
First-Principles Study on the Migration and Release Properties of Xe on the Surface of Uranium Mononitride

 ,
, 
Abstract
1. Introduction
2. Method
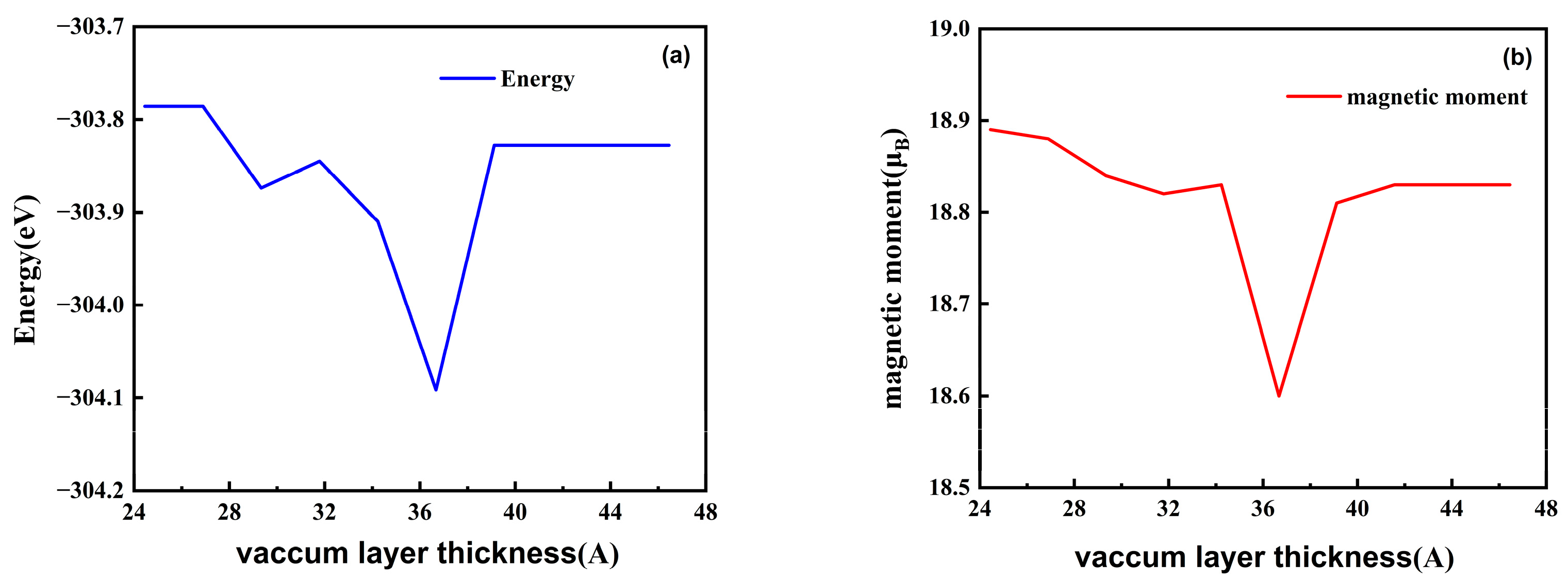
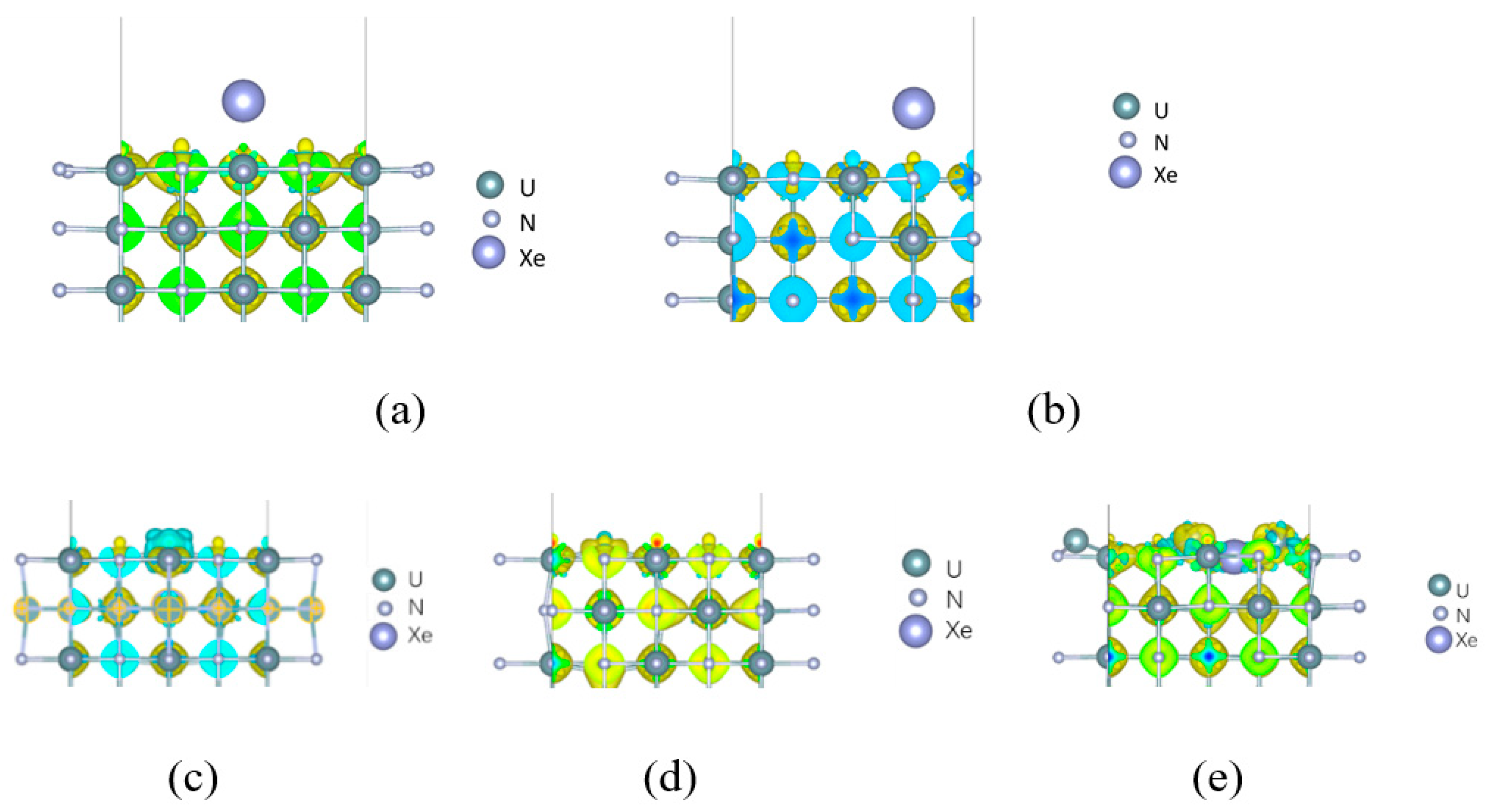
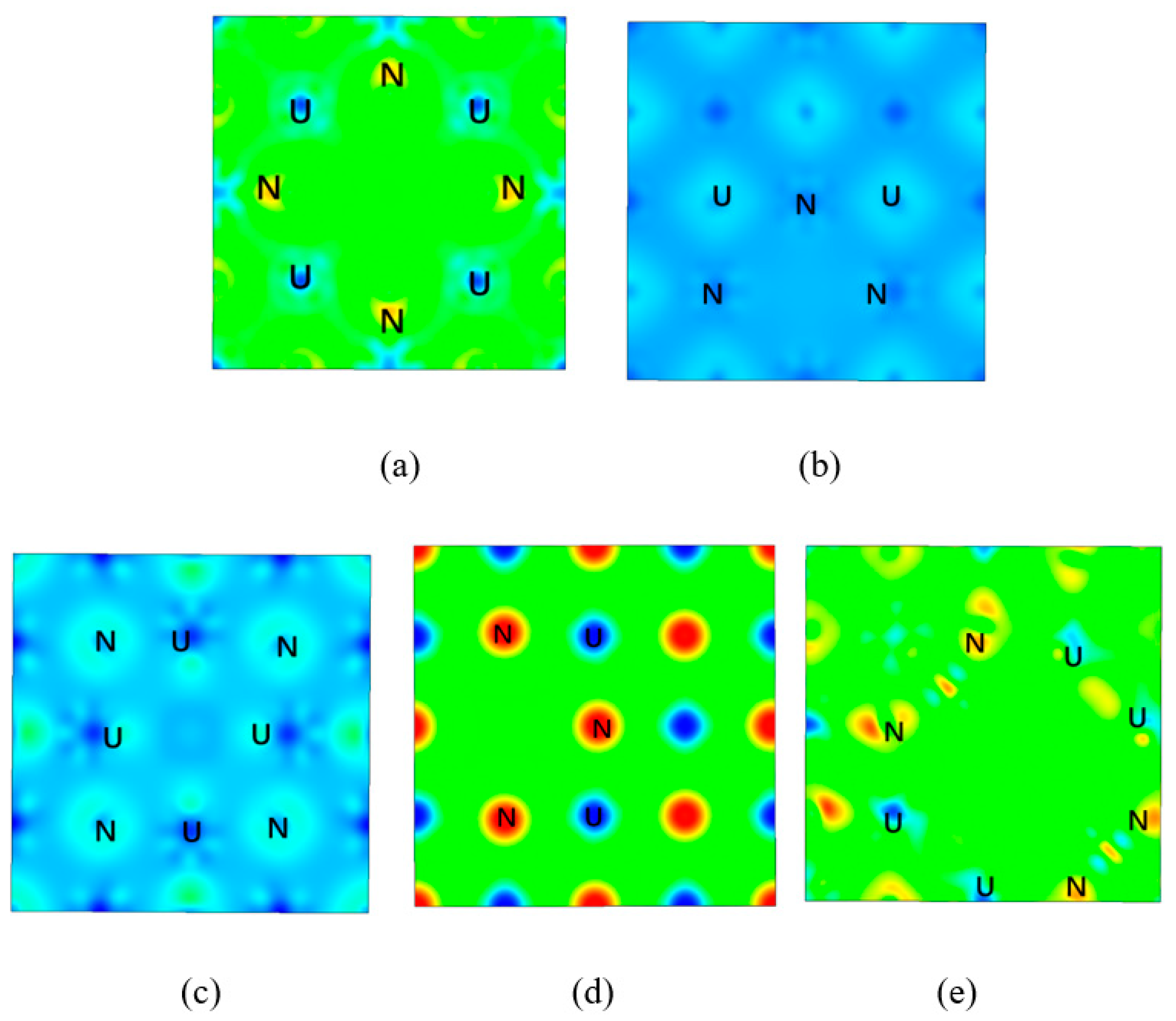
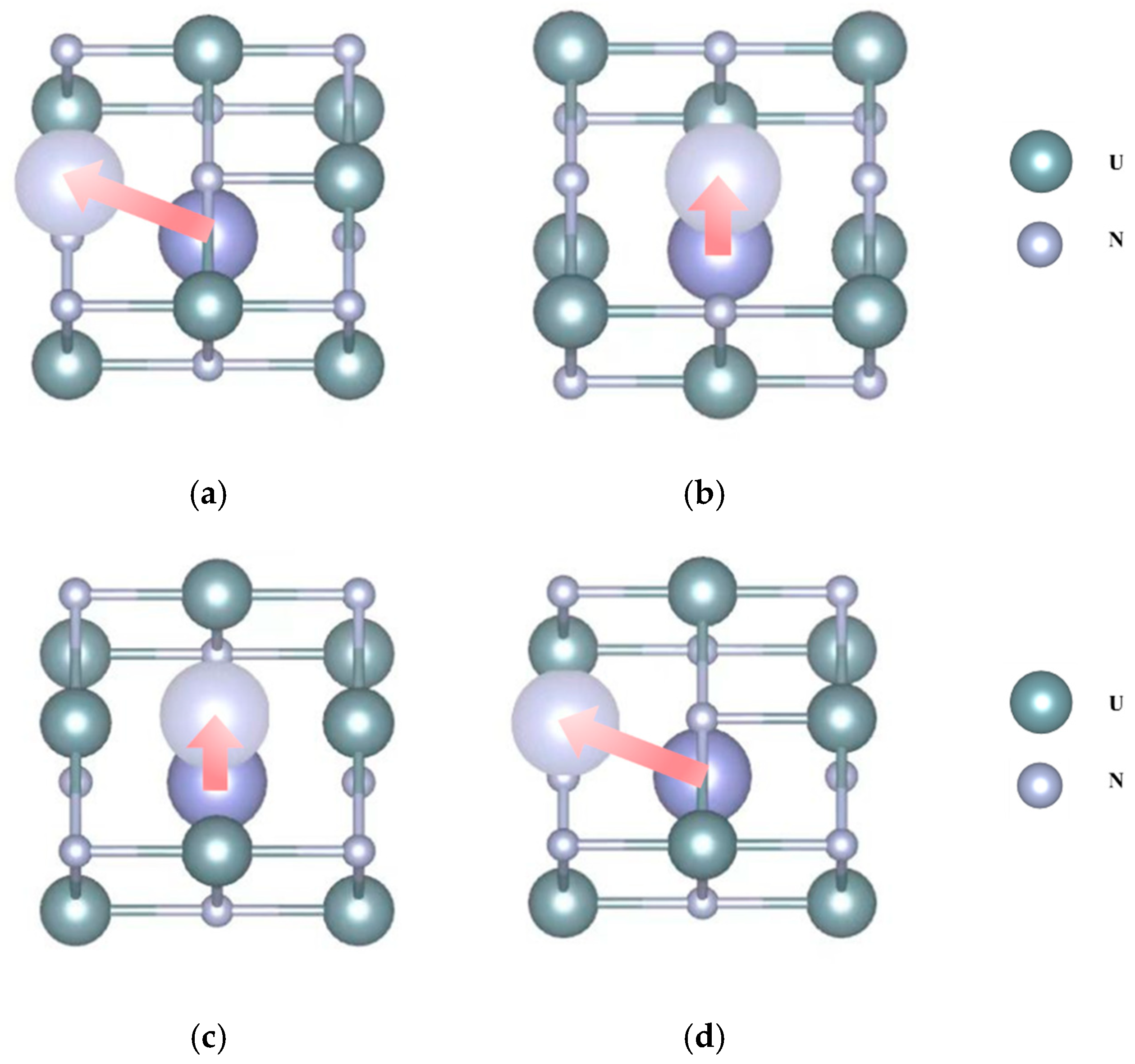
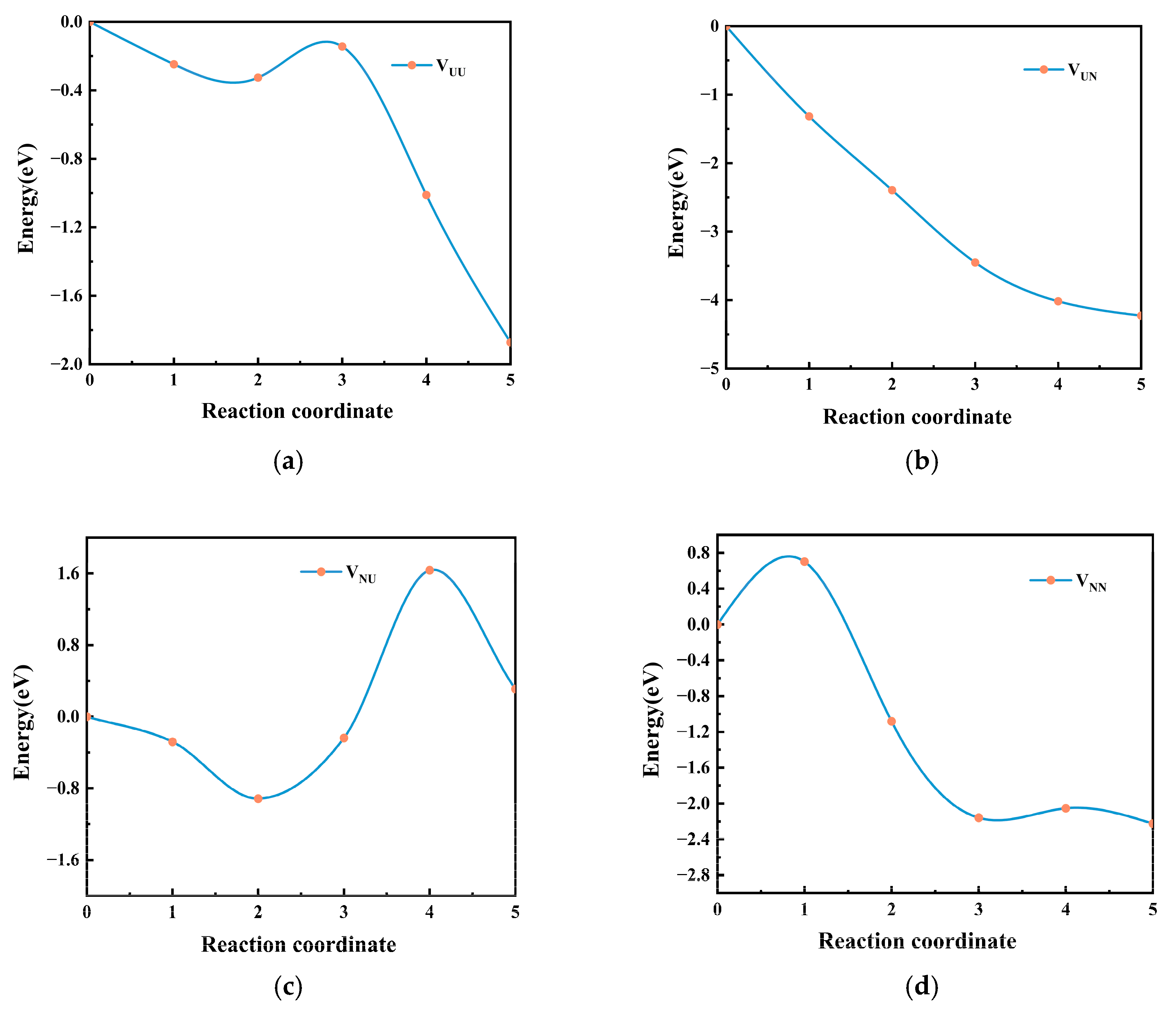
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watkins, J.K.; Gonzales, A.; Wagner, A.R.; Sooby, E.S.; Jaques, B.J. Challenges and opportunities to alloyed and composite fuel architectures to mitigate high uranium density fuel oxidation: Uranium mononitride. J. Nucl. Mater. 2021, 553, 153048. [Google Scholar] [CrossRef]
- Mishchenko, Y.; Johnson, K.D.; Jädernäs, D.; Wallenius, J.; Lopes, D.A. Uranium nitride advanced fuel: An evaluation of the oxidation resistance of coated and doped grains. J. Nucl. Mater. 2021, 556, 153249. [Google Scholar] [CrossRef]
- Gong, B.; Yao, T.; Lei, P.; Cai, L.; Metzger, K.E.; Lahoda, E.J.; Boylan, F.A.; Mohamad, A.; Harp, J.; Nelson, A.T.; et al. U3Si2 and UO2 composites densified by spark plasma sintering for accident-tolerant fuels. J. Nucl. Mater. 2020, 534, 152147. [Google Scholar] [CrossRef]
- Watkins, J.K.; Butt, D.P.; Jaques, B.J. Microstructural degradation of UN and UN-UO2 composites in hydrothermal oxidation conditions. J. Nucl. Mater. 2019, 518, 30–40. [Google Scholar] [CrossRef]
