Abstract
Breast cancer is a prevalent malignancy worldwide. Human MutT homolog 1 (MTH1) is over expressed in breast tumors, and cancer cells rely on MTH1 for survival. This protein ensures the integrity of the nucleotide pool by preventing the integration of oxidized 2′-deoxynucleoside triphosphates (dNTPs) during DNA replication. Therefore, inhibiting MTH1 pharmacologically emerged as a valid target in treating breast cancer. In the present study, we screened biologically active phytochemicals from the NPACT database to discover potential inhibitors of MTH1. Molecular docking analysis was employed to identify the binding conformation and the interaction pattern. The top five compounds were selected for detailed analysis based on their superior binding affinity and interactions with crucial residues (Asn33, Gly36, Tyr7, Phe72, Trp117, Lys23, and Phe27, Glu100) of MTH1. Additionally, the ADMET profile of selected compounds highlighted the high intestinal absorption, low toxicity, and acceptable metabolic stability, exhibiting their potential as drug candidates. Furthermore, in silico validation of selected compounds was performed through molecular dynamics (MD) simulation, which revealed that the resultant complexes are appreciably stable. Compounds revealed RMSD values ranging between 1.0 and 1.5 Å, indicating strong and stable binding conformations. PCA analysis revealed restricted conformational sampling, highlighting stabilization, particularly with ZINC14727630, ZINC14819291, ZINC14781695, and ZINC95099417. MM-GBSA confirmed the stability of the ligand–protein complexes, with ZINC14819291, ZINC14727630, and ZINC95099417 demonstrating the most stable interactions with MTH1, with total binding free energies of −32.46, −45.06, and −33.44 kcal/mol, respectively. Our results support that these natural compounds could act as potential anti-MTH1 for ameliorating the breast cancer. However, experimental validation is required to validate the efficacy of these molecules and robustness of this anticancer approach.
1. Introduction
Cancer remains the leading cause of mortality in both developed and developing countries, despite extensive research into its biological mechanisms, identification of therapeutic targets, and drug development [1]. Cancers include over 200 types, each originating in distinct locations within the body and from various underlying causes [2]. Among these, Breast cancer ranks as the second most prevalent cancer worldwide. Each year, nearly 1.5 million women are diagnosed with breast cancer [3]. Although breast cancer can occur in younger women, those over 50 are at a far higher risk [4]. It is a type of metastatic cancer that can propagate throughout the body, including the bones, liver, brain, and lungs, making it untreatable [5]. Several biological subtypes of breast cancer exist, and these differ from each other in their genetic features, clinical characteristics, and responses to different treatments [6].
A hallmark in the prognosis of breast cancer is oxidative stress, which is promoted by redox imbalance and contributes to its progression. Reactive oxygen species (ROS) are produced by the processes of xanthine oxidase, lipogenesis, cyclooxygenase, and NADPH oxidase (Nox), which produce hydroxyl radicals, hydrogen peroxide, and superoxide radicals. High ROS production triggers tumor inhibition through the activation of DNA damage [7]. ROS levels are frequently high in carcinogen-induced cancer cells, which are recognized by rapid proliferation and metabolic abnormalities [8]. Hence, these mechanisms pose a risk to cancer cells and apoptosis. Studies report that elevated levels of ROS have been found in breast cancer cells and various immune cells in the tumor microenvironment (TME). Signaling pathways, such as MAPK, NF-κB, and PI3K/AKT, can be activated through ROS and promote tumor cell proliferation, likely playing a complex function in tumor progression and metastasis by facilitating two-way communication between diverse components. Furthermore, an excessive amount of ROS can damage the micromoles within the cells and cause mutations in the genetic information within the genome. Hence, cancer cells often increase the expression of various DNA damage repair proteins, including human MutT homolog 1 (MTH1), to modulate the genome stability [9].
MTH1 is an important member of the NUDIX hydrolase superfamily and has a significant role in cancer biology. To combat the inhibitory impacts of high levels of ROS, cancer cells stimulate adaptive mechanisms [10]. According to previous research, cancer cells mostly depend on the MTH1-mediated base excision repair pathway to inhibit the detrimental oxidized nucleotides from getting improperly incorporated into the genome, which can reduce genotoxicity. MuT1 enzyme prevents the integration of oxidized nucleotides, such as 8-oxo-7,8-dihydro-2′-deoxyguanosine triphosphate 8-oxo-dGTP and N6-methyl-dATP, into DNA by hydrolyzing them into their monophosphate forms, thereby regulating genomic stability (Figure 1A) [11]. The MTH1 structure is characterized by its unique α-β-α scaffold, making a monomeric enzyme with a molecular weight of 18 kDa. MTH1 contains a conserved amino acid sequence motif known as the MUT box motif (Gly37-Leu59), essential for its enzymatic activity (Figure 1B).
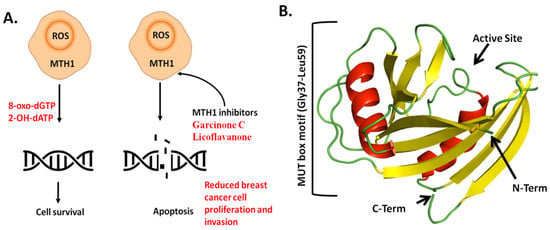
Figure 1.
Role and Structure of MTH1 in cancer cell proliferation. (A) Therapeutic role of MTH1. (B) Overall crystal structure of MTH1. Structure is colored based on secondary structure elements.
The MUT box comprises important residues like Gly37, Glu52, and Leu53, which stabilize the catalytic activity and neutralize oxidative nucleotides [12]. Several studies demonstrate that MTH1 is overexpressed in multiple cancers, including colorectal cancer, lung cancer, gastric cancer, renal cancer, brain tumors, and breast cancer. High MTH1 expression correlates considerably with poor prognosis in distinct malignancies [13,14]. MTH1 was examined in breast cancer subtypes, including luminal, hormone receptor-positive (HR+) tumors, and triple-negative breast cancer (TNBC) [15]. Another study revealed that MTH1 is significantly overexpressed in TNBC, which protects the cancer cells from oxidative DNA damage and apoptosis [16]. Thus, MTH1 is considered an important factor in the progression of breast cancer and a potential target for therapeutic intervention.
Conventional chemotherapeutic medicines effectively suppress breast cancer but also damage normal cells in the body [17]. While targeted therapies are highly effective for certain subsets of breast cancer patients, they are not universally practical due to their restricted specificity. As such, there is a need to identify enzyme inhibitors to address these therapeutic constraints and serve as a promising avenue for cancer treatment. The discovery of small-molecule inhibitors by targeting a specific protein is the trending method of drug discovery [18,19,20,21,22,23]. The inhibition of MTH1 can exert anticancer effects and exhibit broad-spectrum anticancer activity. TH287 and TH588 were discovered as the first small-molecule inhibitors targeting MTH1 [24,25]. However, computational approaches have been widely employed for developing potential therapeutic drug targets for various life-threatening diseases. Many researchers have already utilized computational methods to investigate MTH1 inhibitors, particularly from natural products. Several studies have investigated inhibitors such as aminopyrimidine derivatives, natural extracts, nucleoside analogs, Kettle’s three classes of inhibitors, and nanoparticles, all of which revealed a significant inhibitory effect on MTH1 [26]. Furthermore, Warpman Berglund U revealed that the MTH1 inhibitor TH1579 is an anticancer agent for acute myeloid leukemia by inducing oxidative DNA damage and mitotic arrest [27].
Computer-aided drug discovery (CADD) approaches significantly accelerate drug development while reducing associated costs [28]. Innovations in supercomputing, algorithms, and computational tools have further enhanced the efficiency of lead compound identification in pharmaceutical research [29,30,31,32,33,34,35]. Leveraging these progressions, we employed high-throughput screening integrated with molecular dynamics simulations to identify novel inhibitors, such as ZINC14819291, ZINC14727630, and ZINC95099417, that show stable binding and strong inhibitory potential against MTH1. ADMET analysis and PCA provided insights into their pharmacokinetic features and dynamic behavior. These results provide insight and candidate compounds for the innovations of new therapies and drugs targeting the common abnormal phenotypes in breast cancer.
2. Methodology
The current study is illustrated in a flow chart as shown in Figure 2.
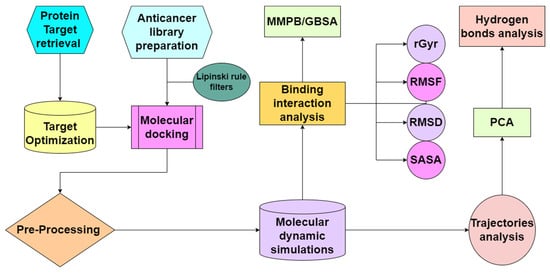
Figure 2.
Complete flow chart of the in silico study for drug designing against the cancer protein target.
