1. Introduction
In the production of high-quality specialty chemicals, which are used in fine chemicals as well as in life science and pharmaceutical products, crystallization is a widely used separation, purification, and preparation method [
1]. Products from these industries often have higher molecular weights and higher melting and boiling points compared to low-molecular-weight basic chemicals, as well as an increased thermal sensitivity. These aspects must be considered in process development for achieving an economical and sustainable process performance [
2].
In fine and specialty chemistry, batch operation is dominant due to the wide product variety and often low product volumes [
3]. A significant disadvantage of batch processes is the potential for variations between individual runs, which can lead to a reduction in product yield and/or quality. Additionally, scaling-up of batch processes often requires adjustments that may compromise product quality or process efficiency. As a result, there is a noticeable trend in both research and industry towards transitioning from batch processes to continuous operations. Continuous processing allows for a high degree of automation and stable operation in steady-state, which helps minimizing quality fluctuations [
4]. Furthermore, continuous systems typically require smaller processing equipment compared to batch processes, thereby enhancing the overall process safety [
5,
6].
In fine chemistry, crystallization is often used as thermal separation process, where a substance is converted from an amorphous (disordered) state into a crystalline, ordered solid [
7]. The most commonly used method in industry is the crystallization from a liquid phase [
8], which is applied in industrial sectors such as fine chemicals, life science, and pharmaceuticals production [
9,
10]. Thus, in the following, crystallization from solutions will be considered. In recent years, the development and improvement of small-scale continuous crystallization processes has gained much relevance, which relates to various factors. One key factor is increasing product quality requirements, often a low amount of product impurities, which must be maintained and ensured for pharmaceutical products. To ensure product quality, variations in the process must be minimized. In addition to purity, other aspects such as crystal shape, size, and morphology play an increasingly important role. These have an influence, for example, on the subsequent further processing in the individual preparation steps of the crystals [
11,
12].
A gentle method for product recovery and purification is cooling crystallization from solutions [
13]. In the literature, many concepts were proposed and characterized ranging from oscillating baffled crystallizer [
14] to tank-in-series. To implement and study cooling crystallization in a continuous plug flow operation on the laboratory scale, coiled flow inverter (CFI) devices can be utilized [
13,
15]. Here, redirected flow counteracts the gravitation [
16] and leads to the continuous suspension of the particles and a narrow residence time distribution of both the continuous liquid and the disperse solid phase [
17]. The goal of cooling crystallization is often a spherical product with a narrow size distribution for optimal solid–liquid separation (purity) and solids handling [
18].
Amino acids are important ingredients in human dietary supplement and animal feeding stuff and are often produced by fermentation. The separation of amino acids from the fermentation broth is laborious and energy-intensive [
19]. Tryptophan, in particular
l-tryptophan (
l-Trp), is an essential amino acid which is industrially produced for use, for example, in food supplements, animal feed, and pharmaceuticals [
20]. It can be synthesized in various ways. One important method of production is fermentative production using microorganisms. However, the crude tryptophan obtained in this way, which contains impurities, must be purified before use in food supplements, animal feed, and pharmaceuticals. Known purification processes for crude tryptophan are based on the principle of pH change [
21,
22]. Like other amino acids,
l-Trp is a weak base and is largely protonated in the acidic pH range of the pH change process, as described in [
23]. The solubility of the resulting cations of
l-Trp in this aqueous solution is significantly increased. In the process of Toshio et al. [
21], solid crude tryptophan consisting of solid
l-Trp and solid secondary components/impurities, is first dissolved in the aqueous solution of a strong inorganic acid, for example, sulfuric acid, at a low pH of 1 to 4, in particular < 2, close to the saturation concentration. The pH value is then shifted to the neutral range of pH = 5–8 by adding strong inorganic bases, for example, sodium hydroxide solution. Thereby, the isoelectric point, i.e., the pH at which the net charge of the system is zero, is approached, and the solubility of the crystallizing compound is minimized. The isoelectric point of
l-Trp in aqueous solution is 5.89 [
24]. This supersaturates the solution and the
l-Trp crystallizes out as a solid. The solid can be separated from the solution, washed, and dried in further process steps. Although it is described that crystallization takes place in the presence of lower alcohols or ketones, neither a lower alcohol nor a ketone is used in the crystallization step in the examples. This well-known pH change process has numerous disadvantages due to the use of inorganic acids and bases. These have a corrosive effect and require systems and equipment made of high-quality materials. The acids and bases are consumed as auxiliary materials in the process and cannot be easily recovered. The resulting salts must first be removed from the product crystals with the mother liquor during the solid/liquid separation following crystallization, washed with suitable solvents and finally disposed. The accumulation of salts also makes it difficult or impossible to work up and return the mother liquor to the process, leading to a loss of
l-Trp. Furthermore, process analysis, process control and quality assurance are made more difficult by the presence of acids, bases and salts.
