.; Li, S.; Sui, L.; Zhao, D.; Yuan, K.; Xiao, G.; Tang, J.; Quan, Z. Pressure-driven reverse intersystem crossing: New path toward bright deep-blue emission of lead-free halide double perovskites. J. Am. Chem. Soc. 2021, 143, 15176–15184. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Wang, M.; Xiao, G.; Zou, B. Thinking about the development of high-pressure experimental chemistry. J. Phys. Chem. Lett. 2020, 11, 7297–7306. [Google Scholar] [CrossRef]
- Yu, Z.-H.; Liu, D.-X.; Ling, Y.-Y.; Chen, X.-X.; Shang, Y.; Chen, S.-L.; Ye, Z.-M.; Zhang, W.-X.; Chen, X.-M. Periodate-based molecular perovskites as promising energetic biocidal agents. Sci. China Mater. 2023, 66, 1641–1648. [Google Scholar] [CrossRef]
- Gagin, A.; Levin, I. Accounting for unknown systematic errors in Rietveld refinements: A Bayesian statistics approach. J. Appl. Crystallogr. 2015, 48, 1201–1211. [Google Scholar] [CrossRef]
- Toby, B.H.; Von Dreele, R.B. GSAS-II: The genesis of a modern open-source all purpose crystallography software package. J. Appl. Crystallogr. 2013, 46, 544–549. [Google Scholar] [CrossRef]
- Von Dreele, R.B. Small-angle scattering data analysis in GSAS-II. J. Appl. Crystallogr. 2014, 47, 1784–1789. [Google Scholar] [CrossRef]
- Greene, R.G.; Luo, H.; Ruoff, A.L. High pressure study of AlP: Transformation to a metallic NiAs phase. J. Appl. Phys. 1994, 76, 7296–7299. [Google Scholar] [CrossRef]
- Klotz, S.; Chervin, J.; Munsch, P.; Le Marchand, G. Hydrostatic limits of 11 pressure transmitting media. J. Phys. D Appl. Phys. 2009, 42, 075413. [Google Scholar] [CrossRef]
- Ciezak, J.A.; Jenkins, T.A.; Liu, Z.; Hemley, R.J. High-pressure vibrational spectroscopy of energetic materials: Hexahydro-1,3,5-trinitro-1,3,5-triazine. J. Phys. Chem. A 2007, 111, 59–63. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, T.; Li, H.; Xi, D.; Liu, L.; Sun, L.; Jin, D. Synthesis and High-Pressure Stability Study of Energetic Molecular Perovskite DAI-X1. Crystals 2025, 15, 530. https://doi.org/10.3390/cryst15060530
Yan T, Li H, Xi D, Liu L, Sun L, Jin D. Synthesis and High-Pressure Stability Study of Energetic Molecular Perovskite DAI-X1. Crystals. 2025; 15(6):530. https://doi.org/10.3390/cryst15060530
Chicago/Turabian StyleYan, Tingting, Han Li, Dongyang Xi, Linan Liu, Lei Sun, and Dinghan Jin. 2025. "Synthesis and High-Pressure Stability Study of Energetic Molecular Perovskite DAI-X1" Crystals 15, no. 6: 530. https://doi.org/10.3390/cryst15060530
APA StyleYan, T., Li, H., Xi, D., Liu, L., Sun, L., & Jin, D. (2025). Synthesis and High-Pressure Stability Study of Energetic Molecular Perovskite DAI-X1. Crystals, 15(6), 530. https://doi.org/10.3390/cryst15060530