- Tonks, M.; Andersson, D.; Devanathan, R.; Dubourg, R.; El-Azab, A.; Freyss, M.; Iglesias, F.; Kulacsy, K.; Pastore, G.; Phillpot, S.R.; et al. Unit mechanisms of fission gas release: Current understanding and future needs. J. Nucl. Mater. 2018, 504, 300–317. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, W.; Prudil, A.; Chan, P.K. Multiphysics modeling of UO2-SiC composite fuel performance with enhanced thermal and mechanical properties. Appl. Therm. Eng. 2016, 107, 86–100. [Google Scholar] [CrossRef]
- Guo, J.; Lai, H.; Zhou, W.; Wei, J. Fission Gas Behaviors and Relevant Phenomena in Different Nuclear Fuels: A Review of Models and Experiments. Front. Energy Res. 2022, 10, 766865. [Google Scholar] [CrossRef]
- Amato, I.; Colombo, R.L.; Grappiolo, G.C. Grain boundary grooving in uranium dioxide. Solid State Commun. 1966, 4, 237–239. [Google Scholar] [CrossRef]
- Reynolds, G.L. The surface self-diffusion of uranium dioxide. J. Nucl. Mater. 1967, 24, 69–73. [Google Scholar] [CrossRef]
- Henney, J.; Jones, J.W.S. Surface-diffusion studies on UO2 and MgO. J. Mater. Sci. 1968, 3, 158–164. [Google Scholar] [CrossRef]
- Maiya, P.S. Surface diffusion, surface free energy, and grain-boundary free energy of uranium dioxide. J. Nucl. Mater. 1971, 40, 57–65. [Google Scholar] [CrossRef]