2.1. Compound Libraries Preparation
The Naturally Occurring Plant-based Anti-Cancer Compound-Activity-Target database (NPACT, http://crdd.osdd.net/raghava/npact/ (accessed on 27 May 2024)) [36], which contains naturally occurring plant-derived anticancer compounds and the MPD3 (Medicinal Plant Database for Drug Discovery) [37] database were employed for virtual screening analysis [38] The retrieved ligands were then energy-minimized using MMFF94 implemented in Openbabel for structural optimization [39], after which they were filtered according to Lipinski’s Rule of Five (MW ≤ 500 Da, LogP ≤ 5, H-bond donors ≤ 5, H-bond acceptors ≤ 10) to retain drug-like candidates.
Ligands were further optimized by adding charges and hydrogens. The prepared ligands were saved in PDBQT format using AutoDock Tools to facilitate virtual screening [40,41]. Redundant compounds were removed to avoid computational redundancy, and the ligand structures were visually inspected to ensure structural integrity [42]. The final ligand library was compiled into a single directory for further analysis.
2.2. Target Selection and Preparation
The MTH1 protein was first identified as a therapeutic target based on its pivotal role in sanitizing oxidized nucleotides and its over expression in several cancer types. The 3D crystal structure of MTH1 in complex with N6-methyl-dAMP (PDB ID: 6QV0) was then retrieved from the Protein Data Bank (https://www.rcsb.org/structure/6QV0 (accessed on 27 May 2024)). All the unnecessary molecules were removed from the structure. Hydrogens were added and the structure was energy-minimized using UCSF Chimera [43] to relieve any steric clashes and optimize geometry prior molecular docking.
2.3. Grid Generation and Molecular Docking
The AutoDock Vina program in the PyRx software was employed for molecular docking experiments. AutoDock Vina employs empirical scoring function. It is extensively used due to its combination of rapid search algorithms and an empirically derived scoring function for accurate hit identification. It efficiently searches for the best possible binding poses by analyzing configurations and scoring them according to binding affinities [44,45,46,47,48]. The docking grid region was defined by centering the grid box at the active site with at x, y, and z centered around −0.508, 10.840, and 27.554, respectively, with dimensions of 40 Å in each direction, ensuring full coverage of the active site for subsequent docking calculations [47].
After grid generations, the ligands and protein structure were uploaded to PyRx and Autodock Vina was run to perform docking. During the virtual screening, each compound underwent 20 iterations, and the best binding pose was predicted with the low-affinity score. After docking, the docked complexes were ranked based on the docking scores and visually inspected using PyMol3.1 [49] (https://pymol.org/ (accessed on 27 May 2024)) and Discovery Studio v.2020 [50]. Compounds displaying the most favorable binding affinities and formed key interactions with the active site residues were shortlisted as candidate molecules for subsequent analyses. To examine the binding potency of the selected compounds with the MTH1 protein and to understand the interactions within the docked complex, the compound N6-methyl-dAMP was also re-docked in the same binding pocket under the identical conditions as the very first virtual screening.
2.4. Molecular Docking Protocol Validation
Molecular docking is an important approach to predict the interaction between a macromolecule and a small molecule, leading to stable complex formation. Redocking of the co-crystallized ligand operates as a validation process for the docking criteria, validating that the in silico model can accurately replicate the experimentally identified orientation of the ligand within the protein active site [51]. The native compound N6-methyl-dAMP was re-docked onto the MTH1 protein (Figure 3). The docking accuracy was assessed by comparing the docked pose with the experimental binding conformation. Consequently, the RMSD valued for the docked ligand superimposed on the crystallized ligand was 0.15 Å, within the generally accepted range for precise pose replication in docking analysis. The native ligand showed a binding energy score of −8.9 kcal/mol and the hydrogen bonding interaction with Leu9, Lys23, Asn33, Asp119, Gly37, Phe72, Trp117, Met81, Glu100, and Glu56, as shown in Table 1. The native ligand formed a sandwich-stabilized structure between phenylalanine A:72 and tryptophan A:117 hydrophobic aromatic amino acid residues. These residues revealed strong pi–pi stacking interactions, which positively affect the ligand affinity within the hydrophobic core of the binding pocket. Furthermore, hydrogen bonding interaction was observed with polar glycine A:37 residue.
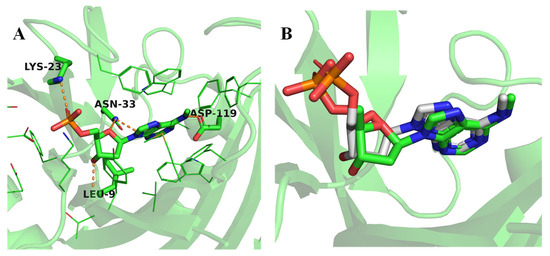
Figure 3.
Molecular docking of native and screened compounds against MTH1 Protein. (A) Interactions of the native ligand N6-methyl-dAMP with the MTH1 active site residues. (B) Re-docked conformation (gray) of ligand N6-methyl-dAMP superimposed to crystallized conformation (green).

Table 1.
Hydrogen bond interactions and binding affinities of the selected compounds.
2.5. ADMET Analysis
The absorption, distribution, metabolism, elimination, and toxicity (ADMET) features are crucial for the efficacy and safety of a therapeutic agent [52]. The characteristics of the two compounds were examined for their physiochemical features, hydrophobicity, lipophilicity, and gastrointestinal environment, which were directly affected by the ADMET profile before the defecation and urination of the drug from the body. This analysis was conducted using the SwissADME [53] (http://www.swissadme.ch/index.php (accessed on 28 May 2024), webserver to facilitate the assessment of physicochemical properties.
2.6. Molecular Dynamics Simulations
Molecular dynamics simulation was conducted using the AMBER16 simulation tool. During the molecular simulation, the dynamic and structural profiles of ligands docked into the binding sites of the target protein of interest were assessed using the AMBER16 program [54], which had the general AMBER force field [55] for ligand preparation and the ff14SB force field [56] for enzyme preparation. Following the initial setup, 500 steps of conjugate gradient minimization and steepest descent were applied to each system. The TIP3P water box [57] was used to submerge the selected docking complexes, with a 12.0 Å adjustment made between the complex and edge box. System neutralization was achieved by adding ions. The system was heated to a target temperature of 310 K by running the NVT ensemble for 200 ps [58]. After that, the system was subjected to the NPT ensemble for around 400 ps to achieve system equilibration. Isotropic position scaling was used to keep the pressure at 1 atm on average. Langevin dynamics was used to adjust the temperature, resulting in a collision frequency of 1 ps−1.The SHAKE method was used to restrict hydrogen bonding [59]. Finally, a production run of 200 ns was performed. A 200 ns molecular dynamics (MD) simulation was selected due to its established efficacy in capturing essential structural and dynamic properties for systems of comparable complexity. Studies show this simulation duration is sufficient for monitoring ligand–protein interactions, analyzing complex dynamics, and exploring conformational spaces without excessive computational demands [60,61,62]. The simulation paths were assessed by using the CPPTRAJ program [63]. RMSD is a measure used to compare the spatial positions of protein C atoms by calculating their average distance during a certain time frame [64].
2.7. PCA Analysis
Furthermore, Principal Component Analysis (PCA) was conducted using the GROMACS program and graphically visualized by R studio program [65]. PCA methodology transforms complex protein dynamics data from high-dimensional to low-dimensional space, generating eigenvectors and eigenvalues that represent global movements in the protein [66]. PCA may be used on any system to analyze the impact of different factors by simplifying the complexity of collective motion. This method is linked to the phase space behavior of protein functions and stability [67]. It is often used to describe various structural changes that occur during protein folding, the opening and closing of ion channels, and the dynamics of molecular conformations [68].
2.8. MMPBSA and MMGBSA
Binding free energy calculations were performed on the MD trajectories with the MM-PB/GBSA module of AMBER 16 to quantify the ligand–protein interaction energy [69]. The binding free energies of the ligand–MTH1 complexes were estimated of the 5000 frames from the last 25 ns of the 200 ns MD trajectory. They calculate free energies by forecasting the free energy difference between the ligand and protein alone and with complex. The binding free energy of the ligand–protein complex is calculated as follows:
The binding free energy is further broken down into various energetic components, expressed as follows:
is the molecular mechanics energy consisting bonded and non-bonded energy terms, is solvation energy comprising polar and non-polar solvation, and is the entropic contribution. The non-polar solvation term is derived from the solvent-accessible surface area (SASA), while the polar solvation term is computed with either the Poisson–Boltzmann (PB) or Generalized Born (GB) models. Collectively, these metrics reveal not just the overall binding strength but also the key forces hydrophobic contacts, electrostatic effects, and entropic penalties that stabilize the ligand–protein complex.
