3.1. EDTA Treatment Results
An aim of this work was to confirm the relationship between phosphate dissolution in oxalic acid and the extent of REE dissolution in EDTA. The duration of contacting the solids with EDTA was also examined, and further testing was conducted over periods varying from 15 to 30 min. Another aim was to develop a greater appreciation of the reactions occurring and reagent usage, again as required for flowsheet definition.
In the first instance, a one-for-one molar requirement for EDTA complexes and chelate formation with the precipitated REE was assumed [
13]. The feed phosphorus content based on XRF analysis results obtained in previous endeavours was also employed (recognised in subsequent solids analyses to reflect low neodymium content). An expected extent of release from phosphate mineralisation as indicated by phosphorus dissolution in oxalic acid treatment at 50 percent was also applied.
On this basis and using Reaction (1), it was predicted that 0.1 M EDTA would provide an equimolar solution for solubilisation of REE content. This also assumed a one-for-one molar requirement for EDTA complex formation for cerium ion, which could be present in a tri- or tetravalent form, given that cerianite mineral, CeO
2, has elsewhere been identified in the ore [
17]. Hence, the testing was working with a deficit of EDTA.
The 0.1 M EDTA contacting was initially allowed to proceed for approximately 16 min, during which time slurry pH and Eh measurements were taken.
Figure 1 summarises selected pH measurements from several tests. The pH measurements were taken within four 4 min periods. Time 0 was prior to the addition of the oxalic acid leach residue. The values shown at 4 min were taken during the period from 0 to 4 min, contributing to the variability in response observed. This measurement pattern was replicated for 4 to 8 min, 8 to 12 min, and 12 to 16 min. After 16 min, the pH had dropped to approximately 5.5 or 5.6 when treating these oxalic acid leach residues. The data also identifies the lack of response for concentrate samples not subjected to oxalic acid treatment.
Contacting concentrate with EDTA solution where this had not received prior oxalic acid treatment resulted in slurry pH exhibiting negligible change. Analytical results also indicated little REE solubilisation, with only minor amounts, at levels of a few ppm (less than 5), detected in solution, this implying the REE minerals remain unaffected by EDTA contacting without prior oxalic acid treatment.
Conversely, a significant slurry pH change was evident for the oxalic acid leach residues. Results stabilised with pH in the order of approximately 5.5 regardless of the extent of prior oxalic acid leaching. Inadequate washing of acidic components from the oxalic acid leach residue was initially suspected as a potential interference and a contributor to the observed pH decreases. Hence, rewashing of residue prior to EDTA treatment was carried out. This rewashing did not make an impact on this acidic final pH position in the EDTA. The pH change was normal behaviour conforming to expectations in terms of Reaction (2).
Oxidation potentials of the initial solutions and the various EDTA slurries were also measured, with these falling in a range from 220 to 450 mV (Ag/AgCl). Different Eh starting points were observed for each test. There was a trend of increasing Eh with test progression, but repeatable measurements were not achieved.
Of concern in these preliminary tests was the occurrence of precipitation of a white substance in filtrates of EDTA-contacted residues from the oxalic acid leaching tests. Initial filtrates free of mineral solids were typically a pale blue-grey but clouded on standing for several hours. Extensive precipitation occurred after several days. Precipitation was slow, with occurrence worsening as standing time increased.
This effect can be seen in the images presented in
Figure 2, where the two images were taken after EDTA treatments yielding solutions C2E1A and C4E1A. The image on the left was taken on completion of Test C4E1, and solution C2E1A had been left standing for approximately one hour. The C2E1A solution was initially clear, the same as C4E1A, but subsequently became decidedly clouded. The image on the right was taken approximately three hours later, and both solutions were clouded.
Precipitation in primary filtrate had an adverse effect on REE concentrations and prevented assessment of the degree of release of REE in comparison with the extent of phosphorus dissolution in the oxalic acid leach step. In the first instance, and using analytical results with the highest recorded levels for a single-stage oxalic acid leach residue, concentrations of EDTA-soluble REE were roughly half of expected. The final pH values of the EDTA solutions were around 5.5, acidic, and at levels that will result in insoluble REE oxalates [
2]. Precipitation worsened on further standing, so the reaction is slow.
Acidic solution conditions are the primary cause for the poor recovery of REE to solution, but it was also suspected that an inadequate availability of EDTA could have contributed to the poor response, in light of the low neodymium content analysis that was used as a basis for calculation and the potential presence of tetravalent cerium and the assumption of a one-to-one EDTA requirement that may have been inadequate.