- Marlowe, M.O.; Kaznoff, A.I. Tracer study of the surface diffusivity of UO2. J. Nucl. Mater. 1968, 25, 328–333. [Google Scholar] [CrossRef]
- Zhou, S.Y.; Olander, D.R. Tracer surface diffusion on uranium dioxide. Surf. Sci. 1984, 136, 82–102. [Google Scholar] [CrossRef]
- Robertson, W.M. Surface diffusion of oxides (A review). J. Nucl. Mater. 1969, 30, 36–49. [Google Scholar] [CrossRef]
- Olander, D.R. Interpretation of tracer surface diffusion experiments on UO2—roles of gas and solid transport processes. J. Nucl. Mater. 1981, 96, 243–254. [Google Scholar] [CrossRef]
- Auskern, A.B.; Belle, J. Uranium ion self diffusion in UO2. J. Nucl. Mater. 1961, 3, 311–319. [Google Scholar] [CrossRef]
- Claisse, A.; Schuler, T.; Lopes, D.A.; Olsson, P. Transport properties in dilute UN(X) solid solutions (X = Xe, Kr). Phys. Rev. B 2016, 94, 174302. [Google Scholar] [CrossRef]
- Muntaha, A.; Chatterjee, S.; Blondel, S.; Aagesen, L.; Andersson, D.; Wirth, B.D.; Tonks, M.R. Impact of grain boundary and surface diffusion on predicted fission gas bubble behavior and release in UO2 fuel. J. Nucl. Mater. 2024, 594, 155032. [Google Scholar] [CrossRef]
- Feng, B.; Kazimi, M.S.; Forget, B. Feasibility of Breeding in Hard Spectrum Boiling Water Reactors with Oxide and Nitride Fuels. Ph.D. Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA, 2011. [Google Scholar]