3. Results
3.1. Molecular Docking Analysis
The present study involved screening of the NPACT database against the MTH1 protein, followed by the ranking of docked compounds based on their binding energies (measured in kcal/mol). Docking scores of the top 10 compounds from both libraries were shown in Table S1. The top five compounds were selected according to their binding affinity scores (kcal/mol), having interactions with the important active site residue and possessing a similar binding orientation to the native ligand. The binding affinity of these compounds exceeded that of the native ligand, as highlighted by their superior binding scores. The superimposed conformation of compounds and the one native ligand showed an almost similar binding confirmation with the MTH1 binding pocket, with minimal deviations, representing structural similarity and compatibility of the compounds with the active site (Figure 4).
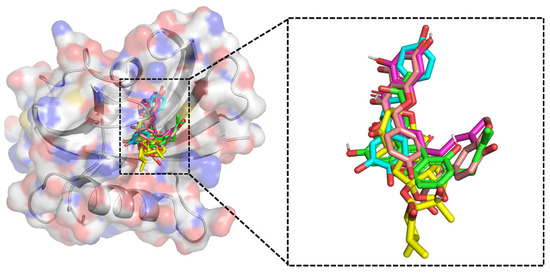
Figure 4.
The binding conformations of the top screened ligands within the MTH1 active site are shown on the left, with the superimposed conformations of the screened ligands and the native ligand displayed on the right. Ligands were shown in colored sticks; ZINC14781695 (cyan), ZINC95099417 (yellow), ZINC1530850 (light pink), ZINC14819291 (pink), and ZINC14727630 (green).
The docking analysis revealed that ZINC14781695 showed the highest binding affinity, with a docking score of −10.7 kcal/mol. It formed hydrogen bonds with important residues Lys23, Glu56, and Gly36, revealing strong interactions within the binding pocket (Figure 5A). Similarly, the ZINC14781695 compound, the ZINC95099417 compound, and the ZINC1530850 compound also showcased significant binding affinities towards the MTH1 with binding energies recorded at −10.3 kcal/mol and −10.2 kcal/mol, respectively. The ZINC95099417 compound interacted with Lys23, Asn33, and Phe27 (Figure 5B), while the ZINC1530850 compounds formed hydrogen interactions with Lys23, Asn33, and Lys38 residues (Figure 5C).
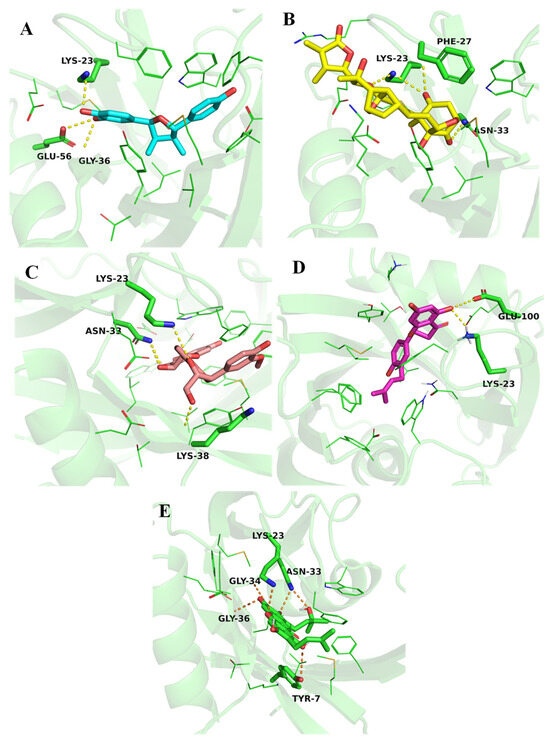
Figure 5.
Binding pose and interactions of the top selected ligands. (A) ZINC14781695, (B) ZINC95099417, (C) ZINC1530850, (D) ZINC14819291, and (E) ZINC14727630.
Additionally, ZINC14819291 also had the highest binding affinity to the MTH1 catalytic site, with a −9.9 kcal/mol binding energy. The side chain carboxylate oxygen atom of Glu100 and side chain amine group of Lys23 was involved in a conventional hydrogen bond interaction with the ligand (Figure 5D). ZINC14727630 binds to the protein with a slightly lower binding energy of −8.8 kcal/mol. The compound establishes a hydrogen bond with Tyr7, Lys23, Asn33, Gly34, and Gly36 (Figure 5E). These interactions highlight the compound’s ability to target specific sites on the protein, potentially leading to unique therapeutic outcomes. In a study, similar interactions of the natural compounds Thymoquinone and Baicalin with the key residues (Asn33, Gly36, Tyr7, Phe72, and Trp117) of the MTH1 binding pocket were reported. The similar interaction patterns revealed that these residues are crucial for structural support and inhibition of the MTH1 enzymes [14].
3.2. ADMET Properties
After the identification of drug-like potential compounds, ADMET analysis was conducted. Interactions with target macromolecules can lead to either beneficial or detrimental pharmacological effects [70] as drug bioavailability relies primarily on the safety and efficacy factors, which depend on the ADME qualities. ADME properties of the selected five compounds were calculated through the in silico pkCSM server (https://biosig.lab.uq.edu.au/pkcsm/ (accessed on 29 May 2024) to observe the pharmacokinetic features or properties, such as blood–brain permeability, absorption, metabolism, clearance, and toxicity [71]. The ADME properties of the five selected compounds are shown in Table 2. Most compounds show high intestinal absorption, exceeding 80%, except for ZINC14727698, which has an absorption rate of 68.725%, suggesting these can be efficiently absorbed into the bloodstream and have potential for oral bioavailability. All compounds are substrates of P-glycoprotein, a transporter protein that plays a role in drug efflux and influences bioavailability and resistance. However, they do not inhibit P-gp, meaning they are unlikely to interfere with its function in drug–drug interactions. Water solubility varies among the compounds, with ZINC95099417 having the lowest solubility at −4.087. The blood–brain barrier (BBB) is a closely regulated barrier that shields the brain from potentially harmful substances in the blood. Multiple studies suggest that brain metastasis occurs in 10 to 15 percent of breast cancer patients based on clinical diagnosis [72]. Blood–brain barrier permeability values indicate that these compounds have limited penetration into the brain, with ZINC1530850 and ZINC14727630 showing the lowest permeability at −1.407 and −1.362, respectively. These negative values suggest limited potential for these compounds to target brain metastases in breast cancer patients. None of the compounds act as CYP2D6 substrates, and only ZINC95099417 is a CYP3A4 substrate, suggesting that most compounds may not undergo significant metabolism through this enzyme, reducing the risk of drug interactions. The cytochrome P450 CYPs serve a multifaceted role in contributing to carcinogenesis, tumor growth, and metastasis [73]. CYP gene polymorphisms are linked with drug responses and sensitivity to breast cancer. Furthermore, findings suggest a generally favorable metabolic profile, with implications for increased metabolic stability and reduced interaction potential in breast cancer therapy. Most compounds inhibit CYP1A2, CYP2C9, and CYP2C19, which may influence metabolic pathways. Total clearance values range from −0.175 for ZINC14727630 to 0.197 for ZINC14819291, indicating variability in metabolic clearance rates. None of the compounds shows hepatotoxicity, supporting their safety profile. These compounds demonstrate good intestinal absorption, low hepatotoxicity risk, and varying metabolic properties. While their interactions with metabolic enzymes should be considered, their overall ADMET profile suggests potential suitability for further drug development.
Table 2.
The ADME properties of the five selected compounds.
3.3. Molecular Dynamic Simulation
Molecular dynamics (MD) simulations were used to assess the dynamics and stability of the complexes. MD simulations enables extensive analyses of structural stability and residue-level flexibility [74].
3.3.1. Root Mean Square Deviations
The reference–MTH1 complex exhibits considerable structural variations, with RMSD values ranging from 2.5 to 3.5 Å. These variations indicate that the structure of MTH1 coupled to reference N6-methyl-dAMP experiences significant conformational changes throughout simulation. The considerably larger RMSD values reflect the intrinsic instability of the MTH1 protein structure, which is dynamic and flexible. In contrast, the MTH1 structure associated with ZINC14819291 demonstrated higher stability than the reference. The RMSD values are steady between 1.5 and 2.5 Å, especially after the first equilibration period (10 ns) (Figure 6). This pattern implies that ZINC14819291 binding produces structural stability in the complex, but with some conformational flexibility, most likely due to interactions or binding modes between ZINC14819291 and the target protein. The system bound with ZINC14727630 has the stable trajectory, with RMSD values ranging from 1.0 to 1.5 Å. The low RMSD values observed throughout the simulation suggest that ZINC14727630 forms a very stable complex with few structural aberrations. This shows that the interaction of ZINC14727630 with the target protein results in a firmly bound and conformationally stable system, possibly due to strong and evenly dispersed intermolecular contacts. The MTH1–ZINC14781695 complex shows higher initial instability, with RMSD values rising above 3.0 Å in the early phase of the simulation. However, after 50 ns, the system stabilizes within the 2.0–3.0 Å range. The ZINC95099417 and ZINC1530850 complexes exhibit the most pronounced fluctuations, indicating higher instability compared to the other systems. Their RMSD values frequently fluctuate between 2.0 and 3.5 Å. To further support these observations, superimposition of the ligand–protein complexes at 0 ns and 200 ns is presented in Figure 7. The ligands remain stably engaged within the binding pocket throughout the simulation, showing that they preserve binding affinity and spatial accommodation within the active site despite certain conformational changes.