The cooling crystallization of
l-Trp from water in the presence of gelatin and in the absence of acid, bases, salts and alcohols is described in [
25], which enables the generation of so-called spherulitic crystals in a batch process. However, the process is complex, comprises harsh conditions and the gelatin has to be removed again in a further step. The process is based on extremely rapid cooling (quenching) of the solution from 90 °C to 20 °C within a single minute on a laboratory scale, which makes technical implementation and scale-up difficult. Such rapid cooling often prevents high purity in industrial processes.
The aim of this study is to develop a combined process involving continuous evaporation and subsequent cooling crystallization for the recovery of the high-value amino acid
l-Trp from raw tryptophan (raw-Trp), which is typically derived from a fermentation process [
26]. Various aqueous multi-component solvent systems, consisting of pure water, water and 1-propanol (1-PrOH) or water and iso-propanol (iPrOH), will be investigated for the cooling crystallization. Additionally, an assessment of the cooling crystallization will be conducted to evaluate its suitability and efficiency by comparing it with existing industrial batch process from industry, which often include a pH-shift crystallization with inorganic acids and basis. This comparison will also involve validation through both batch and continuous experiments on the laboratory scale. The results are expected to provide insights into optimizing the recovery process for
l-Trp while enhancing efficiency and product quality. The task of the present contribution was therefore to provide an improved process for the purification of
l-Trp, which requires fewer steps and/or fewer demands on the apparatus used for this purpose. In particular, a process was to be found which minimizes or completely avoids the use of acids and/or bases.
3. Results
In the previous section, the theoretical and experimental investigations and results are transferred to lab processes to demonstrate the feasibility of the approach.
3.1. Solubility
The measured
l-Trp solubility in pure water is compared with data from the literature and modeled with a simple exponential regression analysis, which yields quite good accuracy. The solubility of
l-Trp in water increases with increasing temperature. The data are given in
Table 2 and show good agreement with the data of Do et al. [
34], slightly higher solubility compared to Dalton and Schmidt [
35], but lower solubility compared to the work of Zhu et al. [
36]. The work of Dalton and Schmidt is from 1933. Since in the original paper, only few details on the experimental methods are provided, slight deviations in solubility might result, e.g., from too short an equilibration time. The higher deviations to the data provided by Zhu et al. are remarkable as they used the same 12 h equilibration time and similar gravimetric analytics as used in this work. These deviations are further discussed in the following based on the mixed-solvent results.
With adding an alcohol, the solubility increased to a certain concentration and decreases with higher alcohol content. The measurement was performed in alcohol-concentration steps for temperatures ranging from 10 to 60 °C. The experimental results are given in
Tables S1 and S2 in the Supporting Information and are graphically shown in
Figure 3.
As expected from the pure water results, the solubility in general increases with increasing temperature, both in the 1-PrOH/water- and in the iPrOH/water system. The solubility of
l-Trp in the 1-PrOH/water mixture is shown in
Figure 3 (lhs) and exhibits a maximum solubility at intermediate solvent composition at roughly 40 wt.-% 1-PrOH in the 1-PrOH/water mixture. This was previously found by Chen et al. [
33] and has qualitatively been reproduced in this work. However, solubilities measured by Chen et al. [
33] tend to be a little lower compared to the results measured in this work, especially for the lower temperatures range of 20–30 °C. In the higher temperature range, some data points of Chen et al. [
33] are in very good agreement with the data measured in this work. The slight deviations in solubility data can be explained by the relatively short equilibration time (20 min) used by Chen et al. [
33], compared to this work (48 h) and a more or less step-wise addition method of Chen et al. [
33] compared to sampling and analyzing the equilibrated solution in this work.