Subsequently, 0.2 M EDTA concentrations were used, and these higher concentration solutions yielded greater REE solubilisation, less decrease in slurry pH, and could be used to achieve solubilisation without incurring significant precipitation issues. The first test utilising 0.2 M EDTA witnessed only a small pH decrease from 10.0 to 9.0 over the entire 30 min period while the formation of white precipitate did not occur. There was still no white precipitate in the primary filtrate two weeks later. An alkaline dilution was carried out with 0.5 M sodium hydroxide solution, thus achieving a clear diluted sample that was sent for analysis. This result supported the use of either higher initial EDTA concentrations, better control of solution pH levels, or utilisation of both these conditions.
Testing did not include an investigation of whether pH manipulation of the lower strength 0.1 M EDTA solution could achieve the required REE solubilisation, so reagent requirements for this oxalic acid-leached ore material remain to be optimised but fall in the range from 0.1 to 0.2 M for treating material at 100 g/L solids loading.
Care needs to be taken to maintain alkaline conditions. Any form of change to acidic conditions resulted in the formation of precipitates. Even dilution with deionised water proved inappropriate. Precipitates when formed were difficult to redissolve. A small amount of white precipitate collected by filtration of a diluted sample that was acidic was subjected to XRD analysis.
Results in
Figure 3 depict diffraction patterns for a sample from the current work and a sample from a previous investigation where synthetic REE oxalate crystals were formed and analysed [
2]. Similar diffraction patterns were realised, confirming REE oxalates reprecipitated where pH decreased to acidic conditions. Slight differences in the patterns are evident, and efforts to identify reasons that probably relate to differences in components present remain to be explored.
The threshold pH level for precipitation was not clearly defined. However, the main focus was determining the interrelationship between phosphorus dissolution in oxalic acid leaching and EDTA recovery of REE, and hence testing examined response where pH was adjusted to between 9 and 10, while alkaline dilutions were used in subsequent endeavours.
For the next set of experiments, 50 mL of 0.2 M EDTA was used to treat approximately 5.0 g of oxalic acid leach residues, with pH adjusted to 9.0 or above using 1 M sodium hydroxide. In these and the earlier tests, pH was seen to decrease, and the solubilisation reaction was not necessarily instantaneous. Sodium hydroxide additions were loosely consistent with different extents of phosphorus dissolution in the oxalic acid leach step, with the amounts added increasing as the extent of prior dissolution increased. The final pH in these tests was stable after 30 min, implying reactions were complete. Precipitates did not form during or after these tests where pH remained above 9.
Various flocculant trials pertaining to solids recovery were also completed after the contacting had finished. This was where the approximately 100 g/t dosing was indicated as adequate for minimising losses to filtrates and was adopted as the norm for further testing.
Aliquots of primary filtrates were taken for dilution, typically with sodium hydroxide solution made up to pH 10. In one instance, 0.5 M sodium hydroxide was used. Both options maintained solubility of the contained REE and avoided precipitate formation. The diluted solutions also remained stable. Dilution with pH 10 sodium hydroxide solution was used in subsequent testing as the norm.
Table 2 summarises select chemical analysis results embracing a range of elements. Significant calcium solubilisation occurred, along with extensive REE dissolution. It could be expected that oxalate also dissolved and entered solution, although this was not specifically tested or demonstrated. Iron and phosphorus were not solubilised to any significant extent, remaining at concentrations below 100 mg/L.
There was broad alignment across the results for specific oxalic acid leach residues. There were slight differences, with these extending to nearly 10 percent for one component, notably C9E3 and C9E6 for lanthanum. Typically, however, differences for the REE were less than 10 percent. These were raw data results with no correction for the small alkali and flocculant additions, which, once applied, should draw results into alignment.