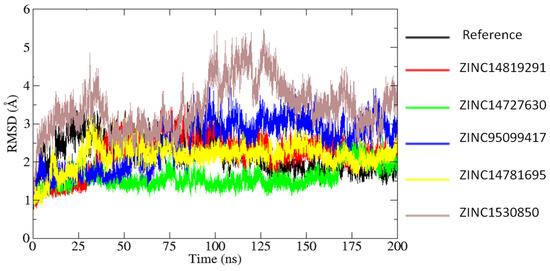
Figure 6.
RMSD analysis of the simulated systems.
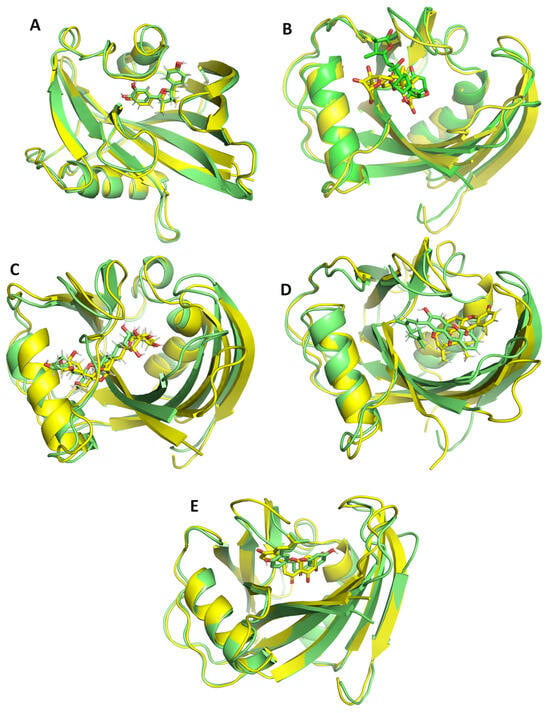
Figure 7.
Snapshots of the ligand protein complexes extracted at 0 ns and 200 ns superimposed to each other. The 0 ns snapshot of the complex is shown in green color and snapshot extracted at 200 ns is shown in yellow color. (A) ZINC14781695, (B) ZINC95099417, (C) ZINC1530850, (D) ZINC14819291, and (E) ZINC14727630.
3.3.2. Root Mean Square Fluctuations
To understand how the candidate ligands affect structural flexibility of the MTH1, the Root Mean Square Fluctuation (RMSF) analysis as performed. RMSF assesses the flexibility of individual residues in of the protein. Figure 8 illustrates the comparative flexibility analysis of different protein–ligand complexes through root mean square fluctuation (RMSF) and beta factor distributions, providing insight into residue-level flexibility. The reference exhibited the highest fluctuations, particularly around residues 50, 100, and 140, indicating highly dynamic regions. This suggests that in the absence of a ligand, the protein structure was more flexible, potentially due to the lack of stabilizing interactions. The ZINC14819291 complex follows a similar trend with slightly lower fluctuations, suggesting that this ligand maintains flexibility in key regions while exerting some stabilizing effects. In contrast, the ZINC14727630, ZINC1530850, and ZINC14781695 complex displayed significantly lower RMSF values, indicating that these ligands contribute to enhanced structural stability. The ZINC95099417 complex exhibited moderate flexibility, suggesting an intermediate stabilization effect. The beta factor illustrates the flexibility changes in the protein upon ligand binding. The reference exhibited the highest flexibility, indicating that these complexes retain significant conformational freedom. All ligand-bound complexes showed a reduction in flexibility to varying degrees. Among them, ZINC14727630 demonstrated the most significant stabilization, with the lowest B-factor values, suggesting strong interactions between the ligand and protein. ZINC14819291 closely follows the reference, indicating a moderate stabilization effect, while ZINC95099417, ZINC14781695, and ZINC1530850 demonstrated intermediate levels of rigidity. The flexible regions with high fluctuations belong to the loops (Figure 8C). These findings indicate that different ligands influence protein flexibility to varying degrees.
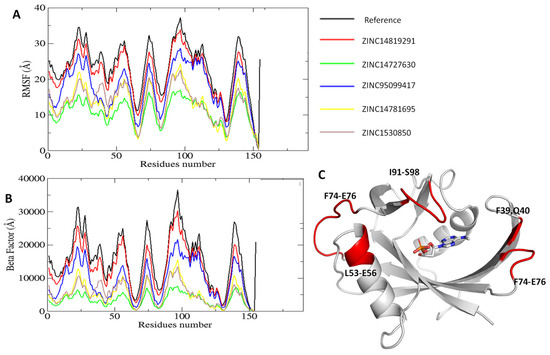
Figure 8.
RMSF analysis on structural flexibility of the MTH1 Protein. (A) Root mean square fluctuation analysis on candidate ligands. (B) Beta factor analysis of the simulated systems. (C) The areas of MTH1 with the high residual fluctuations are highlighted in red color.
3.3.3. Principle Component Analysis
The first two principal components (PCs) derived from the atomic displacement covariance matrix were evaluated to determine the main movements inside the systems. The 2D projections of these components, show considerable variations in the conformational space traversed by the complexes during molecular dynamics simulations (Figure 9). In this figure, Mode1 and Mode2 represent the principal components that describe the conformational flexibility of MTH1 in the presence of different ligands. The density distribution, represented by a color gradient from yellow (low density) to purple (high density), indicates the frequency of specific conformational states. The reference structure (A) shows a well-defined conformational space with distinct high-density clusters, representing the natural flexibility of MTH1 in its unbound state. This demonstrates the protein’s dynamic and unstable character, with significant sampling of different structural states. Upon ligand binding, significant variations are observed in the conformational distributions. ZINC14819291 causes a change in the conformational landscape, resulting in two dominating clusters that differ from the reference. ZINC14727630 changes the conformational space by forming a more linked distribution, implying less flexibility than the reference. ZINC95099417 produces an extended, arch-shaped distribution, indicating a significant change in conformational states. A similar trend is found with ZINC14781695, albeit with significantly less flexibility. ZINC1530850 has the greatest deviation, occupying a broader conformational area and indicating greater flexibility or ligand-induced structural alterations. These observations are consistent with the results of the RMSD and RMSF.
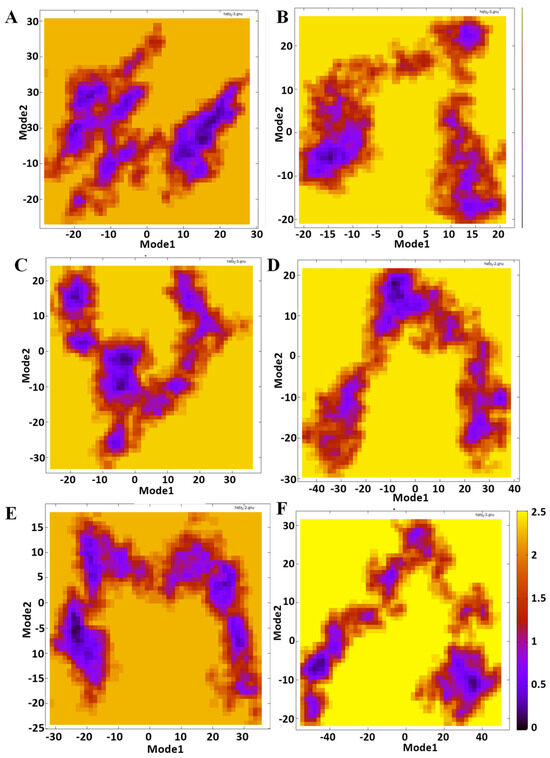
Figure 9.
PCA analysis of MTH1–ligand complexes. Mode1 and Mode2 represent the principal components that describe the conformational flexibility of MTH1 in the presence of different ligands. (A) Reference, (B) ZINC14819291, (C) ZINC14727630, (D) ZINC95099417, (E) ZINC14781695, and (F) ZINC1530850.
Each ligand uniquely affects MTH1’s structural dynamics. ZINC95099417 and ZINC14781695 stabilize specific conformational states, while ZINC14819291 and ZINC14727630 maintain partial similarities to the reference yet introduce notable shifts. Moreover, the observed trends are consistent with RMSD and RMSF analyses, reinforcing the role of PCA in capturing ligand-dependent conformational modulations.