In the iPrOH/water mixture, an even more distinctive solubility behavior was found compared to the 1-PrOH/water system results (see
Figure 3, rhs). The solubility shows a maximum at roughly 40 wt.-% iPrOH in the iPrOH/water mixture and, in addition, a minimum at roughly 20 wt.-% iPrOH in the iPrOH/water mixture at temperatures of 10–20 °C. The absolute solubility of the iPrOH/water system at respective temperatures is slightly lower compared to the 1-PrOH/water system. Do et al. [
34] found comparable minimum/maximum behavior in their measurements at 25 °C. They explained the improved solubility in a certain concentration range with the influence of the indole group within the chemical structure of the amino acid, which improves solute-solvent interactions. The work of Batov et al. [
37,
38] shows negative enthalpies of solution of
l-Trp in water/glycerol mixtures above concentrations of 8 mol-% glycerol in water, indicating a better solubility with increasing alcohol content. Unfortunately, they measured the enthalpies only up to 10 mol-% glycerol in water.
The corresponding data of Zhu et al. [
36] for the iPrOH/water system show a completely different trend, in comparison to a typical solvent/antisolvent behavior with water and iPrOH being the solvent and the antisolvent, respectively. A similarly disparate trend has already been discussed by Zhu et al. [
36], comparing their solubility data of
l-Trp the methanol/water solvent system to the data of Chen et al. [
33]. As the data of Chen et al. [
33] and Do et al. [
34] agree qualitatively very well with this work, showing the same trends in the 1-PrOH/water solvent systems, but the data of Zhu et al. [
36] disagree both with Chen et al. [
33] (methanol/water solvent system), and with this work (iPrOH/water solvent system), which might be explained by a systematical error in the work of Zhu et al. [
36], such as having unknown impurities in their experiments. Hence, their results are excluded in the following investigations.
Samples equilibrated at temperatures >50 °C showed significant yellow discoloration after the equilibration period of 48 h, indicating a chemical change in
l-Trp (e.g., decomposition or oxidation) due to thermal stress. To be sure to have the correct crystal morphology, X-Ray powder diffraction (XRPD) measurements were performed at the Laboratory of Thermodynamics. Crystals from various equilibrated turbid suspensions were analyzed. The results are shown in
Figure 4.
All diffraction patterns of the investigated samples correspond very well with the theoretical pattern of the thermodynamically stable
α-polymorph [
39], proving that mainly typical hexagonal
l-Trp crystals were present in all samples. In the recent literature, two further metastable polymorphs
β [
40] and
α’ [
41] were isolated and characterized, which seem to have no practical relevance for the systems and process conditions of the present study.
Unfortunately, the regression analysis for the influence of iPrOH and 1-PrOH on the solubility of
l-Trp was not successful, since no polynomial, logarithmic, or exponential approach gave sufficient results. Moreover, the complex solubility behavior with maximum (1-PrOH/water systems) or both maximum and minimum (iPrOH/water system) could not be modeled satisfactorily by parameter regression of a standard NRTL model which works well for smaller amino acids in aqueous/alcohol mixed solvent systems [
42], showing a classic solvent/antisolvent behavior.
3.2. Batch Crystallization via Crash Cooling
The crash cooling experiments represent a simple and fast experimental method, leading to a first orientation of the kinetical crystallization behavior of unknown systems. However, the crystallization parameters (cooling rate, mixing) are less controlled.
For samples with alcohol in the solvent mixture, an induction time of approx. 10–15 min was measured. In contrast, it takes approx. 30–45 min for crystals to form in samples with only water as a solvent. This behavior can be seen as an initial indication of the different widths of the metastable zone (MSZW), which appears to be narrower for the solvent mixtures than for pure water as the solvent. The MSZW will be described in more detail in the next section.
The desired crystal habit is in general close to a spherical shape due to their good filterability [
43]. The temperature influence on the crystal shape was investigated in the range from 30 to 50 °C, but no major influence could be found (see
Figure S1 in the Supporting Information). The influence of the alcohol content in the solvent mixture on the crystal shape was already described in the literature, as noted in the introduction. The influence of the two different alcohols on the crystal shape of
l-Trp is shown in
Figure 5 for pure water and three different alcohol concentrations. Plate-like single crystals were obtained in pure water, while increasing alcohol content in the solvent mixture led to progressively stacked agglomeration of the single crystals. No compact three-dimensional or needle crystal habitus were observed in pure water.
The more alcohol is present in the solvent mixture, the more agglomerates are formed, where several platelets form together or overlap. For 1-PrOH, these agglomerates take on a roundish shape (see
Figure 5, lhs b-d). There can be two reasons for the increasing agglomerate formation with increasing 1-PrOH content: on the one hand, the increasing proportion of 1-PrOH decreases the surface tension and coalescence of the particles occurs. On the other hand, the higher solubility
l-Trp in the saturated solvent mixture leads to higher supersaturation during crash cooling, resulting in the formation of more crystals and thus a higher probability of aggregation and subsequent agglomeration.