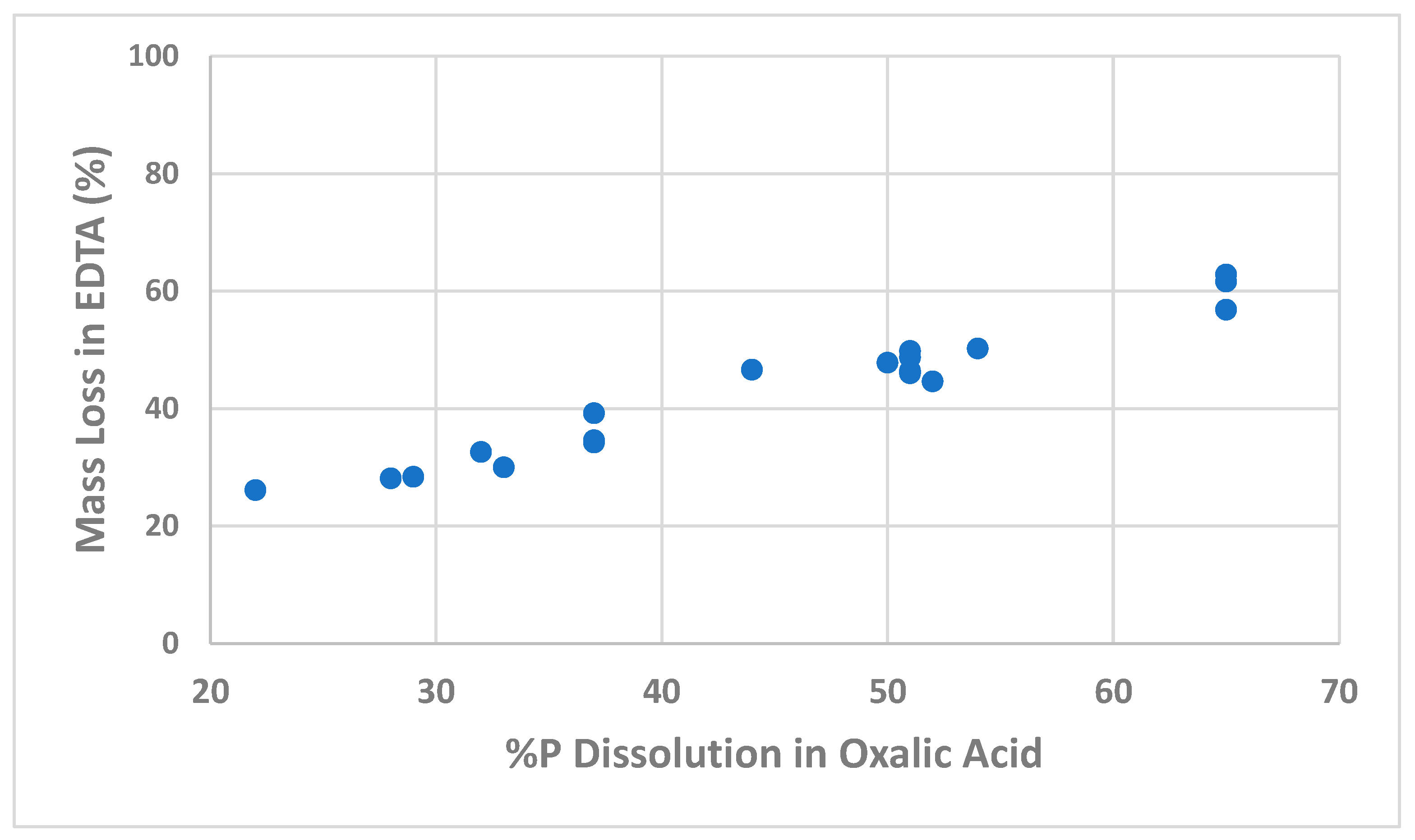
A trend of increasing REE solubilisation with increased phosphorus dissolution in the preceding oxalic acid leach step was evident. Another important factor was highlighted in one test where sodium hydroxide solution was alkaline to pH 10 but without EDTA present. The lack of REE dissolution demonstrated that sodium hydroxide alone is not capable of effecting REE re-dissolution.
Aluminium and magnesium were both solubilised by the EDTA treatment. These were only present in minor amounts in the concentrate and oxalic acid leach residues. Solubilisation in the EDTA solution was extensive.
Another factor emerging from this EDTA experimentation was that allowing the sample to stand overnight when pH was over 9 did not suffer precipitation, and the solution remained stable. This was unlike the situation where pH had dropped to acidic conditions in the first series of EDTA tests and the solution was left to stand with extensive precipitate formation over a period of hours.
