
Structural Study of N-(1,3-Benzothiazol-2-yl)-4-Halobenzenesulfonylhydrazides: Hirshfeld Surface Analysis and PIXEL Calculations


Abstract
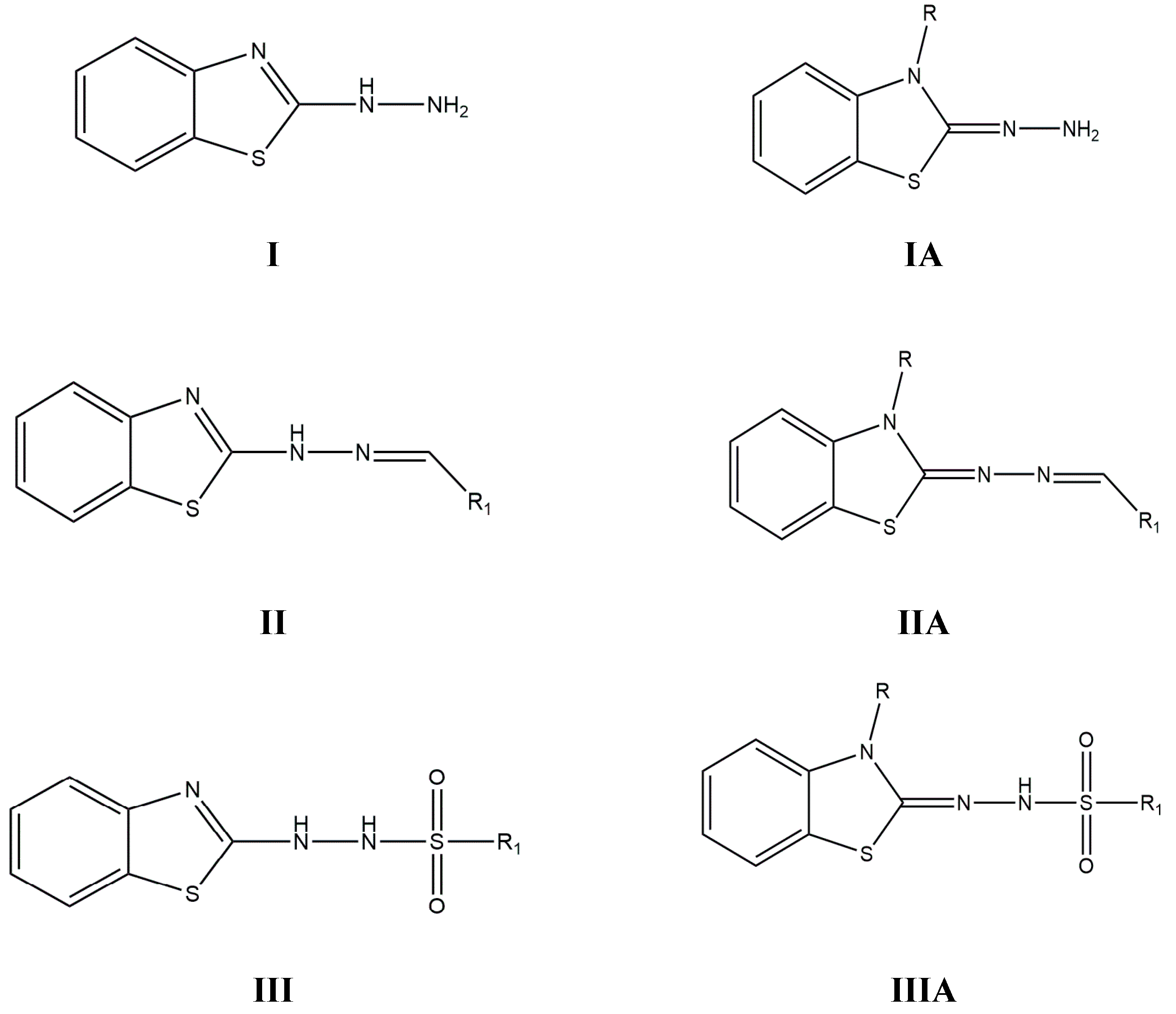
1. Introduction
2. Materials and Methods
2.1. Instruments
2.2. General Synthesis
- (i)
- A solution of 2-hydrazinyl-1,3-benzothiazole(1 mmol) and the arenesulfonyl chloride (1 mmol) in EtOH (20 mL) was refluxed for 1 h. The reaction mixture was washed with water, then the organic layer was collected, dried over magnesium sulphate, and rotary evaporated. The residue was recrystallized from an ethanol solution.
- (ii)
- To a stirred solution of 2-hydrazinobenzothiazole (1 mmol) in dichloromethane (12 mL) were sequentially added 2 drops of Et3N and the appropriate arenesulfonyl chloride (1.20 mmol). The reaction mixture was stirred for 24 h at room temperature and concentrated under reduced pressure. The residue was purified by column chromatography using dichloromethane/methanol (100→95%) as an eluent, resulting inthe benzothiazolyl derivatives in 40–65% yields.