- Deforest, D.L. Transient Fission Gas Behavior in Uranium Nitride Fuel Under Proposed Space Applications. Ph.D. Thesis, Texas A&M University, College Station, TX, USA, 1991. [Google Scholar]
- Klipfel, M.; Uffelen, P.V. Ab initio modelling of volatile fission products in uranium mononitride. J. Nucl. Mater. 2012, 422, 137–142. [Google Scholar] [CrossRef]
- Nie, J.L.; Ao, L.; Zu, X.T.; Huang, H.; Liu, K.Z. First-principles study of oxygen adsorption and diffusion on the UN(001) surface. Phys. Scr. 2015, 90, 125801. [Google Scholar] [CrossRef]
- Bocharov, D.; Gryaznov, D.; Zhukovskii, Y.; Kotomin, E. DFT calculations of point defects on UN(001) surface. Surf. Sci. 2011, 605, 396–400. [Google Scholar] [CrossRef]
- Andersson, D.A.; Uberuaga, B.P.; Nerikar, P.V.; Unal, C.; Stanek, C.R. U and Xe transport in UO2±x: Density functional theory calculations. Phys. Rev. B—Condens. Matter Mater. Phys. 2011, 84, 054105. [Google Scholar] [CrossRef]
- Ono, T.; Hirose, K. Real-space density-functional calculations for transport properties of nanostructures. J. Comput. Theor. Nanosci. 2007, 4, 840–859. [Google Scholar] [CrossRef]
- Ono, T.; Heide, M.; Atodiresei, N.; Baumeister, P.; Tsukamoto, S.; Blügel, S. Real-space electronic structure calculations with full-potential all-electron precision for transition metals. Phys. Rev. B 2010, 82, 205115. [Google Scholar] [CrossRef]