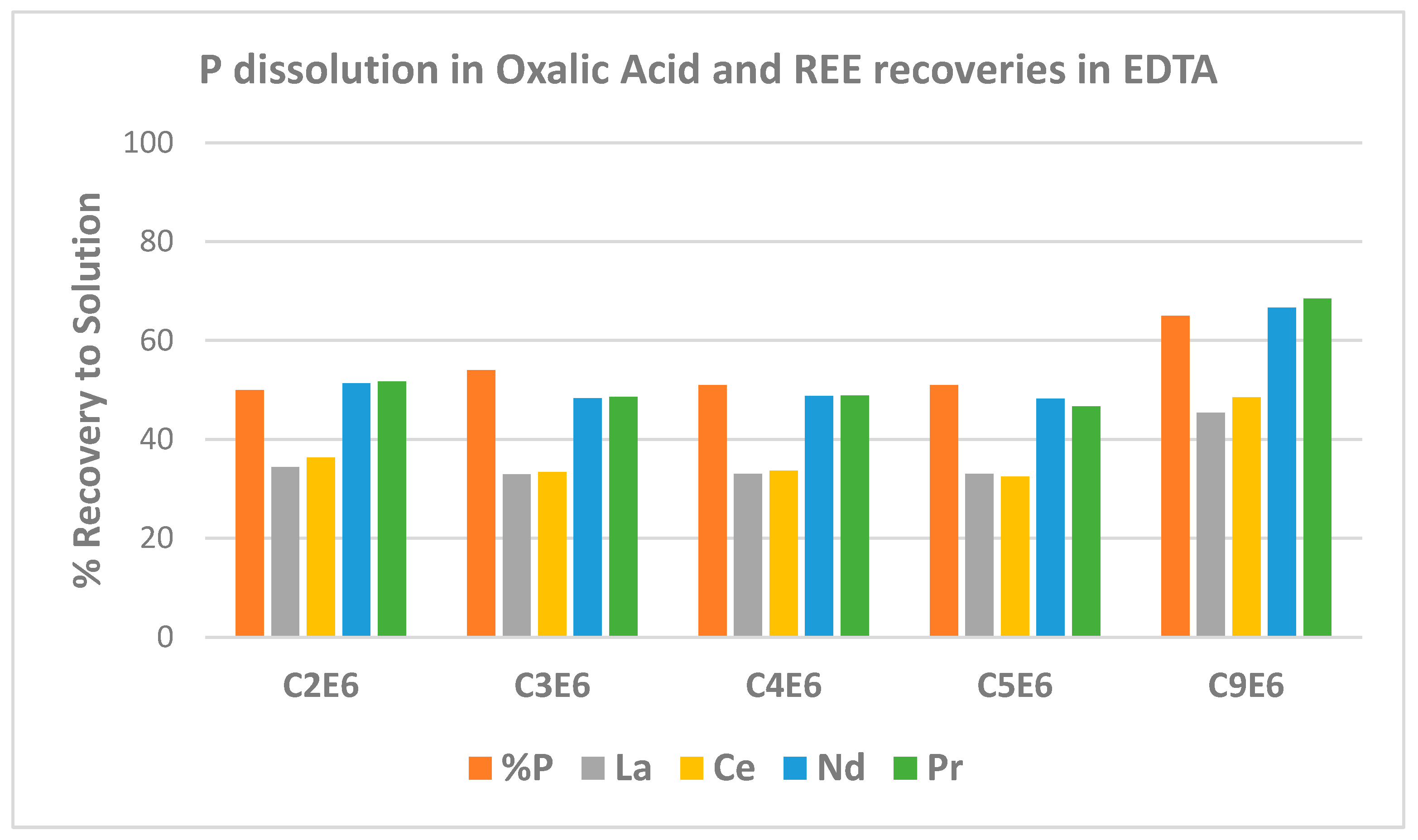
The percentage dissolution of REE and phosphorus achieved across the oxalic acid leach and EDTA treatment steps was calculated, with phosphorus dissolution occurring in oxalic acid and REE dissolving in EDTA. The values determined were based on the solids analyses shown above, where it must be acknowledged that considerable variability in determinations was experienced. However, noting these reservations,
Figure 4 summarises some of the percentage dissolution results. The responses shown were achieved for oxalic acid leach residues where solids loadings of 100 g/L concentrate in oxalic acid were used.
Test C2 was carried out at 65 °C while the others were kept at 45 °C. Tests C3 and C4 were carried out over extended periods of 96 and 72 h, respectively, while Test C5 treated residue contacted with recycled oxalic acid, which was made up with oxalic acid reagent but where no attempt was made to remove phosphorus or iron from solution before it was reused. Test C9 results were after three stages of leaching with fresh oxalic acid used for each stage.
There were only minor differences in results across temperatures and residence times in the oxalic acid leaching in the temperature range from 45 °C to 65 °C, and a minimum of 24 h residence time was utilised. The only significant improvement was seen when a multi-stage treatment approach was utilised where fresh oxalic acid was introduced for each stage, as was the approach for Test C9.
On a molar basis, the amount of phosphorus dissolved by the oxalic acid leach step was typically calculated at levels more than the molar dissolution of the four REE in the EDTA contacting. The pH of the EDTA slurries was not returned to the original levels before introducing the oxalic acid leach residues, only being increased to above 9 and not brought back to 10. The amounts of alkali used were considerably less than the molar amounts of total REE dissolved in these tests. Alkali addition thus only provided no more than an indicator of performance. Hence, the extent of acid generation predicted by reaction 2 remains to be quantified.