2.2.1. 2-[2-(4-Fluorobenzenesulfonyl)Hydrazinyl]-1,3-Benzothiazol, (1: X = F)
2.2.2. 2-[2-(4-Chlorobenzenesulfonyl)Hydrazinyl]-1,3-Benzothiazol, (1: X = Cl)
2.2.3. 2-[2-(4-Bromobenzenesulfonyl)Hydrazinyl]-1,3-Benzothiazol (1: X = Br)
2.3. X-ray Data Collection and Structure Refinement
2.4. Lattice Energy and Intermolecular Interaction Energy Calculations
2.5. Hirshfeld Surface Analysis and Two-Dimensional Fingerprint (FP) Plots
3. Results
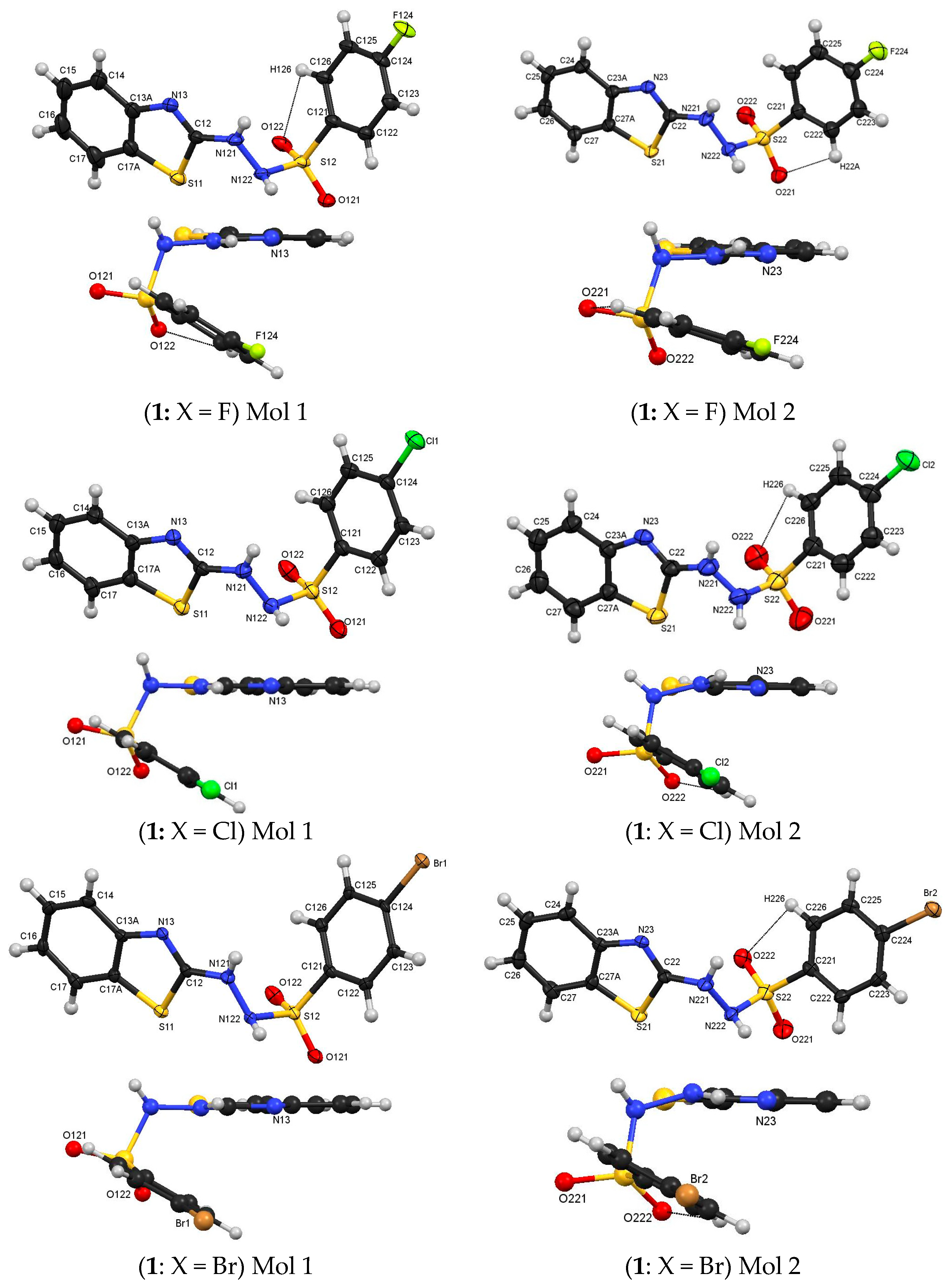
3.1. Molecular Structures
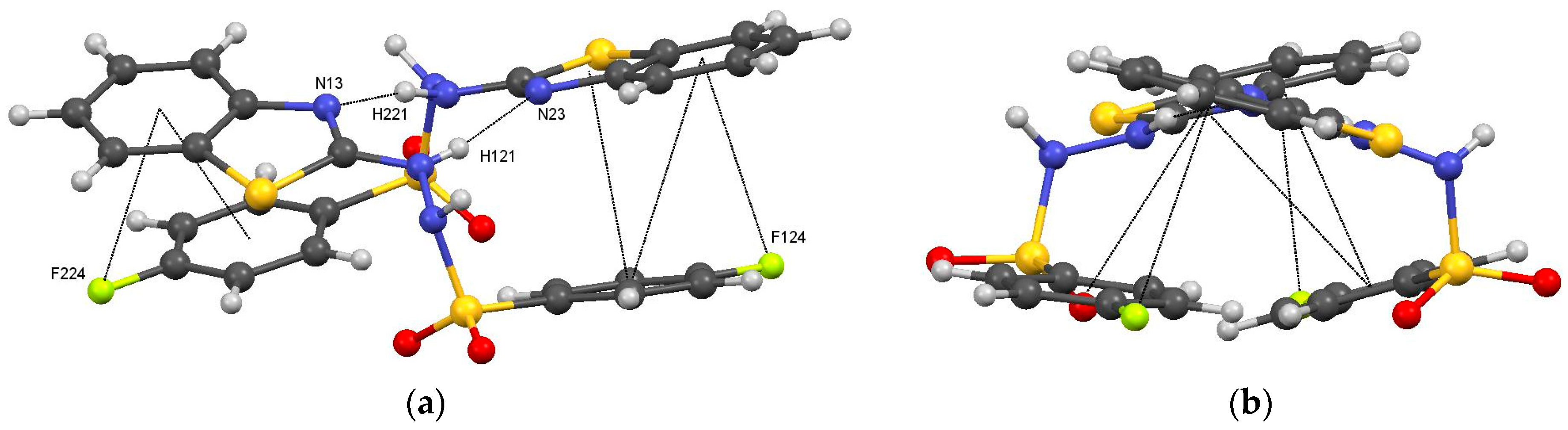
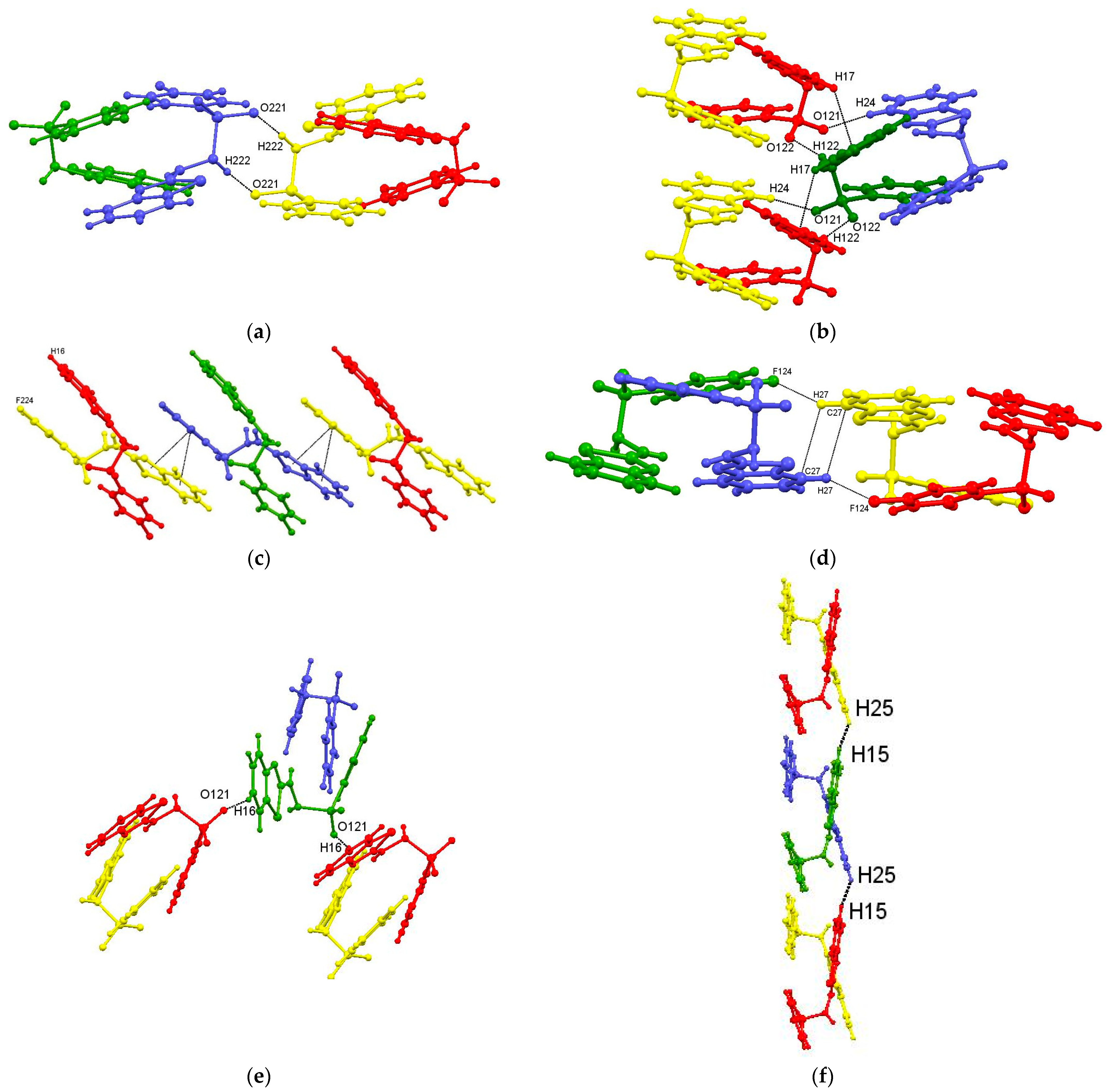
3.2. Intermolecular Interactions
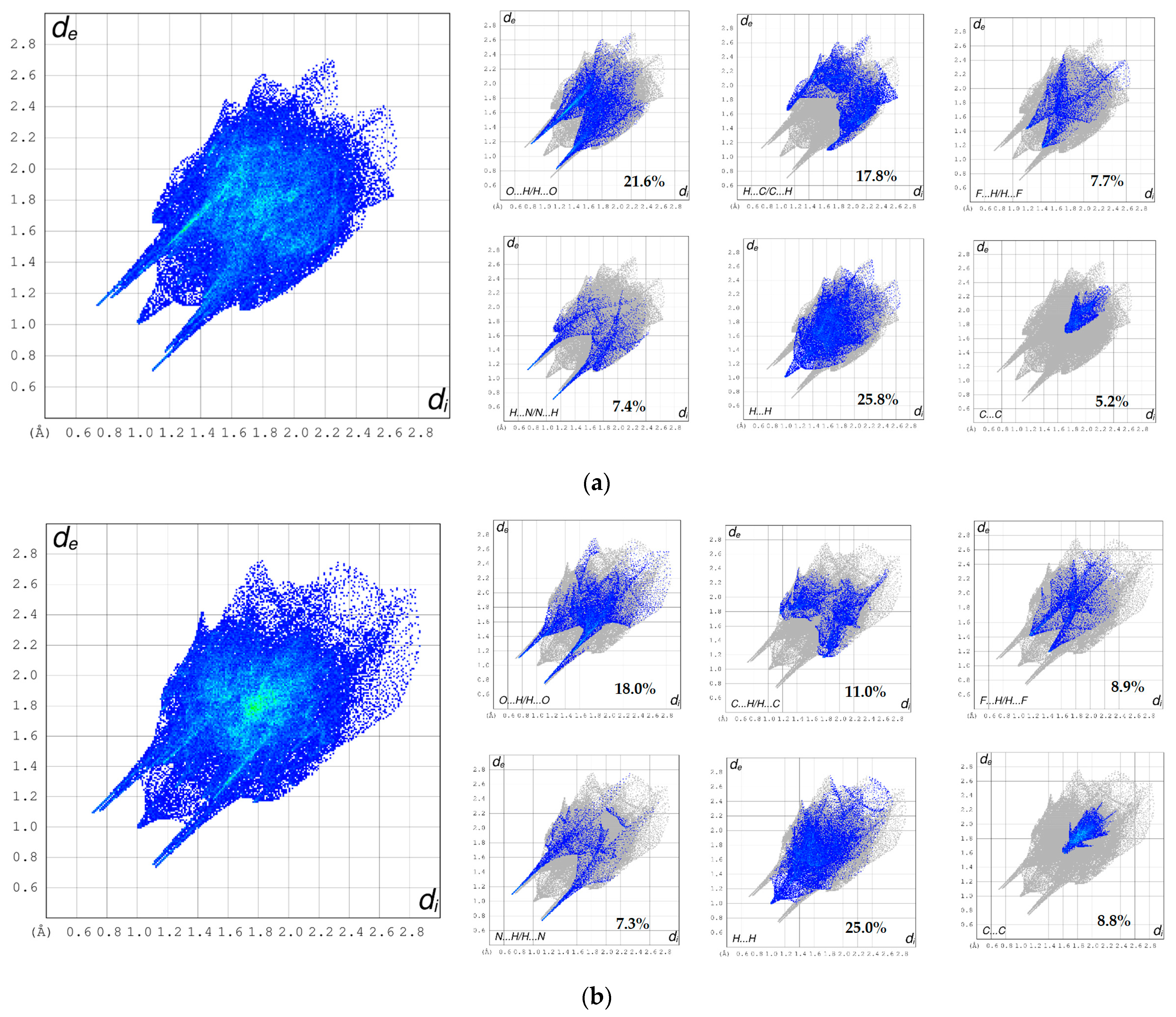