3.4. Binding Free Energies Calculations
The results of the molecular dynamics simulations provide valuable insights into the binding affinities and interaction characteristics of the five candidate compounds ZINC14781695, ZINC1530850, ZINC14819291, ZINC14727630, and ZINC95099417 with the reference N6-methyl-dAMP molecule, based on energy parameters derived from MM-GBSA calculations (Table 3). Among the selected compounds, ZINC14727630 exhibits the strongest binding affinity, with total binding energies of −45.06 kcal/mol. It has van der Waals energy of −42.58 kcal/mol, indicating stronger non-covalent interactions such as hydrophobic contacts. Although its electrostatic energy (−12.90 kcal/mol) is slightly weaker than the reference compound (−15.38 kcal/mol), and other four compounds but its overall stronger gas–phase energy (−53.35 kcal/mol) and total binding energy highlight its superior stability within the binding site. ZINC1530850 also shows good binding potential, with a total binding energy of −32.70 kcal/mol. It has the best gas–phase interaction (−73.41 kcal/mol) among all compounds, showing significant van der Waals and electrostatic contributions. ZINC95099417 also emerges as a strong candidate having a total binding energy of −33.44 kcal/mol. It has a van der Waals energy of −38.12 kcal/mol and an electrostatic energy of −31.30 kcal/mol, totaling a gas–phase energy of −69.42 kcal/mol. Despite its solvation energy (35.98 kcal/mol), it shows to have a promising binding profile. ZINC14819291, while also shows strong binding potential, with total −32.46 kcal/mol. Its van der Waals (−30.67 kcal/mol) and electrostatic (−13.24 kcal/mol) energies contribute to a gas–phase energy of −43.67 kcal/mol, suggesting high stability within the binding site. Among all, ZINC14781695 exhibits the lowest total binding energy (−18.70 kcal/mol) between the selected compounds. Despite a strong van der Waals energy (−31.28 kcal/mol) and electrostatic interactions (−25.76 kcal/mol), its high solvation energy (38.34 kcal/mol) weakens its overall binding affinity.
Table 3.
Binding energy calculation for the selected five complexes along the control reference compound.
4. Discussion
Accelerated metabolism in cancer cells generates significantly more ROS than normal cells. MTH1, which cleanses oxidized nucleotide pools, is essential for the survival of cancer cells but not necessary for the growth of normal cells [75]. Many MTH1 inhibitors have been researched for cancer treatment development by increasing oxidative damage in cancer cells. Although some inhibitors were successful, others were ineffective in destroying cancer cells, which complicates the potential of MTH1 as a target for eradicating cancer [13]. MTH1 inhibition as a therapeutic strategy faces complexities due to redundancy in cancer cells’ antioxidant systems, off-target effects that compromise selectivity and safety, and variability in MTH1 expression requiring patient-specific approaches. These challenges emphasize the need for precise targeting and better understanding of MTH1’s role in cancer [76,77]. This study provides an overview of the progress in creating MTH1 inhibitors isolated from medicinal plants as possible drug candidates, in which they were categorized based on structure, as well as discusses their potential in cancer treatment by targeting MTH1 [78]. Natural compounds offer structural diversity, biological relevance, lower toxicity, and multiple pathways, making them effective enzyme inhibitors, particularly valuable in complex diseases like cancer, and excellent drug development starting points. Their multi-target capabilities and favorable safety profiles may help overcome resistance mechanisms and improve therapeutic efficacy [79,80,81].
To identify potential inhibitors against MTH1, two libraries (NPACT and MPD3) of natural compounds were screened against the MTH1 protein using PyRx. After molecular docking, we ranked all candidates by binding energy (kcal /mol). The 15 top scoring ligands were then subjected to detailed interaction profiling against MTH1. We retained only those molecules that engaged the greatest number of critical residues within the MTH1 active site, ensuring that the candidate ligands not only exhibit a favorable binding affinity but also formed key interactions essential for enzymatic inhibition.
In this study, the native ligand N6-methyl-dAMP bound with MTH1 (PDB ID 6QVO) was also included for comparison. This ligand forms hydrogen bond interactions with Leu9, Lys23, Asn33, and Asp119 and pi–pi interactions with Phe72 and Trp117. The phosphate tail is involved in attractive charge interactions with Glu100 and Lys23 [82]. The compounds ZINC14819291, ZINC14727630, ZINC95099417, ZINC14781695, and ZINC1530850 exhibited potential interactions with the MTH1 active site residue, showing good binding affinity compared to the reference compound. All the ligands formed hydrogen bonds with the critical residues such as Lys23, Asn33, Gly34, and Glu100. Further, MD simulations were conducted, providing a comprehensive understanding of the dynamic behavior, stability, and binding interactions within protein–ligand complexes [83]. Through the analysis of various structural metrics, including RMSD, RMSF, and Rg, critical insights into the structural integrity and conformational dynamics of the complexes were collected [84]. The consistent stability observed in RMSD profiles, coupled with the fluctuations in RMSF, indicates the robustness and adaptability of the protein–ligand systems over the simulation period. Additionally, Principal Component Analysis (PCA) offers a systematic approach to dissecting the complex structural dynamics of protein–ligand interactions [85]. Through PCA, major modes of motion and conformational changes within the complexes have been visualized, providing insights into the dynamic behavior of the systems and the structural determinants of ligand recognition and binding. The congruence between PCA-derived motion trajectories and other structural metrics further validates the significance of PCA in capturing the dynamic behavior and structural integrity of protein–ligand complexes [85]. Further MM-GBSA analyses revealed the strong stability and binding affinity between the candidate ligand and MTH1 protein. The compounds ZINC14819291, ZINC14727630, and ZINC95099417 had greater stability than the reference compound N6-methyl-dAMP suggesting that these compounds could be strong candidates against breast cancer through targeting MTH1 for further investigation. Among these ZINC14819291 is a flavonoid compound. Flavonoids are widely researched for their function in cancer prevention and treatment. These compounds can cause apoptosis, limit cell growth, and affect critical signaling pathways such PI3K/Akt/mTOR and NF-κB in different cancer types [86]. ZINC14819291 has been explored for various biological activities and has demonstrated promising potential across multiple conditions.
Liu et al. studied the effect of ZINC14819291 against nasopharyngeal carcinoma. ZINC14819291 inhibited the mTOR/PI3K/AKT signaling pathway, causing anticancer effects in human nasopharyngeal carcinoma cells (HK1) [87]. ZINC14819291 treatment resulted in a substantial drop in phosphorylated mTOR, PI3K, and AKT levels, indicating inhibition of this pathway, which is essential for cancer cell proliferation and differentiation [87]. Another study discovered the therapeutic potential of ZINC14819291 against cancer by targeting VEGFR-2. ZINC14819291 inhibits the proliferation, cycle, apoptosis, migration, invasion, and EMT of VEGF-stimulated MKN-45 cells by targeting VEGFR-2 and blocking the PI3K/AKT and MEK/ERK signaling pathways [88]. ZINC14727630 natural compound is derived from Garcinia oblongifolia Champ have cytotoxic properties against some malignancies. ZINC14727630 exhibits a broad bioactivity profile, showing potent antioxidant, anti-inflammatory, and anticancer effects through ROS scavenging, Nrf2 pathway activation, and apoptosis modulation in multiple cell and animal models. ZINC14727630 has been studied as an inhibitor of a variety of diseases, including cancer (HCC, lung, gastric, breast, leukemia, pancreatic), inflammatory conditions, bacterial and fungal infections, metabolic disorders, and neurodegenerative diseases [89]. Yudui et al. investigated the potential of ZINC14727630 as an anti-tumor drug by regulating the expression of ATR/Stat3/4E BP1 in nasopharyngeal cancer cells [90,91]. ZINC14727630 dramatically reduced the cell survival of the human NPC cell lines CNE1, CNE2, HK1, and HONE1. This inhibition occurred in a time and dosage-dependent way. Alpha-amylase is an enzyme that converts starch to sugars, and inhibiting it is used to treat postprandial hyperglycemia in type 2 diabetes. ZINC14727630 inhibits alpha-amylase, which reduces glucose synthesis from dietary starch [92]. In a recent study, researchers explored the inhibitory effects of ZINC14727630 on osteoclast development and bone resorption. ZINC14727630 reduces RANKL-induced osteoclast development and bone resorption by blocking oxidative stress and the NF-κB signaling pathway [93].
Moreover, given the strong in silico affinity of these compounds for MTH1, these compounds could be explored as adjuvants to conventional chemotherapy as chemo-herbal combination therapy. Chemo-herbal combination therapy has the potential to boost tumor cell kill, overcome drug resistance, and lower dose-limiting toxicities through synergistic or protective mechanisms [94,95].
In silico approaches, such as molecular docking, dynamics simulations, and machine learning, significantly aid in accelerating the drug discovery process by predicting potential hits, understanding binding interactions, and reducing the number of compounds for experimental testing, however they cannot consistently predict biological activity. They cannot fully capture the complexity of biological systems. These in silico findings require experimental validation (in vitro and in vivo) to confirm biological activity, efficacy, and safety.