- Iwata, J.I. First-principles calculations for extremely large systems by parallel computations based on the order-N3 real-space density-functional theory. J. Comput. Theor. Nanosci. 2009, 6, 2514–2520. [Google Scholar] [CrossRef]
- Wang, Y.C.; Jiang, H. Local screened Coulomb correction approach to strongly correlated d-electron systems. J. Chem. Phys. 2019, 150, 154116. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B Condens. Matter 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, B.; Zhang, Q.; Liu, H.; Song, H. Theoretical insights into the hydroxyl-promoted H2 releasing reaction after H2O splitting on Pu-oxide surfaces. J. Nucl. Mater. 2023, 585, 154642. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, B.; Zhang, Q.; Liu, H.; Song, H. The reaction mechanism of CO2 on PuO2 and α-Pu2O3 surfaces. J. Nucl. Mater. 2025, 603, 155451. [Google Scholar] [CrossRef]
- South, C.J.; Roy, L.E. Insights into the thermal decomposition of plutonium(IV) oxalate—A DFT study of the intermediate structures. J. Nucl. Mater. 2021, 549, 152864. [Google Scholar] [CrossRef]
- Andersson, D.; Wang, G.; Yang, P.; Beeler, B. KCl-UCl3 molten salts investigated by Ab Initio Molecular Dynamics (AIMD) simulations: A comparative study with three dispersion models. J. Nucl. Mater. 2024, 599, 155226. [Google Scholar] [CrossRef]