3.3. Hirshfeld Surface Analyses and Fingerprint Plots
3.4. Calculated Energies for (1: X = 4-F)
4. Discussion
4.1. Molecular Structures
4.2. Crystal Structures
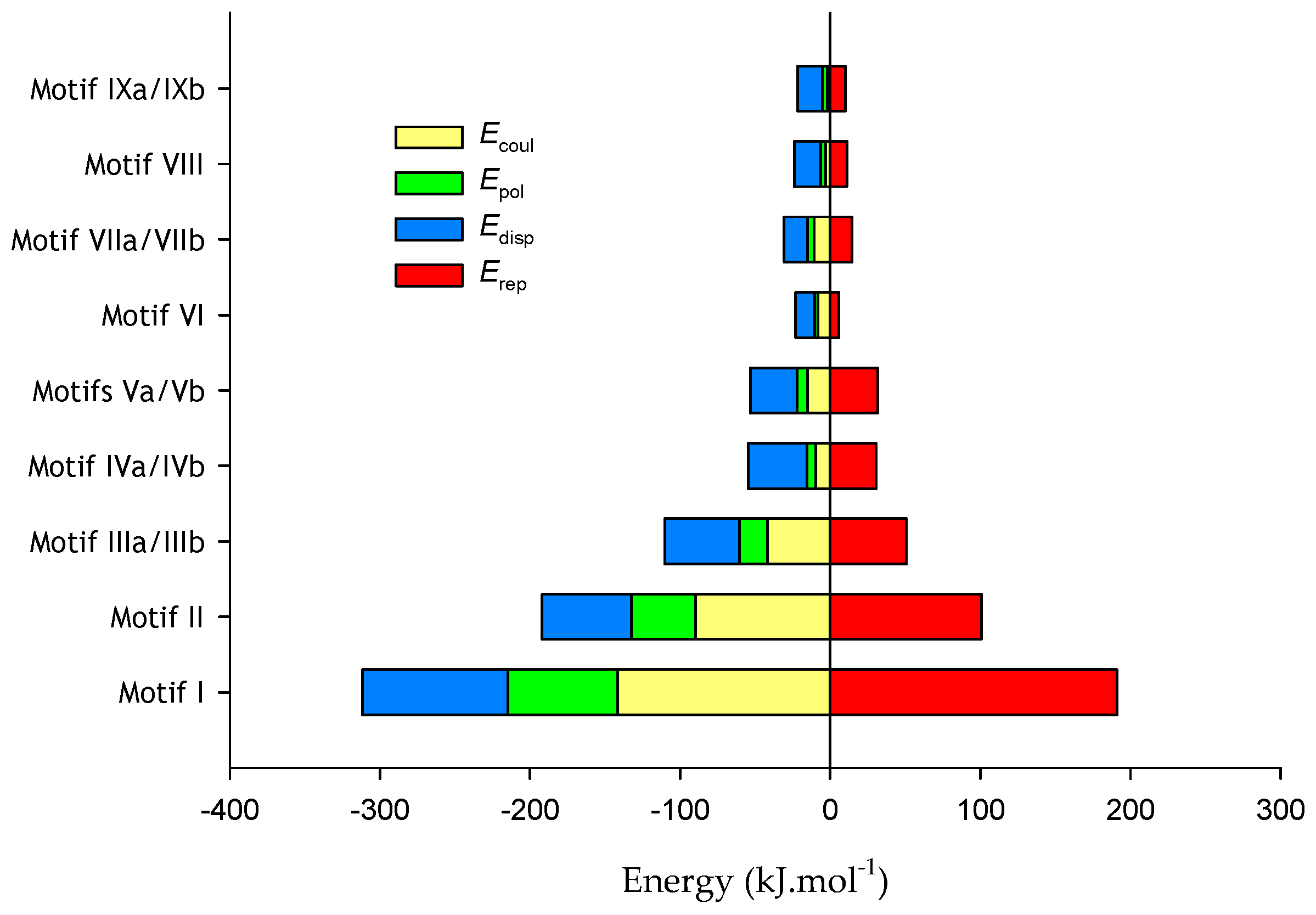
4.2.1. Supramolecular Motifs and Lattice Energies for 1: X = 4-F
Intermolecular Contacts in 1: X = F
1: X = F Motifs
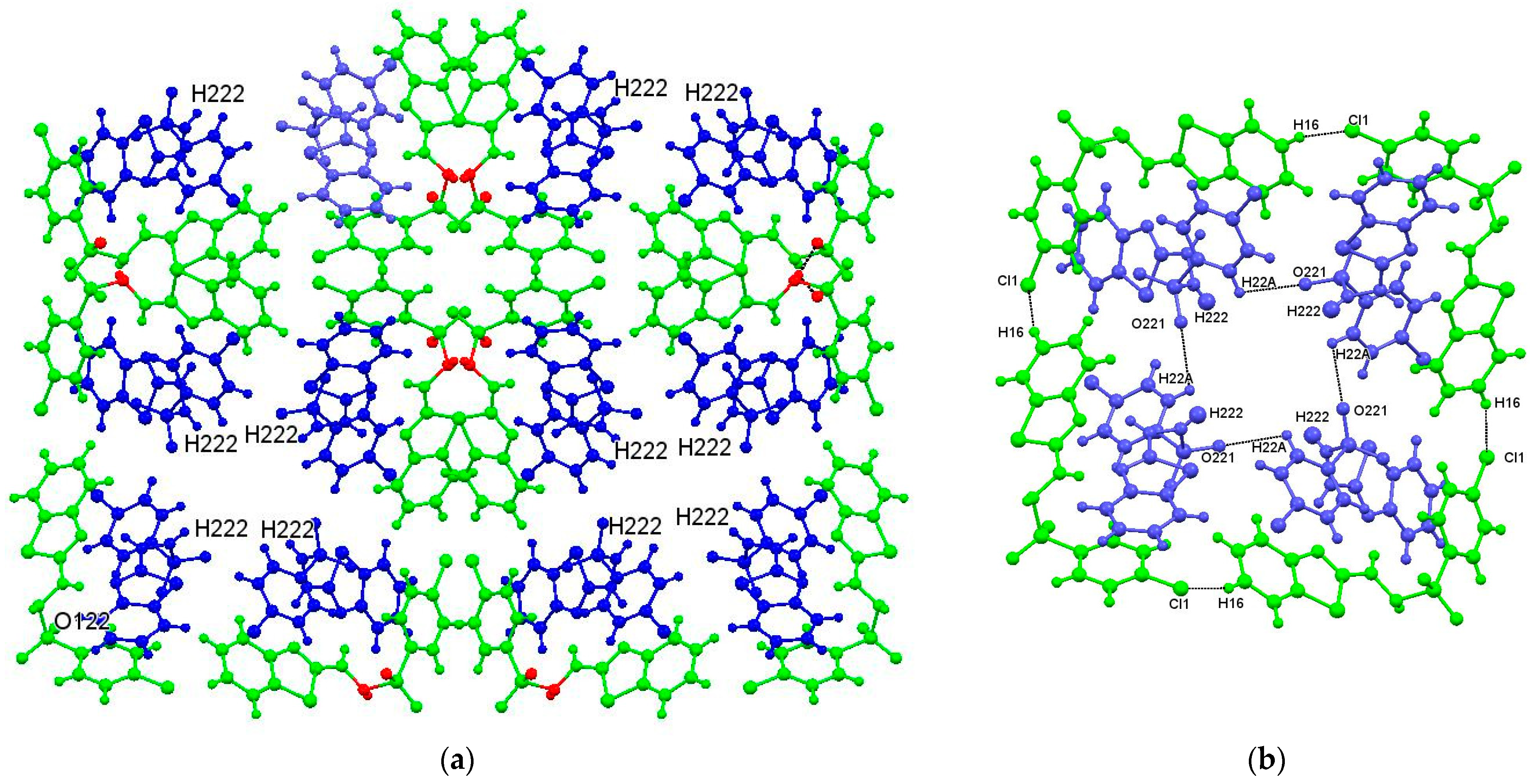
4.2.2. Intermolecular Interactions in (1: X = Cl) and (1: X = Br)
- the motifs of type I in (1: X = F), with N121–H121···N23 and N221–H221···N13 hydrogen bonds, C124–X···π6 interactions, and face-to-face offset π···π interactions, have equivalents in (1: X = Cl and Br),
- the motifs of type III in (1: X = F), with N122–H122···O122 hydrogen bonds and C17–H17···π(1), have equivalents in (1: X = Cl and Br),
- the motifs of type IV in (1: X = F), with face-to-face, offset π···π interactions have equivalents in (1: X = Cl and Br),
- the interactions present in the weaker, lower-energy motifs, IV to IX, in (1: X = F), were not identified by PLATON in (1: X = Cl and Br),
- the equivalent of motif II in (1: X = F) is not present in (1: X = Cl and Br). The links between the molecules in motif II in (1: X = F) are a pair of N222–H222···O221 hydrogen bonds, while in (1: X = Cl and Br), the N222–H222 atoms, while not confirmed, are apparently hydrogen bonded to disordered water molecules residing in voids in the crystal structure.