5. Conclusions
MTH1 is known to break down oxidized nucleotide triphosphates as part of DNA repair. It may prevent the incorrect insertion of nucleotides during DNA replication and reduce cell death. High levels of ROS in a cancer cell may cause significant DNA damage and mutations via base pairing mismatch. MTH1 remove oxidized dNTP and inhibit cancer cells from undergoing apoptosis. Thus, targeting MTH1 activity is seen as a potential strategy for anticancer treatment. This work used high-throughput screening methods to screen a library of compounds to identify potential inhibitor for MTH1. Five compounds with MTH1 inhibitory properties were evaluated along with the reference compound N6-methyl-dAMP. Further molecular dynamics (MD) simulations and MM-PBSA/GBSA analysis were carried out to corroborate the complexes’ stability and binding affinities. The results show that among the screened compounds, ZINC14819291, ZINC14727630, and ZINC95099417 formed stable complexes with reduced flexibility, indicating a strong interaction with MTH1. These findings indicate that ZINC14819291, ZINC14727630, and ZINC95099417 have the potential to be used as therapeutic inhibitors of MTH1. Because these three ligands combine the strongest binding energies, favorable ADMET profiles, and consistent interactions with key catalytic residues, they are the most credible leads for progression into in vitro, cell-based, and eventual preclinical studies. Moreover, these are plant based natural compounds; they do not require complex chemical synthesis and can be directly extracted from the plants, facilitating their availability for further experimental evaluation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst15060507/s1, Table S1: Docking scores of the top 10 compounds from both libraries. The compounds chosen as potential candidate for further analysis are highlighted in yellow color.
Author Contributions
Conceptualization, M.A.A.; Data curation, M.A.A.; Formal analysis, A.S.A.; Funding acquisition, M.A.A.; Investigation, A.S.A.; Methodology, A.S.A.; Project administration, M.A.A.; Resources, M.A.A.; Software, A.S.A. and M.A.A.; Supervision, M.A.A.; Validation, M.A.A.; Visualization, A.S.A.; Writing—original draft, A.S.A.; Writing—review and editing, M.A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Prince Sattam bin Abdulaziz University grant number [PSAU/2024/03/31118].
Data Availability Statement
Data are provided within the manuscript.
Acknowledgments
The authors extend their appreciation to Prince Sattam bin Abdulaziz University for funding this research.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Hashim, D.; Boffetta, P.; La Vecchia, C.; Rota, M.; Bertuccio, P.; Malvezzi, M.; Negri, E. The global decrease in cancer mortality: Trends and disparities. Ann. Oncol. 2016, 27, 926–933. [Google Scholar] [CrossRef]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 2005, 115, 1503–1521. [Google Scholar] [CrossRef]
- Torre, L.A.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer in women: Burden and trends. Cancer Epidemiol. Biomark. Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Black, W.C.; Nease Jr, R.F.; Tosteson, A.N. Perceptions of breast cancer risk and screening effectiveness in women younger than 50 years of age. J. Natl. Cancer Inst. 1995, 87, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Stamenkovic, I. Metastatic cancer cell. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 221–247. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, L.; Fanotto, V.; Pelizzari, G.; Agostinetto, E.; Puglisi, F. Do platinum salts fit all triple negative breast cancers? Cancer Treat. Rev. 2016, 48, 34–41. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Heber, D.; Byerly, L.O.; Chlebowski, R.T. Metabolic abnormalities in the cancer patient. Cancer 1985, 55, 225–229. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Bialkowski, K.; Kasprzak, K.S. A profile of 8-oxo-dGTPase activities in the NCI-60 human cancer panel: Meta-analytic insight into the regulation and role of MTH1 (NUDT1) gene expression in carcinogenesis. Free Radic. Biol. Med. 2020, 148, 1–21. [Google Scholar] [CrossRef]
- Lin, J.-F.; Hu, P.-S.; Wang, Y.-Y.; Tan, Y.-T.; Yu, K.; Liao, K.; Wu, Q.-N.; Li, T.; Meng, Q.; Lin, J.-Z. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct. Target. Ther. 2022, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Shimokawa, H.; Sekiguchi, M.; Nakabeppu, Y. Functional significance of the conserved residues for the 23-residue module among MTH1 and MutT family proteins. J. Biol. Chem. 1999, 274, 38251–38259. [Google Scholar] [CrossRef] [PubMed]
- Berglund, U.W.; Sanjiv, K.; Gad, H.; Kalderen, C.; Koolmeister, T.; Pham, T.; Gokturk, C.; Jafari, R.; Maddalo, G.; Seashore-Ludlow, B. Validation and development of MTH1 inhibitors for treatment of cancer. Ann. Oncol. 2016, 27, 2275–2283. [Google Scholar] [CrossRef]
- Taiyab, A.; Choudhury, A.; Haidar, S.; Yousuf, M.; Rathi, A.; Koul, P.; Chakrabarty, A.; Islam, A.; Shamsi, A.; Hassan, M.I. Exploring MTH1 inhibitory potential of Thymoquinone and Baicalin for therapeutic targeting of breast cancer. J. Biomed. Pharmacother. 2024, 173, 116332. [Google Scholar] [CrossRef]
- Zhang, X.; Song, W.; Zhou, Y.; Mao, F.; Lin, Y.; Guan, J.; Sun, Q. Expression and function of MutT homolog 1 in distinct subtypes of breast cancer. Oncol. Lett. 2017, 13, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Y.; Mao, F.; Lin, Y.; Shen, S.; Sun, Q. lncRNA AFAP1-AS1 promotes triple negative breast cancer cell proliferation and invasion via targeting miR-145 to regulate MTH1 expression. Sci. Rep. 2020, 10, 7662. [Google Scholar] [CrossRef]
- Duarte, I.L.; da Silveira Nogueira Lima, J.P.; Lima, C.S.P.; Sasse, A.D. Dose-dense chemotherapy versus conventional chemotherapy for early breast cancer: A systematic review with meta-analysis. Breast 2012, 21, 343–349. [Google Scholar] [CrossRef]
- Islam, S.; Hosen, M.A.; Ahmad, S.; ul Qamar, M.T.; Dey, S.; Hasan, I.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M. Synthesis, antimicrobial, anticancer activities, PASS prediction, molecular docking, molecular dynamics and pharmacokinetic studies of designed methyl α-D-glucopyranoside esters. J. Mol. Struct. 2022, 1260, 132761. [Google Scholar] [CrossRef]
- Ahmed, B.; Ashfaq, U.A.; ul Qamar, M.T.; Ahmad, M. Anticancer potential of phytochemicals against breast cancer: Molecular docking and simulation approach. Bangladesh J. Pharmacol. 2014, 9, 545–550. [Google Scholar] [CrossRef]
- Bashir, Y.; Noor, F.; Ahmad, S.; Tariq, M.H.; Qasim, M.; Tahir ul Qamar, M.; Almatroudi, A.; Allemailem, K.S.; Alrumaihi, F.; Alshehri, F.F. Integrated virtual screening and molecular dynamics simulation approaches revealed potential natural inhibitors for DNMT1 as therapeutic solution for triple negative breast cancer. J. Biomol. Struct. Dyn. 2024, 42, 1099–1109. [Google Scholar] [CrossRef]
- Muneer, I.; Ahmad, S.; Naz, A.; Abbasi, S.W.; Alblihy, A.; Aloliqi, A.A.; Aba Alkhayl, F.F.; Alrumaihi, F.; Ahmad, S.; El Bakri, Y. Discovery of novel inhibitors from medicinal plants for v-domain ig suppressor of t-cell activation. Front. Mol. Biosci. 2021, 8, 716735. [Google Scholar] [CrossRef] [PubMed]
- Sadaqat, M.; Qasim, M.; ul Qamar, M.T.; Masoud, M.S.; Ashfaq, U.A.; Noor, F.; Fatima, K.; Allemailem, K.S.; Alrumaihi, F.; Almatroudi, A. Advanced network pharmacology study reveals multi-pathway and multi-gene regulatory molecular mechanism of Bacopa monnieri in liver cancer based on data mining, molecular modeling, and microarray data analysis. Comput. Biol. Med. 2023, 161, 107059. [Google Scholar] [CrossRef] [PubMed]