Similar results as for 1-PrOH were found for the crystallization and agglomeration in iPrOH/water solvent mixtures (shown in
Figure 5, rhs). With 20 wt.-% of iPrOH, only few agglomerates were found. With higher iPrOH content, the agglomerates became more compact. A major difference between the two alcohol/water systems could not be detected, although the maximum solubility of
l-Trp in a 40% iPrOH/water mixture at 50 °C is only 2/3 compared to the solubility in a 1-PrOH/water mixture with the same conditions. Hence, agglomerate formation seems to be similar in both alcohol/water systems.
These differences in the crystal shapes are relevant for the further downstream processing, i.e., solid–liquid separation, crystal washing, and drying. The coarse particles from the mixed solvents might be easier to separate compared to the small and thin platelets from the pure water system, but agglomerates are usually prone to mother liquor inclusions which might result in a lower purity. However, the detailed experimental investigation and optimization of the further downstream process steps is not in the focus of this work.
3.3. Batch Crystallization via Controlled Cooling
In the experiments involving controlled cooling within a stirred double-jacket glass vessel, we measured the temperature at which the first visible crystals appeared following spontaneous nucleation (the point at which they could be seen with the naked eye). The results are presented in
Figure 6 and
Figure 7. In
Table S3 in the Supporting Information, the data are given from the experiments.
The MSZW was found be depending on the cooling rates with faster cooling leading to wider MSZW or lower nucleation temperatures, respectively. The iPrOH/water system shows the narrowest MSZW, pure water as a solvent provides the widest MSZW in this comparison.
The optical observation of the first visible crystals after nucleation and the crystallization process revealed an unusual behavior of
l-Trp in pure water in perfect accordance with the findings of Kadam et al. [
44]. In pure water, individual relatively large primary crystals, so called ‘parent crystals’, can be observed (see
Figure 8, lhs, indicated with red circles). These few crystals exist and grow for a significant period before a secondary nucleation shower can be observed, probably being generated by the attrition of the parent crystals. In the mixed iPrOH/water solvent system, a more classic nucleation behavior was found with numerous primary crystals forming simultaneously. In the following secondary nucleation, attrition, and growth cannot be further differentiated.
The relative yield is shown in
Table 3. It was found that particularly in the PrOH/water system, a yield close to the theoretical yield was achieved, even though the theoretical yield for this system is higher than that in water or in the 1-PrOH/water system. This suggests an unexpectedly high crystal growth rate in this system.
Similarly to the findings in
Section 3.2, the stacked agglomeration of single crystals was also observed during crystallization from the alcohol/water mixtures, while non-agglomerated single platelet crystals were observed during crystallization from water, as shown in
Figure S2 in the Supporting Information. The cooling rate was found not to be very sensitive to the crystal habitus in the investigated range.
3.4. Continuous Crystallization
To exploit the different solubilities of
l-Trp in the different alcohol/water systems, continuous processing is beneficial for high yields due to the combination of crystallization and solvent evaporation in successive steps. Hence, continuous crystallization is a major process steps, which will be presented in the following based on the experimental setup described in
Section 2.5 with
Figure 2. The 20.0 wt.-% iPrOH/water system was chosen, based on the previous results. In addition to the highest possible yield of approx. 59%, which was confirmed experimentally, the solvent system has the narrowest metastable range of Δ
T = 12.2 K at a cooling rate of 1.0 K min
−1 of the three solvent systems investigated in the batch experiments.
Figure 9 (lhs) shows exemplary temperature profiles of the continuous cooling crystallization runs 1 and 3 without seeding. It is noticeable that relatively large temperature differences appear at feed inlet (0 m) and the product outlet (6.54 m) of the crystallizer. In contrast, there is a smaller temperature difference in the middle of the crystallizer. Thus, the axial temperature profile was not perfectly linear, but slightly cubic parabola or s-shaped. A comparable profile was found in run 2. As an overall result from the three experiments without seeding, it was found that the
l-Trp in the water/iPrOH solution cannot be continuously operated in the CFI.
At the low throughput (experimental run 1), primary nucleation occurred on the inner tube surface in the later coil segments 8 and 9 of the CFI crystallizer. The nuclei were stagnant on the wall and grew, resulting in crystallization fouling and complete blockage 20 min after start-up of the run. This experiment was reproduced twice, and the same result was found within 15–20 min after start-up.