4.3. Related Compounds
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heijstek, A. Benzothiazole: Preparation, Structure and Uses, 1st ed.; Nova Science Publishers Inc.: New York, NY, USA, 2020. [Google Scholar]
- Elamin, M.B.; Elaziz, A.A.E.S.A.; Abdallah, E.M. Benzothiazole moieties and their derivatives as antimicrobial and antiviral agents: A mini-review. Int. J. Res. Pharm. Sci. 2020, 11, 3309–3315. [Google Scholar] [CrossRef]
- Pathak, N.; Rathi, E.; Kumar, N.; Kini, S.G.; Rao, C.M. A Review on Anticancer Potentials of Benzothiazole Derivatives. Mini Rev. Med. Chem. 2020, 20, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.; Imran, M. A Mini-Review on Pharmacological Importance of Benzothiazole Scaffold. Mini Rev. Org. Chem. 2021, 9, 1086–1097. [Google Scholar] [CrossRef]
- Abrol, S.; Bodla, R.B.; Goswami, C. A comprehensive review on benzothiazole derivatives for their biological activities. Int. J. Pharm. Sci. Res. 2019, 10, 3196–3209. [Google Scholar]
- Keri, R.S.; Patil, M.R.; Patil, S.A.; Budagumpi, S.A. Comprehensive Review in Current Developments of Benzothiazole-Based Molecules in Medicinal Chemistry. Eur. J. Med. Chem. 2014, 89, 207–251. [Google Scholar] [CrossRef] [PubMed]
- Law, C.S.W.; Yeong, K.Y. Current trends of benzothiazoles in drug discovery: A patent review (2015–2020). Exp. Opin. Ther. Pat. 2022, 32, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Kikelj, T.D.; Masic, L.P. Benzothiazole-based Compounds in Antibacterial Drug Discovery. Curr. Med. Chem. 2018, 25, 5218–5236. [Google Scholar] [CrossRef]
- Maliyappa, M.R.; Keshavayya, J.; Mallikarjuna, N.M.; Pushpavathi, I. Novel substituted aniline based heterocyclic dispersed azo dyes coupling with 5-methyl-2-(6-methyl-1, 3-benzothiazol-2-yl)-2, 4-dihydro-3H-pyrazol-3-one: Synthesis, structural, computational and biological studies. J. Mol. Struct. 2020, 1205, 127576. [Google Scholar] [CrossRef]
- Thekkeppat, N.P.; Lakshmipathi, M.; Jalilov, A.S.; Das, P.; Malik, A.; Peedikakkal, P.; Ghosh, S. Combining Optical Properties with Flexibility in Halogen-Substituted Benzothiazole. Cryst. Growth Des. 2020, 20, 3937–3943. [Google Scholar] [CrossRef]
- Nootem, J.; Daengnern, R.; Sattayanon, C.; Wattanathana, W.; Wannapaiboon, S.; Rashatasakhon, P.; Chansaenpak, K. The synergy of CHEF and ICT toward fluorescence ‘turn-on’ probes based on push-pull benzothiazoles for selective detection of Cu2+ in acetonitrile/water mixture. J. Photo. Chem. Photobiol. A Chem. 2021, 415, 113318. [Google Scholar] [CrossRef]
- Foo, K.-L.; Ha, S.-T.; Yeap, G.-Y. Synthesis and phase transition behavior of calamitic liquid crystals containing heterocyclic core and lateral ethoxy substituent. Phase Trans. 2002, 95, 178–192. [Google Scholar] [CrossRef]
- Lioa, C.; Kim, U.J.; Kannan, K. A Review of Environmental Occurrence, Fate, Exposure, and Toxicity of Benzothiazoles. Environ. Sci. Technol. 2018, 52, 5007–5026. [Google Scholar] [CrossRef] [PubMed]
- Le Bozec, L.; Moody, C.J. Naturally Occurring Nitrogen–Sulfur Compounds. The Benzothiazole Alkaloids. Aust. J. Chem. 2009, 62, 639–647. [Google Scholar] [CrossRef]
- Khalil, M.I.; Khalal, Q.Z. Synthesis and characterization of new compounds derived from 2-hydrazinobenzothiazole and evaluated their antibacterial activity. J. Phys. Conf. Ser. 2021, 1853, 012007. [Google Scholar] [CrossRef]
- Badr, M.Z.A.; Mahmoud, A.M.; Mahgoub, S.A.; Hozien, Z.A. Condensation and Cyclization Reactions of 2-Hydrazinobenzimidazole, -benzoxazole, and -benzothiazole. Bull. Chem. Soc. Jpn. 1988, 61, 1339–1344. [Google Scholar] [CrossRef]
- Elsayed, S.A.; Saad, E.A.; Mostafa, S.I. Development of New Potential Anticancer Metal Complexes Derived from 2-Hydrazinobenzothiazole. Mini Rev. Med. Chem. 2019, 19, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Low, J.N.; Pinheiro, A.C.; de Souza, M.V.N.; Wardell, J.L. Crystal structure, Hirshfeld surface analysis and PIXEL calculations of the three isomeric (E)-2-((pyridinylmethylidene)hydrazinyl)benzo[d]thiazoles: Occurrence of stacking interactions. Journal of Molecular Structure. J. Mol. Struct. 2021, 1230, 129907. [Google Scholar] [CrossRef]