Solids analyses were erratic on occasion, and repeatable results proved challenging. However, it is evident that REE solubilisation follows phosphorus dissolution relatively closely. The values determined do not entirely correspond, however, with less REE being solubilised. Leaching of other REE components such as samarium, europium, gadolinium, yttrium, and dysprosium remains to be investigated and will contribute to some extent. Yet another explanation is the presence of a phosphorus mineral low in REE content that also leaches in the oxalic acid. Apatite, or alumino-phosphate of low REE content, for instance, could fit this observation.
Neodymium and praseodymium recoveries were greater than the extent of phosphorus dissolution in oxalic acid, with the trade-off appearing to be less dissolution of lanthanum and cerium. This is likely beneficial given the higher values of the neodymium and praseodymium components. Again, caution is expressed in terms of using definitive statements regarding the extent of leaching given solids analysis challenges. However, results indicate selectivity for neodymium and praseodymium components could be engendered through appropriate selection of operating conditions in the oxalic acid leach step.
While not shown, thorium followed REE. However, actinide analysis is subject to further refinement in terms of repeatable and accurate analytical determinations for solids. Both thorium and uranium were present only at ppm levels and were thus subject to difficult analytical determinations. Analysis using ICP-MS techniques, which are reportedly sensitive to ppb levels, is currently under investigation.
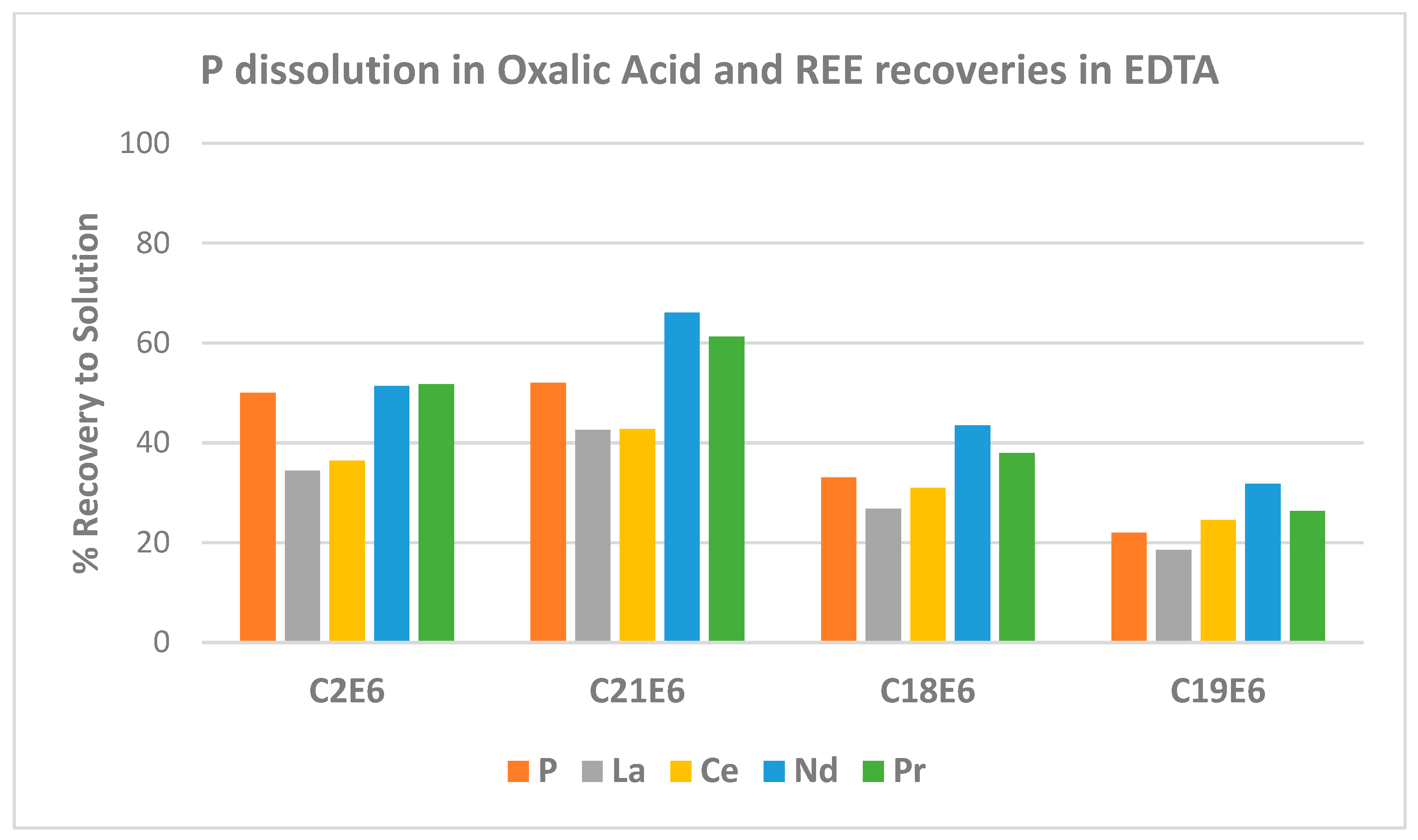
Solids loadings in the oxalic acid leach step had a noticeable effect on the recovery of REE in the EDTA step.