- Altharawi, A.; Ahmad, S.; Alamri, M.A.; ul Qamar, M.T. Structural insight into the binding pattern and interaction mechanism of chemotherapeutic agents with Sorcin by docking and molecular dynamic simulation. Colloids Surf. B Biointerfaces 2021, 208, 112098. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Ma, J.; Hu, Y.; Di, R.; Wang, L.; Zhang, X.; Lai, Y.; Liu, Y. Targeting MutT homolog 1 (MTH1) for breast cancer suppression by a novel MTH1 inhibitor MA-24 with tumor-selective toxicity. Pharmaceuticals 2024, 17, 291. [Google Scholar] [CrossRef]
- Garutti, M.; Pelizzari, G.; Bartoletti, M.; Malfatti, M.C.; Gerratana, L.; Tell, G.; Puglisi, F. Platinum salts in patients with breast cancer: A focus on predictive factors. Int. J. Mol. Sci. 2019, 20, 3390. [Google Scholar] [CrossRef]
- Kettle, J.G.; Alwan, H.; Bista, M.; Breed, J.; Davies, N.L.; Eckersley, K.; Fillery, S.; Foote, K.M.; Goodwin, L.; Jones, D.R.; et al. Potent and selective inhibitors of MTH1 probe its role in cancer cell survival. J. Med. Chem. 2016, 59, 2346–2361. [Google Scholar] [CrossRef]
- Sanjiv, K.; Calderón-Montaño, J.M.; Pham, T.M.; Erkers, T.; Tsuber, V.; Almlöf, I.; Höglund, A.; Heshmati, Y.; Seashore-Ludlow, B.; Nagesh Danda, A.J.C.R. MTH1 inhibitor TH1579 induces oxidative DNA damage and mitotic arrest in acute myeloid leukemia. Cancer Res. 2021, 81, 5733–5744. [Google Scholar] [CrossRef]
- Niazi, S.K.; Mariam, Z.J.P. Computer-aided drug design and drug discovery: A prospective analysis. Pharmaceuticals 2023, 17, 22. [Google Scholar] [CrossRef]
- Singh, P.; Kaur, J.; Singh, G.; Bhatti, R. Triblock conjugates: Identification of a highly potent antiinflammatory agent. J. Med. Chem. 2015, 58, 5989–6001. [Google Scholar] [CrossRef]
- Singh, P.; Kaur, S.; Kaur, J.; Singh, G.; Bhatti, R. Rational design of small peptides for optimal inhibition of cyclooxygenase-2: Development of a highly effective anti-inflammatory agent. J. Med. Chem. 2016, 59, 3920–3934. [Google Scholar] [CrossRef]
- Kaur, J.; Kaur, B.; Singh, P. Rational modification of semaxanib and sunitinib for developing a tumor growth inhibitor targeting ATP binding site of tyrosine kinase. Bioorganic Med. Chem. Lett. 2018, 28, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Arti, S.; Kaur, K.; Kaur, J.; Ghosh, T.K.; Banipal, T.S.; Banipal, P.K. Host-guest interaction of trimethoprim drug with cyclodextrins in aqueous solutions: Calorimetric, spectroscopic, volumetric and theoretical approach. J. Mol. Liq. 2021, 329, 115431. [Google Scholar] [CrossRef]
- Kaur, J.; Kaur, S.; Singh, P. Rational modification of the lead molecule: Enhancement in the anticancer and dihydrofolate reductase inhibitory activity. Bioorganic Med. Chem. Lett. 2016, 26, 1936–1940. [Google Scholar] [CrossRef]
- Noor, F.; Junaid, M.; Almalki, A.H.; Almaghrabi, M.; Ghazanfar, S.; Tahir ul Qamar, M. Deep learning pipeline for accelerating virtual screening in drug discovery. Sci. Rep. 2024, 14, 28321. [Google Scholar] [CrossRef]
- Majeed, A.; Tahir ul Qamar, M.; Maryam, A.; Mirza, M.U.; Alhussain, L.; Al Otaibi, S.O.; Almatroudi, A.; Allemailem, K.S.; Alrumaihi, F.; Aloliqi, A.A. Structural insights into the mechanism of resistance to bicalutamide by the clinical mutations in androgen receptor in chemo-treatment resistant prostate cancer. J. Biomol. Struct. Dyn. 2024, 42, 1181–1190. [Google Scholar] [CrossRef]
- Mangal, M.; Sagar, P.; Singh, H.; Raghava, G.P.; Agarwal, S.M. NPACT: Naturally occurring plant-based anti-cancer compound-activity-target database. Nucleic Acids Res. 2013, 41, D1124–D1129. [Google Scholar] [CrossRef] [PubMed]
- Mumtaz, A.; Ashfaq, U.A.; ul Qamar, M.T.; Anwar, F.; Gulzar, F.; Ali, M.A.; Saari, N.; Pervez, M.T. MPD3: A useful medicinal plants database for drug designing. Nat. Prod. Res. 2017, 31, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, M.; Chen, L.C.; Gordon, T.; Ito, K.; Thurston, G.D. National Particle Component Toxicity (NPACT) Initiative: Integrated epidemiologic and toxicologic studies of the health effects of particulate matter components. Res. Rep. 2013, 177, 5–13. [Google Scholar]
- Halgren, T.A. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J. Comput. Chem. 1999, 20, 730–748. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinf. 2008, 24, 8.14.1–8.14.40. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Forli, S. Using AutoDock 4 and AutoDock vina with AutoDockTools: A tutorial. Scripps Res. Inst. Mol. Graph. Lab. 2012, 10550, 1000. [Google Scholar]
- Cang, Z.; Mu, L.; Wei, G.-W. Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening. PLOS Comput. Biol. 2018, 14, e1005929. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Sarkar, A.; Concilio, S.; Sessa, L.; Marrafino, F.; Piotto, S. Advancements and novel approaches in modified AutoDock Vina algorithms for enhanced molecular docking. Results Chem. 2024, 7, 101319. [Google Scholar] [CrossRef]
- Zhou, X.; Ling, M.; Lin, Q.; Tang, S.; Wu, J.; Hu, H. Effectiveness analysis of multiple initial states simulated annealing algorithm, a case study on the molecular docking tool AutoDock vina. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 3830–3841. [Google Scholar] [CrossRef]
- Pawar, R.P.; Rohane, S.H. Role of autodock vina in PyRx molecular docking. Asian J. Res. Chem. 2021, 14, 132–134. [Google Scholar]
- Che, X.; Liu, Q.; Zhang, L. An accurate and universal protein-small molecule batch docking solution using Autodock Vina. Results Eng. 2023, 19, 101335. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.S.; Hu, Z. Using PyMOL as a platform for computational drug design. WIREs Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Pawar, S.S.; Rohane, S.H. Review on Discovery Studio: An Important Tool for Molecular Docking. Asian J. Res. Chem. 2021, 14, 1–3. [Google Scholar] [CrossRef]
- Mateev, E.; Valkova, I.; Angelov, B.; Georgieva, M.; Zlatkov, A. Validation through re-docking, cross-docking and ligand enrichment in various well-resoluted MAO-B receptors. Int. J. Pharm. Sci. Res. 2022, 13, 1000–1007. [Google Scholar]
- Lin, J.; Sahakian, D.C.; De Morais, S.; Xu, J.J.; Polzer, R.J.; Winter, S.M. The role of absorption, distribution, metabolism, excretion and toxicity in drug discovery. Curr. Top. Med. Chem. 2003, 3, 1125–1154. [Google Scholar] [CrossRef] [PubMed]
- Bakchi, B.; Krishna, A.D.; Sreecharan, E.; Ganesh, V.B.J.; Niharika, M.; Maharshi, S.; Puttagunta, S.B.; Sigalapalli, D.K.; Bhandare, R.R.; Shaik, A.B. An overview on applications of SwissADME web tool in the design and development of anticancer, antitubercular and antimicrobial agents: A medicinal chemist’s perspective. J. Mol. Struct. 2022, 1259, 132712. [Google Scholar] [CrossRef]
- Lee, T.-S.; Cerutti, D.S.; Mermelstein, D.; Lin, C.; LeGrand, S.; Giese, T.J.; Roitberg, A.; Case, D.A.; Walker, R.C.; York, D.M.; et al. GPU-accelerated molecular dynamics and free energy methods in Amber18: Performance enhancements and new features. J. Chem. Inf. Model. 2018, 58, 2043–2050. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Pang, Y.P. FF12MC: A revised AMBER forcefield and new protein simulation protocol. Proteins Struct. Funct. Bioinform. 2016, 84, 1490–1516. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Brooks, C.L., III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Collier, T.A.; Piggot, T.J.; Allison, J.R. Molecular dynamics simulation of proteins. In Protein Nanotechnology; Springer: Berlin/Heidelberg, Germany, 2020; pp. 311–327. [Google Scholar]
- Moritsugu, K.; Smith, J.C. Langevin model of the temperature and hydration dependence of protein vibrational dynamics. J. Phys. Chem. B 2005, 109, 12182–12194. [Google Scholar] [CrossRef]
- Guterres, H.; Im, W. Improving protein-ligand docking results with high-throughput molecular dynamics simulations. J. Chem. Inf. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef]