- Nogueira, A.; Vasconcelos, T.R.A.; Wardell, J.L.; Wardell, S.M.S.V. Crystal structures of hydrazones, 2-(1,3-benzothiazolyl)-NH—N=CH—Ar, prepared from arenealdehydes and 2-hydrazinyl-1,3-benzothiazole. Z. Für Krist. 2011, 226, 846–860. [Google Scholar] [CrossRef]
- Lindgren, E.B.; Yoneda, J.D.; Leal, K.Z.; Nogueira, A.F.; Vasconcelos, T.R.A.; Wardell, J.L.; Wardell, S.M.S.V. Structures of hydrazones, (E)-2-(1,3-benzothiazolyl)-NHNCHAr, [Ar=4-(pyridin-2-yl)phenyl, pyrrol-2-yl, thien-2-yl and furan-2-yl]: Difference in conformations and intermolecular hydrogen bonding. J. Mol. Struct. 2013, 1036, 19–27. [Google Scholar] [CrossRef]
- Hünig, S.; Kaupp, G. Azofarbstoffedurchoxydativekupplung—XXVIII: Kinetik der kupplungheterocyclischer carbonyl- und sulonylazo-quartärsalzemitphenolenzudiazamerocyaninen. Tetrahedron 1967, 23, 1411–1439. [Google Scholar] [CrossRef]
- Hünig, S.; Kießlich, G.; Oette, K.-H.; Quast, H. AzofarbstoffedurchoxydativeKupplung, XXIX. Derivate der 3-Methyl-1.2-benzisothiazolon-(2)-, 1.3-Benzdithiolon-(2)- und 1.2-Benzdithiolon-(3)-hydrazone. Justus Liebigs Ann. Chem. 1972, 754, 46–55. [Google Scholar] [CrossRef]
- Agfa-Gevaert-AG. Sulphonylamidrazone Prepn–Single Stage Process Using Oxidation Agents. DE2105063 A1, 24 August 1972. [Google Scholar]
- HENKEL ag & Co. KGAA. Sulfonylhydrazines, Metal Complexes Thereof, and Solutions Containing such Compounds for Use in Extraction of Metal Values. USA 4252959, 13 October 1981. [Google Scholar]
- Institut khimiko-fotograficheskoj promyshlennosti. Method of Preparing Heterocyclic Sulfonyl- or Acylhydrazones1. SU702016A1, 5 December 1979. [Google Scholar]
- EISAI Co., Ltd. A1 Aryl and Heteroaryl Compounds Useful as Fibroblast Growth Factor Antagonists. WO2000/30632, 2 June 2000. [Google Scholar]
- Norman, D.D.; Ibezim, A.; Scott, W.E.; White, S.; Parrill, A.L.; Baker, D.L. Autotaxin inhibition: Development and application of computational tools to identify site-selective lead compounds. Bioorg. Med Chem. 2013, 21, 5548–5560. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shan, J.; Chang, W.; Kim, I.; Bao, J.; Lee, H.J.; Zhang, X.; Samuel, V.T.; Shulman, G.I.; Liu, D.; et al. Chemical and genetic evidence for the involvement of Wnt antagonist Dickkopf2 in regulation of glucose metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 11402–11407. [Google Scholar] [CrossRef] [PubMed]
- Noha, S.M.; Jazzer, B.; Kuehni, S.; Rollinger, J.M.; Stuppner, H.; Schaible, A.M.; Wetz, O.; Wolber, G.; Stupper, D. Pharmacophore-based discovery of a novel cytosolic phospholipase A2α inhibitor. Bioorg. Med. Chem. Lett. 2012, 22, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Peretyazhko, M.Z.; Pel’kis, P.S. Unsymmetrically substituted benzothiazolyl-hydrazines. Chem. Heterocycl. Compd. 1971, 7, 713–714. [Google Scholar] [CrossRef]
- Baddeley, T.C.; de Souza, M.V.N.; Wardell, J.L.; Jotani, M.M.; Tiekink, E.R.T. N′-(1,3-Benzo thia zol-2-yl) benzene sulfono hydrazide: Crystal structure, Hirshfeld surface analysis and computational chemistry. Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, E75, 523–526. [Google Scholar]
- Gomes, L.R.; Low, J.N.; Pinheiro, A.C.; Wardell, J.L. Crystal packing of structurally similar and strong dimeric subunits of isomeric N/-(1,3-benzothiazol-2-yl)nitrobenzenesulfonylhydrazides, influenced by the position of the nitro group. J. Mol. Struct. 2024, 1298, 136957. [Google Scholar] [CrossRef]
- Gomes, L.R.; Fruchtl, H.; Low, J.N.; van Mourik, T.; Pinheiro, A.C.; de Souza, M.V.N.; Wardell, J.L. Crystal structure of N-(1,3-benzothiazol-2-yl)-4-iodobenzene-1-sulfonohydrazide: The unexpected importance of l N-H⋅⋅⋅π and I⋅⋅⋅π interactions on the supramolecular arrangement. Z. Anorg. Allg. Chem. 2022, 648, e202200087. [Google Scholar] [CrossRef]
- Morscher, A.; de Souza, M.V.N.; Wardell, J.L.; Harrison, W.T.A. Expected and unexpected products of reactions of 2-hydrazinylbenzo-thia-zole with 3-nitro-benzene-sulfonyl chloride in different solvents. Acta Crystallogr. E Crystallogr. Commun. 2018, 74, 673–677. [Google Scholar] [CrossRef]
- CrysAlisPro Software System; Version 1.171.38.41; Rigaku Oxford Diffraction: The Woodlands, TX, USA, 2015.