At the higher throughput (shorter mean residence time) in experiment 2, no crystals were observable within 60 min inside the CFI crystallizer. Visible crystals appeared in the receiving flask behind at the outlet, even though the solution was widely subcooled with respect to the MSZW found in the batch experiments (see
Figure 6). This result agrees well with previous findings of our group with the
l-alanine/water system in the same CFI crystallizer setup [
13]. The significantly increased cooling rate (r ≈ 3 K/min) compared to the batch processes, combined with the extremely smooth surface of the fluorinated ethylene propylene (FEP) tubing of the crystallizer, the absence of a gas–liquid interface and the well-defined laminar flow regime result in enlarged MSZW, allowing for significantly higher supersaturations compared to batch. In experiment 3, the same behavior as that in experiment 1 was found, but the nucleation and growth of crystals on the tube wall was shifted to segments 3–4. Again, the blockage of the tube occurred after 20 min.
The decision to start with a saturated, seeded solution was clear after the experience with the unstable operation with primary nucleation. In experiment 4, an inlet temperature of 50 °C was applied. The growth of seed crystals was observed during continuous crystallization optically by the increasing turbidity along the crystallizer length. The suspension flow was homogenous and without crystal settling in the entire crystallizer (see
Figure 10).
After an operation time of approx. 10 min, crystal adhesion occurred also in segments 8 and 9, as well as in the outlet drain, which caused blocking of the crystallizer within a further 10 min. To counteract this, the cryostat for the cooling air was set to a temperature of 25 °C in experiment 5 resulting in a gas inlet temperature of approx. 25.8 °C, resulting in a temperature difference of >10 K between crystal suspension and cooling agent at the outlet of the tube crystallizer, which again led to crystal adhesions and fouling. After approximately 5 min after the first adhesions formed in the outlet, the adhesions also spread to segments 8 and 9 until blockages occurred after a total of 30 min and the experiment had to be stopped. This behavior was reproduced three times in an identical manner.
As a result of the preliminary continuous experiments, the entire temperature profile in experiment 6 was shifted to lower temperatures, including the temperature in the feed vessel. The temperature difference in the outlet between the solution and the cooling air was adjusted to a range of approx. 5–6 K to prevent adhesions.
With these optimized settings an average cooling rate of 1.13 K min−1 was obtained. It was possible to operate the CFI crystallizer stably, consuming the entire feed vessel content with a total mass of approx. 3 kg through the tubular crystallizer over a period of approx. 2.5 h without facing critical adhesion leading to blockages. Only small deposits occurred in segments 8 and 9 in the lower area of the horizontal tube coils after approx. 1.5 h, but these did not affect the robust operation of the crystallizer. As these sedimentations were only observed in the gravity-facing area of the tube, they can be explained by settling of large product crystals.
The installed operation conditions for the experiment 6 with seed suspension as a feed material. Through a thermogravimetric analysis of both the seed crystal solution and product solution (mother liquor), the relative yield of the continuous crystallization process in experiment 6 based on the solubility data at the exit temperature of 29.1 °C was determined to be
ΦTrp = 36.3%. This relatively low yield compared to the batch results, see
Table 3 can be explained by the relatively low mean residence time of
tL= 4.4 min in this lab-scale CFI crystallizer at the given flow rate, see
Table 1 and the significantly smaller Δ
Tin,out = 4.9 K compared to 30 K in the corresponding batch experiments (45 min annealing time; see
Section 2.5). From the quasi-linear temperature profile (see
Figure 10) and the mean residence time, a cooling rate of
rCFI = 1.1 K min
−1 can be calculated, which also exceeds the investigated cooling rates in the batch mode. In a technical continuous process crystallization process of
l-Trp, a crystallizer with a higher mean residence time at a given throughput and Δ
T would be required.