- Duemmler, K.; Andersson, D.; Beeler, B. First-principles investigation of the thermophysical properties of NaCl, PuCl3, and NaCl-PuCl3 Molten salts. J. Nucl. Mater. 2024, 591, 154902. [Google Scholar] [CrossRef]
- Andersson, D.A.; Beeler, B.W. Ab initio molecular dynamics (AIMD) simulations of NaCl, UCl3 and NaCl-UCl3 molten salts. J. Nucl. Mater. 2022, 568, 153836. [Google Scholar] [CrossRef]
- Yang, L.; Kaltsoyannis, N. Incorporation of Kr and Xe in Uranium Mononitride: A Density Functional Theory Study. J. Phys. Chem. C 2021, 125, 26999–27008. [Google Scholar] [CrossRef]
- Liu, F.; Ding, X.; Sun, J. High local oxygen coverage causes initial oxidation of UN(001) surface, J. Nucl. Mater. 2023, 574, 154171. [Google Scholar] [CrossRef]
- Fathurrahman, F.; Kasai, H. Density functional study of hydrazine N-N bond cleaving on 3d metal surfaces. Surf. Sci. 2015, 641, 191–197. [Google Scholar] [CrossRef]
- Chen, C.; Niu, J.; Huang, H.; Zhu, D.; Nie, J.-F.; Yuan, G. Basal-plane stacking fault energies of biodegradable Zn-based alloys: A first-principles study of alloying effects. Mater. Lett. 2022, 309, 131413. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Liu, Y.; Wang, Z.; Wärnå, J.; Xu, Z.; Zhang, P. Investigating the solution and diffusion properties of hydrogen in α-Uranium by first-principles calculations. Prog. Nucl. Energy 2020, 122, 103268. [Google Scholar] [CrossRef]