Figure 5 summarises several results obtained, these at an oxalic acid leaching temperature of 65 °C. Both C2 and C21 were oxalic acid leaches carried out with solids loadings of 100 g/L. Test C2 was carried out over 24 h while the other tests were conducted over 48 h. The solids loading was increased to 200 and 300 g/L, respectively, for tests C18 and C19. The same trends were also evident in the curves generated for tests conducted at 45 °C and 55 °C that also examined the three solids loading scenarios.
The relative extent of release of phosphorus decreased significantly using the higher solids loadings of 200 and 300 g/L. This ties in with a limitation attributed to an oxalate availability issue during oxalic acid leaching [
3]. In the high solids loading tests, less REE dissolution was effectively realised, aligning with reduced phosphorus release in the earlier oxalic acid leach step.
The extent of dissolution of REE in EDTA at the high solids loadings also follows the same pattern of greater solubilisation of neodymium and praseodymium as was seen at the 100 g/L solids loadings. The main difference was the diminishing returns for phosphorus solubilisation in oxalic acid and REE solubilisation in EDTA resulting from increasing the solids loading.
A significant finding was an improvement in REE solubilisation in EDTA in Test C21E6 than evident for Test C2E6. The oxalic acid leach in Test C21 included rod milling mid-test with the original solid and liquid components returned to the leach reactor for a further 24 h of leaching. There were insignificant differences between the two results in terms of phosphorus dissolution in the oxalic acid leach step. Phosphorus concentration after 24 h was 3.2 g/L in Test C2, increasing to 3.4 g/L in C21 after 48 h. This increase could be ascribed to the increased duration from 24 to 48 h. However, the REE appear more amenable to dissolution in EDTA with the milling step included. Neodymium and praseodymium dissolution in this EDTA treatment step nearly matched the dissolution achieved after three stages of oxalic acid in Test C9E6.
Replication of this milling step to ensure repeatability is required. However, the superior response after milling could evidence destruction of an ash layer build-up that passivated the mineral surface or release from material where different REE distributions exist in various size fractions that come into play through the milling. Differences in mineralogy and variations in the distribution of the REE in the monazite warrant further investigation.
XRD analyses of concentrate, oxalic acid leach residue, and final residue after EDTA contacting were also completed. A comparison of the diffractograms for these samples with the REE oxalate precipitate is shown in
Figure 6. The peaks for various species at 2-theta angles have been identified: O noted for REE oxalate precipitate [
1,
2]; M for monazite; G for goethite; F against small peaks aligning with the presence of florencite; and U for an as-yet-unidentified species that appear associated with REE oxalates.
These diffractograms show that as the oxalic acid leach progresses, formation of REE oxalate occurs. The peaks for the REE oxalate then disappear in the EDTA contacting as expected with the resolubilisation of REE. A significant issue identified was that the final residue after the oxalic acid and EDTA treatment was indicated to contain significant monazite and goethite, with essentially the same peaks occurring as were found for the initial concentrate.
This was significant as it indicated only partial decomposition of the contained monazite. Hence, restricted phosphorus dissolution in the oxalic acid leaching does not appear to be due to the presence of a different type of REE-bearing mineralisation but simply monazite that remained unaffected by the oxalic acid leach.
Mineralogical phase identifications using TIMA and reported previously indicated monazites with at least two different signatures, one that appeared to preferentially leach while the other remained relatively inert. The disappearing “monazite” more susceptible to dissolution may have contained greater neodymium and praseodymium, which then led to elevated dissolution of these two elements. Continued phase identification and definitions of mineralogy to further explore this observation are warranted.
Washing of residue after the EDTA contact was also investigated.
Table 3 presents select solution concentrations in wash solutions that were affected up to three times using sodium hydroxide at pH 10 and with cationic flocculant added to the wash solution to provide a constant flocculant dosing of approximately 100 g/t solids.
By the third wash, residual soluble REE species were typically removed to low levels, with these decreasing to approximately 3 percent or less of the concentrations present in the original primary filtrate. Of note was that concentrations in the third wash of the test, where excessive flocculant was used, contained the highest residual concentrations of the various elements. It was observed that greater solution entrainment as flocculant dosing increased, albeit to excessive levels, that then hindered removal during washing.