- McArdle, P.; Gilligan, K.; Cunningham, D.; Dark, R.; Mahon, M. OSCAIL: A method for the prediction of the crystal structure of ionic organic compounds—The crystal structures of o-toluidinium chloride and bromide and polymorphism of bicifadine hydrochloride. CrystEngComm 2004, 6, 303–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. 2015, A7, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXL-2018 (2018) Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 2018. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Gavezzotti, A. Calculation of intermolecular interaction energies by direct numerical integration over electron densities. 2. An improved polarization model and the evaluation of dispersion and repulsion energies. J. Phys. Chem. 2003, B107, 2344–2353. [Google Scholar] [CrossRef]
- Gavezzotti, A. Calculation of lattice energies of organic crystals: The PIXEL integration method in comparison with more traditional methods. Z. Für Krist. Cryst. Mater. 2005, 220, 499–510. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. 2004, B60, 627–668. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer; The University of Western Australia: Crawley, Australia, 2012. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Azzam, R.A.; Elgemeie, G.H.; Elsayed, R.E.; Jones, P.G. Crystal structure of N′-[2-(benzo[d]thia-zol-2-yl)acet-yl]-4-methyl-benzene-sulfono-hydrazide. Acta Crystallogr. 2017, E73, 1041–1043. [Google Scholar] [CrossRef]
- Pai, N.; Foro, S.; Gowda, B.T. Crystal structure and Hirshfeld surface analysis of (Z)-4-chloro-N′-(4-oxo-thia-zol-idin-2-yl-idene)benzene-sulfono-hydrazide monohydrate. Acta Crystallogr. Sect. E Crystallogr. Commun. 2018, 74, 1569–1573. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.B.; Jin, Z.M.; Wang, H.B.; Jiang, B. N′-(Benzenesulfonyl)-4-methylthiazole-5-carbohydrazide. Acta Crystallog. 2004, E60, o1292–o1293. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cn2-Nn21 | Nn21-Nn22 | Nn22-Sn2 | Sn2-Cn21 |
|---|---|---|---|---|
| (1: X = F) | ||||
| Mol A: n = 1 | 1.351(3) | 1.393(2) | 1.6565(17) | 1.7555(19) |
| Mol B: n = 2 | 1.343(3) | 1.391(3) | 1.6691(18) | 1.750(2) |
| (1: X = Cl) | ||||
| Mol A: n = 1 | 1.350(5) | 1.401(4) | 1.659(3) | 1.749(3) |
| Mol B: n = 2 | 1.364(6) | 1.396(4) | 1.645(4) | 1.757(2) |
| (1: X = Br) | ||||
| Mol A: n = 1 | 1.358(7) | 1.396(6) | 1.663(4) | 1.756(5) |
| Mol B: n = 2 | 1.356(7) | 1.405(7) | 1.647(5) | 1.776(5) |
| Compound | D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|---|
| (1: X = F) | C222–H22A···O221 | 0.95 | 2.52 | 2.900(3) | 104 |
| (1: X = F) | C126–H126···O122 | 0.95 | 2.60 | 2.945(2) | 102 |
| (1: X = Cl) | C226–H226···O222 | 0.95 | 2.56 | 2.922(6) | 103 |
| (1: X = Br) | C226–H226···O222 | 0.95 | 2.56 | 2.922(6) | 103 |
| Compound | Angle between Bt & Ph Planes | Angle between SO2 and | |
|---|---|---|---|
 | Ph | Bt | |
| (1: X = F) | |||
| Mol 1 | 30.93 (8) | 40.82 | 48.36 |
| Mol 2 | 16.64 (9) | 48.96 | 47.66 |
| (1: X = Cl) | |||
| Mol 1 | 30.53 (16) | 42.31 | 46.35 |
| Mol 2 | 29.21 (18) | 50.24 | 55.26 |
| (1: X = Br) | |||
| Mol 1 | 29.7 (2) | 41.64 | 46.67 |
| Mol 2 | 28.3 (2) | 51.46 | 56.34 |
| Cpd | D–H···A | D–H | H····A | D···A | D–H···A | Symmetry Code |
|---|---|---|---|---|---|---|
| 1: X = 4-F | N121–H121···N23 | 0.78(3) | 2.08(3) | 2.835(2) | 165(3) | x, y, z |
| N221–H221···N13 | 0.86(3) | 1.94(3) | 2.775(2) | 165(3) | x, y, z | |
| N122–H122···O122 | 0.85(3) | 2.15(3) | 2.989(2) | 168(2) | x, 1/2 − y, 1/2 + z | |
| N222–H222···O221 | 0.99(2) | 1.88(2) | 2.862(3) | 177(2) | −x, 1 − y, 1 − z | |
| C16–H16···O121 | 0.95 | 2.44 | 3.360(3) | 162 | −1 + x, 1/2 − y, −1/2 + z | |
| C24–H24···O121 | 0.95 | 2.55 | 3.315(3) | 138 | x, 1/2 − y, 1/2 + z | |
| C25–H25···O122 | 0.95 | 2.62 | 3.232(3) | 123 | x, y, 1 + z | |
| 1: X = 4-Cl | N121–H121···N23 | 0.88 | 2.00 | 2.861(5) | 166 | x, y, z |
| N122–H122···O122 | 0.97 | 2.11 | 2.971(4) | 147 | 1/2 − y, 1/2 − x, −1/2 + z | |
| N221–H221···N13 | 0.88 | 2.07 | 2.855(5) | 149 | x, y, z | |
| C26–H26···Cl1 | 0.95 | 2.81 | 3.649(5) | 148 | −1/2 + y, 1/2 + x,−1/2 + z | |
| 1: X = 4-Br | N121–H121···N23 | 0.88 | 2.00 | 2.864(6) | 166 | x, y, z |
| N122–H122···O122 | 0.88 | 2.37 | 2.979(6) | 127 | 3/2 − y, 3/2 − x, 1/2 + z | |
| N221–H221···N13 | 0.88 | 2.10 | 2.868(6) | 145 | x, y, z |
 | ||||||
|---|---|---|---|---|---|---|
| Cpd | Y–X···π | X···Cg(π) | Xperp | Y–X···Cg(π) | Y···Cg(π) | Symmetry Code |
| 1: X = 4-F | C17–H17···Cg1 | 2.84 | 2.77 | 132 | 3.542(3) | x, 1/2 − y, −1/2 + z |
| C124–F124···Cg6 | 3.7122(16) | 3.684 | 76.07(10) | 3.632(3) | x, y, z | |