- Yoon, K.S.; Hwang, C.O.; Won, T. Determination of the KMC parameters for indium diffusion in silicon substrates via an ab-initio calculation. J. Korean Phys. Soc. 2007, 50, 1651–1655. [Google Scholar] [CrossRef]
- Yoon, K.S.; Won, T. Ab-initio study with transition state theory (TST) for the calculation of the barrier height of migration energy of neutral indium in silicon. Solid State Phenom. 2007, 124-126, 1681–1684. [Google Scholar] [CrossRef]
- Mei, Z.G.; Liang, L.; Yacout, A.M. First-principles study of fission gas incorporation and migration in zirconium nitride. Comput. Mater. Sci. 2017, 133, 175–184. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functions | Lattice Constants (Å) | Magnetic Moment (μB) |
|---|---|---|
| PW91 | 4.87 | 1.17 |
| PBE | 4.868 | 1.25 |
| Model | This Work | Reference [23] |
|---|---|---|
| Surface U vacancy | 1.273 | 1.44 |
| Surface N vacancy | 3.753 | 3.70 |
| Subsurface U vacancy | 2.756 | 3.09 |
| Subsurface N vacancy | 4.413 | 4.33 |
| Models | Incorporation Energies (eV) |
|---|---|
| Xe occupies a surface U atom vacancy | 1.19 |
| Xe occupies a surface N atom vacancy | 4.10 |
| Xe occupies an interstitial site between the surface and subsurface layers | 8.63 |