REE concentrations in the third wash solution were typically reduced to low levels. However, there was no specific measure of the possibility of a bypass occurring during the filtration, which could result in solution being retained with the solids and which was not effectively drawn or flushed from the solids in this wash process. Although reasonable evidence of effective washing exists, evaluation of washing from the perspective of ensuring pregnant solution was effectively removed from the solids in all cases still requires further experimentation.
While REE was removed, iron, phosphorus, and magnesium, at low levels in primary filtrate, may have been solubilised from the solids to a minor extent during the washing process. For example, in one test, neodymium was reduced from 3012 mg/L in primary filtrate to 1164 mg/L in the first wash, 75 mg/L in the second wash, and 10 mg/L in the third and final wash water. The iron concentrations for the same wash filtrates only dropped from 67 mg/L to 33 mg/L to 47 mg/L to 20 mg/L. Phosphorus concentrations increased in the first and second wash filtrates before reducing in the third wash filtrate. Magnesium concentrations hardly changed at all, although these were at low levels in all filtrates, primary as well as wash.
Adjustment of pH with 1 M sodium hydroxide was initially only carried out at varying intervals and with alkali addition only to the extent necessary for the slurry to remain above 9.0. Alkali additions were typically made from 5 to 15 min from the commencement of the test. Reduction of this reaction time was possible, and in the next series of tests, 0.5 M sodium hydroxide solution was used along with a goal to bring pH back close to the pH of the EDTA prior to the addition of ore.
In several tests, the sodium hydroxide to raise pH was added within 10 min, with pH remaining stable for the remainder of the 30 min test. One was even stable after 5 min. This confirms only short residence times are necessary for the solubilisation of REE.
A 30 min residence time is an overestimate, and it appears possible to safely reduce this to 10 min as a design parameter. However, testing is warranted, particularly the option of using an automated dosing system. Specifically, evaluation of continuous addition to evaluate further reduction in residence times should be attempted. Molar usage rates were not sufficiently distinct to allow comparison with the extent of solubilisation.
Further testing could also establish the lower pH limit where solubility decreases since careful pH control and adjustment will be necessary to ensure solubilisation of available REE. Another option, that of effecting separation of mineral solids from the EDTA solution, with this followed by the addition of oxalate and pH, decrease to promote precipitation as a potential solution cleaning step, not examined here, could also be explored to determine whether any benefits are available. This, however, does seem unlikely given the difficult filtration as well as difficult REE redissolution once the solution is returned to alkaline conditions.
The use of pH control during EDTA contacting using lower strength 0.1 M concentration EDTA could also be tested, thereby establishing requirements with greater certainty. Results did not clearly demonstrate a one-for-one molar ratio between REE content, EDTA, and alkali usage. In particular, the EDTA requirements for cerium, which can exist as a tetravalent species, remain to be established. Nevertheless, the use of excess EDTA, as used here with 0.2 M concentration, was successful. Hence, 0.2 M EDTA with pH adjusted to between 9 and 10 is regarded as suitable as design parameters, subject to further refinement.
While 1 M sodium hydroxide was used for pH adjustment, it remained to be seen whether this was the most suitable option. Sodium ions may influence behaviour across the entire leach system, and a sodium control mechanism will be needed. These factors will need investigation.
3.3. Solid-Liquid Separation
Solids at times passed through the filter requiring recycling with filtrates returned to the filter several times before an accumulation of solids allowed a solids-free filtrate to be obtained. A significant impact on recovery was the loss of fines during the washing of solids. Typical practice in early tests was to rinse residual solids after EDTA contacting in the beaker into the filter and then wash the solids in the filter with deionised water. While collection of a primary filtrate that was free of mineral solids could be realised, when rinse and wash water was added, solids often started to pass through the filter paper, with a fine sediment subsequently observed in wash filtrates. In later tests, sodium hydroxide solution at pH 10 was more appropriately used for washing, with this also experiencing similar solids release.
EDTA is recognised as an anti-coagulant in medical applications [
18]. Hence, dispersant properties could be expected, promoting dissociation of fine particles, and these smaller than the filter paper grading size were then free to pass through the filter papers. While it remained impossible to discern individual particles, often an apparent solution colour change from a reddish-brown to a pale blue-grey was accompanied by sedimentation for filtrates from EDTA treatment of oxalic acid-leached residues. This slow settling over periods ranging from days to weeks implies a colloidal nature, this in turn conforming to the presence of particles with sizes of 1 µm or less [
19]. Sedimentation was in accord with the presence of fine particulate matter, not immediately observable, that simply passed through the filter paper [
20].