| C224–F224···Cg2 | 3.853(2) | 3.622 | 71.24(13) | 3.542(3) | x, y, z | |
| 1: X = 4-Cl | C17–H17···Cg1 | 2.83 | 2.72 | 131 | 3.526(4) | 1/2 − y, 1/2 − x, 1/2 + z |
| C124–Cl1···Cg6 | 3.718(2) | 3.471 | 131 | 3.632(3) | x, y, z | |
| 1: X = 4-Br | C17–H17···Cg1 | 2.79 | 2.72 | 135 | 3.526(4) | 1/2 − x, 1/2 − y, 1/2 + z |
| C124–Br1···Cg6 | 3.824(2) | 3.556 | 66.37(16) | 3.522(6) | x, y, z | |
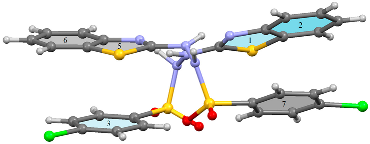 | |||||||
|---|---|---|---|---|---|---|---|
| Cpd | Cg(I)···Cg(J) | Cg···Cg | α | CgIperp | CgJperp | Sym Code | Slippage |
| 1: X = 4-F | Cg2···Cg7 | 3.9448(15) | 9.35(12) | 3.4387(10) | 3.5588(10) | x, y, z | 1.702 |
| Cg3···Cg5 | 3.9711(11) | 7.95(9) | 3.3440(8) | 3.5291(8) | x, y, z | 1.852 | |
| Cg3···Cg6 | 4.0113(12) | 8.15(10) | 3.3376(8) | 3.6097(8) | x, y, z | 1.749 | |
| Cg6···Cg7 | 3.9447(15) | 9.35(12) | 3.3387(10) | 3.4387(10) | x, y, z | 1.933 | |
| Cg5···Cg7 | 3.6345(12) | 16.76(10) | 3.4741(8) | 3.5748(9) | x, y, 1 + z | 0.656 | |
| Cg7···Cg6 | 3.9187(12) | 16.53(11) | 3.5720(9) | 3.1023(9) | x, y, −1 + z | 2.394 | |
| 1: X = 4-Cl | Cg2···Cg7 | 3.818(2) | 3.0(2) | 3.4291(16) | 3.3890(19) | x, y, z | 1.758 |
| Cg3···Cg5 | 3.900(2) | 0.3(2) | 3.4217(17) | 3.4267(16) | x, y, z | 1.863 | |
| Cg3···Cg6 | 3.893(3) | 1.5(2) | 3.4059(17) | 3.4530(19) | x, y, z | 1.798 | |
| Cg5···Cg7 | 3.831(8) | 29.3(2) | 3.4471(16) | 3.8135(19) | x, y, −1 + z | ||
| 1: X = 4-Br | Cg2···Cg7 | 3.788(3) | 2.3(3) | 3.410(2) | 3.378(2) | x, y, z | 1.713 |
| Cg3···Cg5 | 3.947(3) | 1.9(3) | 3.419(2) | 3.438(2) | x, y, z | 1.938 | |
| Cg3···Cg6 | 3.909(3) | 3.1(3) | 3.399(2) | 3.498(2) | x, y, z | 1.744 | |
| Cg5···Cg7 | 3.797(3) | 28.5(3) | 3.385(2) | 3.780(2) | x, y,1 + z | ||
| Cg7···Cg2 | 3.788(3) | 2.3(3) | 3.378(2) | 3.410(2) | x, y, z | 1.649 | |
| Motif | Symmetry Code | Interactions (Close Contacts) | Distance (Å): Angle (°) | Motif Form | Etot |
|---|---|---|---|---|---|
| Motif I Mol 1···Mol 2 Asymmetric unit | x, y, z | N121–H121···N23 | 2.08(3); 165(3) | Asymmetric R22(8)dimer Figure 6 | −120.5 |
| N221–H221···N13 | 1.94(3); 165(3) | ||||
| C124–F124···π6 | 3.7122(16); 76.07(10) | ||||
| C224–F124···π2 | 3.853(2); 71.24(13) | ||||
| π2···π7 | 3.9448(15) | ||||
| π3···π5 | 3.9711(11) | ||||
| π3···π6 | 4.0113(12) | ||||
| π5···π3 | 3.9710(11) | ||||
| π6···π3 | 4.0112(12) | ||||
| π6···π7 | 3.9447(15) | ||||
| Motif II Mol 2···Mol 2 | −x, 1 − y, 1 − z | N222–H222···O221 O221···H222–N222 | 1.88(2); 177(2) | Symmetric dimer: R22(8) Figure 7a | −91.4 |
| Motifs IIIa/IIIb Mol 1···Mol 1 | x, 0.5 − y, 0.5 + z x, 0.5 − y, −0.5 + z | N122–H122···O122 C17–H17···π(1) | 2.15(3); 168(2) 2.84; 132 | C(4) chain Figure 7b | −59.6 |
| Motifs IVa/IVb Mol 2···Mol 2 | x, y, 1 + z x, y, −1 + z | π5···π7 π7···π6 | 3.6345(12) 3.9187(12) | Chain Figure 7c | −23.9 |
| Motifs Va/Vb Mol 1···Mol 2 Mol 2···Mol 1 | 1 − x, 1 − y, 2 − z | F124···H27 | 2.68 | Pair of acyclic dimers Figure 7d | −21.5 |
| Motif VI Mol 2···Mol 2 | 1 − x, 1 − y, 2 − z | C27···H27 | 2.96 | acyclic dimer Figure 7d | −17.1 |
| Motifs VIIa/VIIb Mol 1···Mol 1 | 1 + x, 0.5 − y, −0.5 + z −1 + x, 0.5 − y, −0.5 + z | C16–H16···O121 O121···H16–C16 | 2.44; 162 | C(10) chain Figure 7e | −16.2 |
| Motif VIII Mol 1···Mol 2 | x, y, −1 + z x, y, 1 + z | H25···H15 | 2.20 | Pair of H–H contacted dimers Figure 7f | −12.7 |
| Motifs IXa/IXb Mol 1···Mol 2 Mol 2··· Mol 1 | x, 0.5 − y, 0.5 + z x, −y, −0.5 + z | C24–H24···O121 | 2.55; 138 | Two pairs of acyclic dimers Figure 7b | −11.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, L.R.; Low, J.N.; Pinheiro, A.C.; Wardell, J.L. Structural Study of N-(1,3-Benzothiazol-2-yl)-4-Halobenzenesulfonylhydrazides: Hirshfeld Surface Analysis and PIXEL Calculations. Crystals 2024, 14, 330. https://doi.org/10.3390/cryst14040330
Gomes LR, Low JN, Pinheiro AC, Wardell JL. Structural Study of N-(1,3-Benzothiazol-2-yl)-4-Halobenzenesulfonylhydrazides: Hirshfeld Surface Analysis and PIXEL Calculations. Crystals. 2024; 14(4):330. https://doi.org/10.3390/cryst14040330
Chicago/Turabian StyleGomes, Ligia R., John N. Low, Alessandra C. Pinheiro, and James L. Wardell. 2024. "Structural Study of N-(1,3-Benzothiazol-2-yl)-4-Halobenzenesulfonylhydrazides: Hirshfeld Surface Analysis and PIXEL Calculations" Crystals 14, no. 4: 330. https://doi.org/10.3390/cryst14040330
APA StyleGomes, L. R., Low, J. N., Pinheiro, A. C., & Wardell, J. L. (2024). Structural Study of N-(1,3-Benzothiazol-2-yl)-4-Halobenzenesulfonylhydrazides: Hirshfeld Surface Analysis and PIXEL Calculations. Crystals, 14(4), 330. https://doi.org/10.3390/cryst14040330