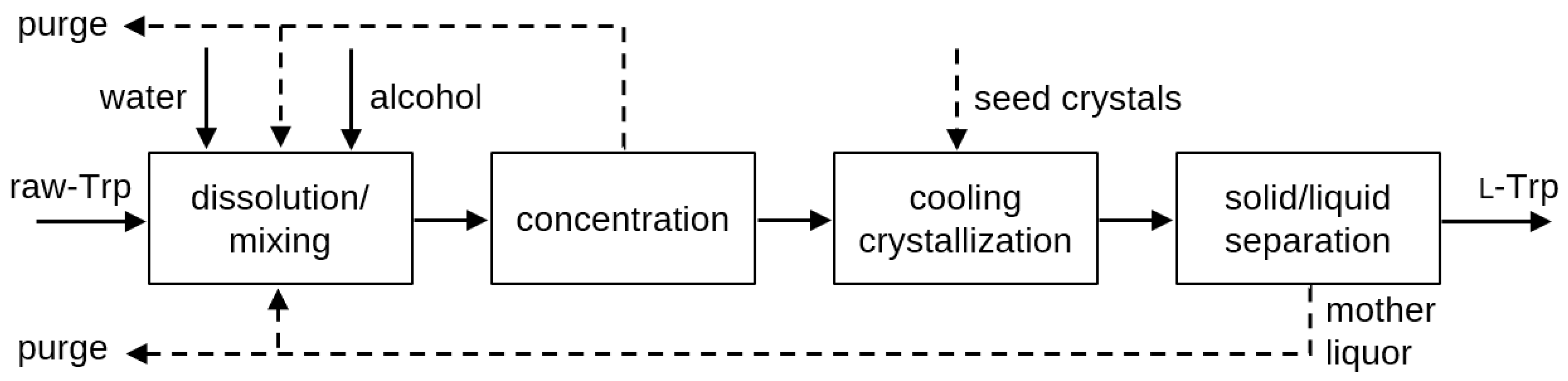
3.5. Design of Continuous Downstream Process
The presented findings on solubility and crystallization characteristics of
l-Trp were utilized to propose a process for continuous downstream processing and purification of
l-Trp from raw tryptophan (raw-Trp), in particular by raw-Trp obtained by fermentation and the concentration of the fermentation broth [
45]. The suggested process is depicted in
Figure 11 and comprises the following steps:
- (1)
Dissolving raw-Trp in a solvent system that includes water and alcohols at a temperature of up to 50 °C to avoid thermal decomposition of l-Trp: A saturated solution should be obtained, e.g., by a CSTR-type setup with a clear solution withdrawal.
- (2)
Optionally concentrating the solution obtained in step (1) at a temperature of up to 50 °C: This concentration step could be either realized by evaporation/distillation under reduced pressure or the use of suitable membranes.
- (3)
Performing cooling crystallization of the l-Trp from the solution to a temperature below 20 °C: The optional dosing of seed crystals (either dry or dispersed in liquid) depends on the actual crystallizer setup, but is recommended based on the presented experimental results to achieve a robust and reproduceable process; and
- (4)
Separating the solid l-Trp in crystalline form from the liquid phase, e.g., by filtration, sedimentation or centrifugation: After mechanical separation of the crystals, further purification, e.g., by crystal washing or drying can be conducted.
In principle, such a purification process can be conducted either continuously or as a batch process. However, in a continuous process, the mother liquor of the mechanical separation step (4) can be recycled directly to maximize the overall yield of the process. The recycling of mother liquor could be limited by an accumulation of impurities from the fermentation, which must be determined for the individual sources of the raw-Trp. The detection of the impurities depends on the upstream process and cannot be discussed here in detail. Thus, a purge of mother liquor might be necessary, depending on the concentration of individual impurities in the raw-Trp and their impact, e.g., on the crystallization process and purity of the crystals. However, the separated stream in the concentration step (2) and withdrawal of mother liquor in the residual moisture of the mechanically separated l-Trp product crystals in step (4) represent a potential outlet for impurities.
The recyclability of the mother liquor in such a continuous cooling crystallization-based purification process of l-Trp is a major measure to overcomes the typical disadvantages of classic batch pH-shift crystallization-based processes as described in the introduction.
Regarding the solvent selection, the iPrOH/water system appears to be the most advantageous. Additionally to the highest possible yield of approx. 59%, the solvent system has the narrowest metastable range of ΔT = 12.2 K at a cooling rate of 1.0 K min−1 of the three solvent systems investigated in the batch experiments. The unusual solubility behavior of the iPrOH/water system could be utilized to maximize the yield even more and minimize the solvent consumption. To do so, the dissolution step (1) of raw-Trp could be performed at wiPrOH = 40% (maximum solubility), whereas in the concentration step, the solvent composition could be adjusted to wiPrOH ≈ 20% (minimum solubility at lower temperatures) to maximize the yield in the crystallization. Alternatively, the iPrOH/water system could be operated without the concentration and solvent composition shifting step (2), resulting in a lean production process. To minimize the solvent consumption, a composition of w1-PrOH = 40% (maximum solubility) would be beneficial as well.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