Both cationic and non-ionic flocculants were able to induce settling of solids. Use of cationic flocculant allowed successful filtration with Whatman No. 1 and No. 2 filter papers in some instances. Flocculant strength ranging from 0.25 percent down to 0.015 percent solution was used. Dosage levels were excessive, typically over 100 g/t in most instances. Optimisation will be required to improve this to lower levels for commercial applications. On balance, however, flocculant use offered potential for obtaining mineral-free filtrates.
Occasionally in initial testing, mineral-solids-free primary filtrates could be obtained without using flocculant. However, this was typically for tests where solutions had become acidic and REE oxalate precipitation had occurred. The acidic conditions likely resulted in improved filtration performance, while the precipitate could have also provided some agglomeration behaviour, further promoting this performance.
Filtrations were typically completed using a 47 mm diameter Buchner funnel. Indicative filtration rates were between 0.3 and 1.0 L/min/m
2 for the collection of primary filtrates using a filter membrane graded to 0.8 µm. Extremely slow filtering material was implied [
21]. Filtration rates for EDTA alkaline slurries were essentially an order of magnitude slower than those pertaining to the recovery of oxalic acid leach residues [
3]. Rates also deteriorated significantly during residue washing.
Testing also explored wash efficiencies after EDTA contact with cationic flocculant added to the wash water. This was relatively successful, with only minor concentrations of various elements detected in the final wash solutions. A three-wash protocol sufficed. Successfully used wash ratios for each wash cycle were approximately 20 percent of the primary filtrate volume. The addition of flocculant to wash solution prior to use stopped or at least limited the extent to which fines passed through the filter media on introduction of the wash water.
The first and second wash filtrates of the tests, including EDTA, were typically more laden with sedimentation than the primary filtrates and typically had a brown colouration. Filtrates from a third wash, where completed, typically had a pale brown colouration with trace or no sedimentation. Only one wash was completed for the solids from the test with pH 10 sodium hydroxide and no EDTA, and a mineral-solids-free wash filtrate was obtained, lending support to the claim of dispersant properties of EDTA.
An observation during these tests was the curling of the filter paper as the vacuum was disconnected and a lifting of the filter paper. In some instances, this lifting behaviour was even observed on the initial introduction of alkaline solution to the filter system. This lifting allowed passage of solids to filtrate by direct bypass, and along with the observed passage of fines through filter paper or membrane, contributed to the difficulties in obtaining comprehensive solids recovery from solution with conventional laboratory filtration equipment.
Another key factor was highlighted in the test with sodium hydroxide solution made alkaline to pH 10 but without EDTA present. The filtration was also problematic and difficult. Filter paper curling and lifting also occurred, and filtration rates remained slow. Difficult filtration of iron oxy hydroxide forms at elevated pH levels and ambient temperatures has been reported elsewhere [
22]. This in turn aligns with the observed move away from alkaline systems for processing high-iron-content rare earth ore materials [
23].
Difficulties in isolating and recovering residues after filtration led to the adoption of a method whereby the filter pot assembly with the filter paper or filter membrane and pre-filter in place was weighed before and after filtration, with the filter assembly allowed to dry at ambient conditions. While still relatively small at the laboratory scale of operation, losses may have a significant impact on mass balancing predictions. Passage of fines through the filter papers, which happened in most but not all experiments, was relatively restricted by using a cationic flocculant, with a loss of around 6 percent to primary filtrate indicated in one test.
Predictions of mass flows for developing a processing flowsheet using experimental mass recovery information should be satisfactory, provided loss occurrence is acknowledged. The determination of particle size of the feed to the EDTA system must be a reported parameter, while further testing should include an assessment of the extent of loss. In practice, it is conceivable that fines losses through the filter media over the entire filtration cycle will occur with the final loss outcome corresponding with the sub-size component of the ore feed material. A separation and recovery option to deal with this sub-size material will be required.
Fine solids lost may be of a somewhat different composition than the bulk of the slurry being filtered. Often, solids passing through filter media were typically of a paler colouration than the bulk solids. This may be consistent with losses of certain types of iron oxides, goethite as opposed to hematite or magnetite, for instance. It could also imply differences in REE phosphate content. Insufficient quantities of this type of material have been collected to date to allow characterisation, but this should be an area of further investigation.
Another factor observed during the filtrations was that the Erlenmeyer filter flask containing wash water often became cold to touch. In one test, the temperature of the wash filtrate, combined from three washes, dropped from 22 °C to 15 °C. The most likely cause was water evaporative cooling under vacuum, conditions which continued for considerable time.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}