- Sultana, T.; Mou, S.I.; Chatterjee, D.; Faruk, M.O.; Hosen, M.I. Computational exploration of SLC14A1 genetic variants through structure modeling, protein-ligand docking, and molecular dynamics simulation. Biochem. Biophys. Rep. 2024, 38, 101703. [Google Scholar] [CrossRef]
- Shahab, M.; Zheng, G.; Khan, A.; Wei, D.; Novikov, A.S. Machine learning-based virtual screening and molecular simulation approaches identified novel potential inhibitors for cancer therapy. Biomedicines 2023, 11, 2251. [Google Scholar] [CrossRef]
- Wang, N.; Gao, J.-G.; Wu, M.-W. Molecular docking and molecular simulation studies for N-degron selectivity of chloroplastic ClpS from Chlamydomonas reinhardtii. Comput. Biol. Chem. 2023, 103, 107825. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O. How root-mean-square distance (rmsd) values depend on the resolution of protein structures that are compared. Appl. Crystallogr. 2003, 36, 125–128. [Google Scholar] [CrossRef]
- Khan, M.K.A.; Alouffi, S.; Ahmad, S. Identifying potential inhibitors of CXC motif chemokine ligand10 against vitiligo: Structure-based virtual screening, molecular dynamics simulation, and principal component analysis. J. Biomol. Struct. Dyn. 2024, 42, 8045–8062. [Google Scholar] [CrossRef]
- Moradi, S.; Nowroozi, A.; Nezhad, M.A.; Jalali, P.; Khosravi, R.; Shahlaei, M. A review on description dynamics and conformational changes of proteins using combination of principal component analysis and molecular dynamics simulation. Comput. Biol. Med. 2024, 183, 109245. [Google Scholar] [CrossRef] [PubMed]
- Sittel, F.; Stock, G. Perspective: Identification of Collective Variables and Metastable States of Protein Dynamics. J. Chem. Phys. 2018, 149, 150901. [Google Scholar] [CrossRef]
- Tieleman, D.P.; Biggin, P.C.; Smith, G.R.; Sansom, M.S.P. Simulation approaches to ion channel structure–function relationships. Q. Rev. Biophys. 2001, 34, 473–561. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N. Affinity calculations of cyclodextrin host–guest complexes: Assessment of strengths and weaknesses of end-point free energy methods. J. Chem. Inf. Model. 2018, 59, 421–440. [Google Scholar] [CrossRef]
- Gandla, K.; Islam, F.; Zehravi, M.; Karunakaran, A.; Sharma, I.; Haque, M.A.; Kumar, S.; Pratyush, K.; Dhawale, S.A.; Nainu, F.; et al. Natural polymers as potential P-glycoprotein inhibitors: Pre-ADMET profile and computational analysis as a proof of concept to fight multidrug resistance in cancer. Heliyon 2023, 9, e19454. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Arshad, F.; Wang, L.; Sy, C.; Avraham, S.; Avraham, H.K. Blood-brain barrier integrity and breast cancer metastasis to the brain. Pathol. Res. Int. 2011, 2011, 920509. [Google Scholar] [CrossRef]
- Luo, B.; Yan, D.; Yan, H.; Yuan, J. Cytochrome P450: Implications for human breast cancer. Oncol. Lett. 2021, 22, 548. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Bao, L.; Feng, C.; Huang, Q.; Zhang, F.; Gao, X.; Han, R. Accurate prediction of protein structural flexibility by deep learning integrating intricate atomic structures and Cryo-EM density information. Nat. Commun. 2024, 15, 5538. [Google Scholar] [CrossRef]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, Q. Targeting the nucleic acid oxidative damage repair enzyme MTH1: A promising therapeutic option. Front. Cell Dev. Biol. 2024, 12, 1334417. [Google Scholar] [CrossRef] [PubMed]
- Gul, N.; Karlsson, J.; Tängemo, C.; Linsefors, S.; Tuyizere, S.; Perkins, R.; Ala, C.; Zou, Z.; Larsson, E.; Bergö, M.O. The MTH1 inhibitor TH588 is a microtubule-modulating agent that eliminates cancer cells by activating the mitotic surveillance pathway. Sci. Rep. 2019, 9, 14667. [Google Scholar] [CrossRef]
- Gad, H.; Koolmeister, T.; Jemth, A.-S.; Eshtad, S.; Jacques, S.A.; Ström, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.O. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.R.; Kucuk, O.; Khuri, F.R.; Shin, D.M. Perspectives for cancer prevention with natural compounds. J. Clin. Oncol. 2009, 27, 2712–2725. [Google Scholar] [CrossRef]
- Tsuda, H.; Ohshima, Y.; Nomoto, H.; Fujita, K.-I.; Matsuda, E.; Iigo, M.; Takasuka, N.; Moore, M.A. Cancer prevention by natural compounds. Drug Metab. Pharmacokinet. 2004, 19, 245–263. [Google Scholar] [CrossRef]
- Scaletti, E.R.; Vallin, K.S.; Bräutigam, L.; Sarno, A.; Berglund, U.W.; Helleday, T.; Stenmark, P.; Jemth, A.-S. MutT homologue 1 (MTH1) removes N6-methyl-dATP from the dNTP pool. J. Biol. Chem. 2020, 295, 4761–4772. [Google Scholar] [CrossRef]
- Shukla, R.; Tripathi, T. Molecular dynamics simulation of protein and protein–ligand complexes. In Computer-Aided Drug Design; Springer: Berlin/Heidelberg, Germany, 2020; pp. 133–161. [Google Scholar]
- Das, R.P.; Behera, S.K.; Sahoo, B.; Arakha, M.; Pradhan, A.K. Comparative Analysis of Backbone Atom Cross-Correlation Matrices and Folding Dynamics of Amyloid Fibril and Its Complexes with Novel Biosurfactants Isolated from Bacillus Strain: A Binding Free Energy Calculation (MM-PBSA) and MD Simulation Approach. J. Biomol. Struct. Dyn. 2024, 42, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Maity, A.; Majumdar, S.; Priya, P.; De, P.; Saha, S.; Ghosh Dastidar, S. Adaptability in protein structures: Structural dynamics and implications in ligand design. J. Biomol. Struct. Dyn. 2015, 33, 298–321. [Google Scholar] [CrossRef] [PubMed]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Büsselberg, D. Flavonoids in cancer and apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef]
- Liu, J.; Song, C.; Liang, Z.; Long, X.; Guo, M.; Xu, J. Licoflavanone exerts anticancer effects on human nasopharyngeal cancer cells via caspase activation, suppression of cell migration and invasion, and inhibition of m-TOR/PI3K/AKT pathway. Trop. J. Pharm. Res. 2021, 20, 1387–1393. [Google Scholar] [CrossRef]
- Hongxia, G.; Xiaojie, J.; Guangxian, L.; Min, Z.; Shiwei, N.; Wangjie, C.; Han, Z.; Yuanding, Z.; Chenghao, L.; Yaling, L. Licoflavone A Suppresses Gastric Cancer Growth and Metastasis by Blocking the VEGFR-2 Signaling Pathway. J. Oncol. 2022, 2022, 5497991. [Google Scholar] [CrossRef]
- Aravind, A.A.; Menon, L.N.; Rameshkumar, K.B. Structural diversity of secondary metabolites in Garcinia species. In Diversity of Garcinia Species Western Ghats: Pythochemical Perspective; Jawaharlal Nehru Tropical Botanic Garden and Research Institute: Kerala, India, 2016; pp. 19–75. [Google Scholar]
- Si, Y.; Xu, J.; Meng, L.; Wu, Y.; Qi, J. Role of STAT3 in the pathogenesis of nasopharyngeal carcinoma and its significance in anticancer therapy. Front. Oncol. 2022, 12, 1021179. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, X.; Zou, C.; Feng, S.; Guo, H.; Yang, Y.; Lei, Y.; Zhang, J.; Lu, Y. Garcinone C exerts antitumor activity by modulating the expression of ATR/Stat3/4E-BP1 in nasopharyngeal carcinoma cells. Oncol. Rep. 2018, 39, 1485–1493. [Google Scholar] [CrossRef]
- Li, X.; Chen, H.; Jia, Y.; Peng, J.; Li, C. Inhibitory effects against alpha-amylase of an enriched polyphenol extract from pericarp of mangosteen (Garcinia mangostana). Foods 2022, 11, 1001. [Google Scholar] [CrossRef]
- Ji, H.; Pan, Q.; Cao, R.; Li, Y.; Yang, Y.; Chen, S.; Gu, Y.; Qian, D.; Guo, Y.; Wang, L. Garcinone C attenuates RANKL-induced osteoclast differentiation and oxidative stress by activating Nrf2/HO-1 and inhibiting the NF-kB signaling pathway. Heliyon 2024, 10, e25601. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, P.; Li, J.; Zhou, Y.; Wang, B.; Lu, Z. The effect of doxorubicin curcumin co-loaded lipid nanoparticles and doxorubicin on osteosarcoma before surgery. Cancer Nanotechnol. 2024, 15, 11. [Google Scholar] [CrossRef]
- Lou, C.; Lu, H.; Ma, Z.; Liu, C.; Zhang, Y. Ginkgolide B enhances gemcitabine sensitivity in pancreatic cancer cell lines via inhibiting PAFR/NF-κB pathway. Biomed. Pharmacother. 2019, 109, 563–572. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).