| Xe occupies a subsurface U atom vacancy | 7.78 |
| Xe occupies a subsurface N atom vacancy | 12.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rui, T.; Lan, Y.; Ma, Z.; Lu, L.; Wang, Y.; Yu, Y.; Deng, M.; Lan, T.; Zhao, Z.; Wang, J.; et al. First-Principles Study on the Migration and Release Properties of Xe on the Surface of Uranium Mononitride. Crystals 2025, 15, 409. https://doi.org/10.3390/cryst15050409
Rui T, Lan Y, Ma Z, Lu L, Wang Y, Yu Y, Deng M, Lan T, Zhao Z, Wang J, et al. First-Principles Study on the Migration and Release Properties of Xe on the Surface of Uranium Mononitride. Crystals. 2025; 15(5):409. https://doi.org/10.3390/cryst15050409
Chicago/Turabian StyleRui, Tianhao, Yulin Lan, Zhuangzhuang Ma, Linyuan Lu, Yunhao Wang, Yang Yu, Mingxuan Deng, Tianxing Lan, Zhekang Zhao, Junjie Wang, and et al. 2025. "First-Principles Study on the Migration and Release Properties of Xe on the Surface of Uranium Mononitride" Crystals 15, no. 5: 409. https://doi.org/10.3390/cryst15050409
APA StyleRui, T., Lan, Y., Ma, Z., Lu, L., Wang, Y., Yu, Y., Deng, M., Lan, T., Zhao, Z., Wang, J., Li, C., & Zhang, H. (2025). First-Principles Study on the Migration and Release Properties of Xe on the Surface of Uranium Mononitride. Crystals, 15(5), 409. https://doi.org/10.3390/cryst15050409