



Effect of Oxygen in Mo-TM (TM = Ti, Zr, Hf) Solid Solutions as Studied with Density Functional Theory Calculations

Abstract
1. Introduction
- Usage of high-purity raw materials [16].
2. Materials and Methods
3. Results and Discussion
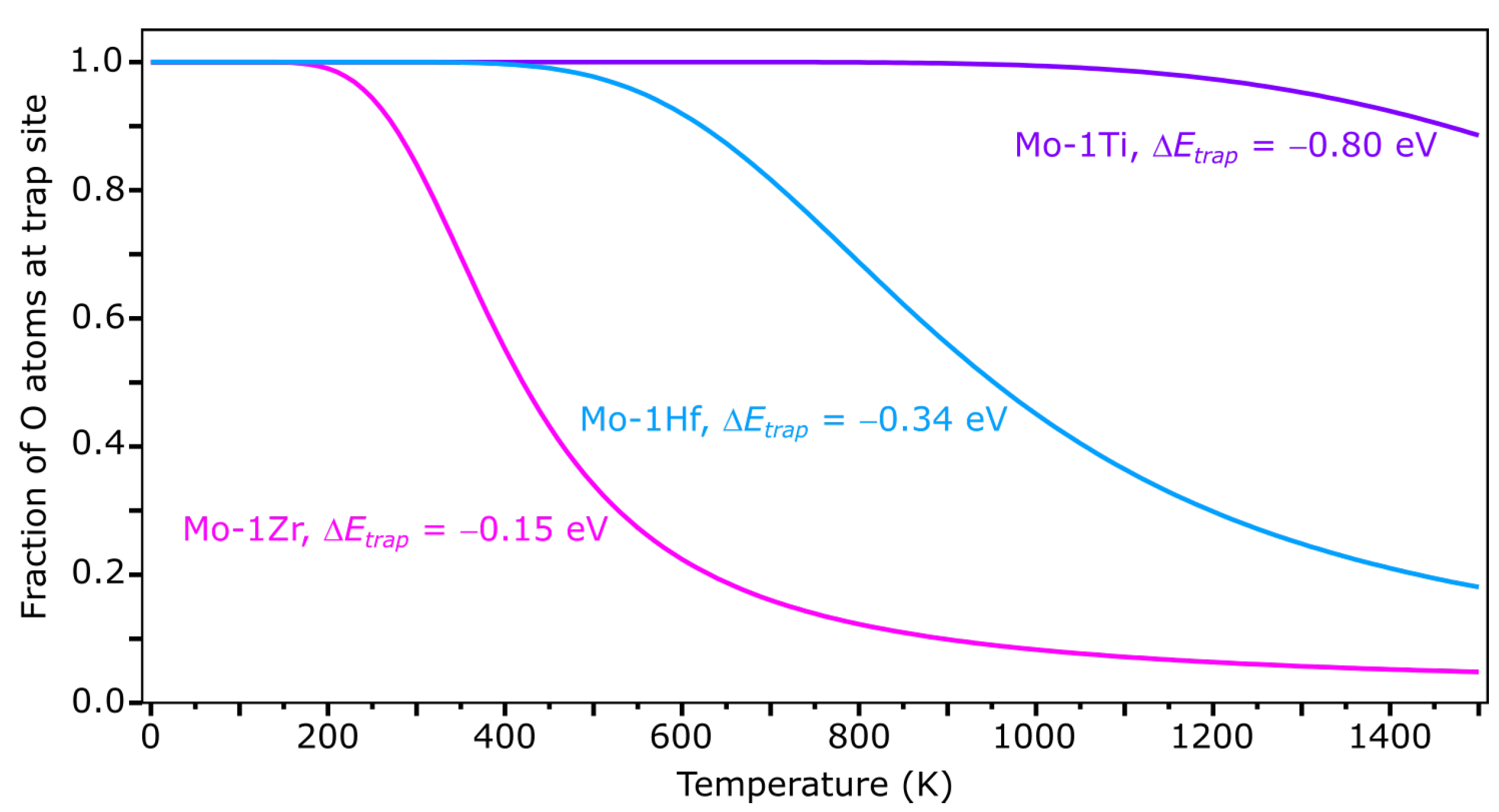
3.1. Temperature-Dependent Trapping of Oxygen Atoms
3.2. Formation Enthalpies and Substitutional and Interstitial Energies
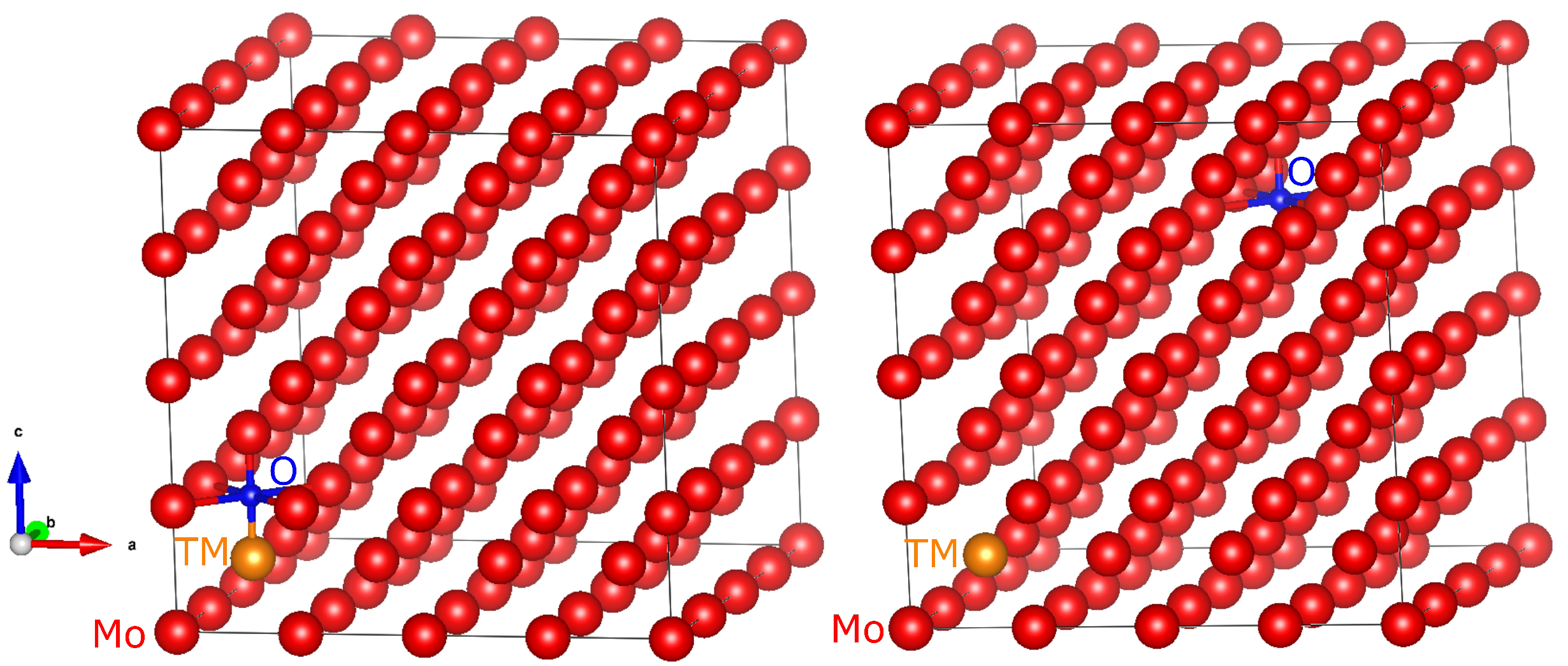
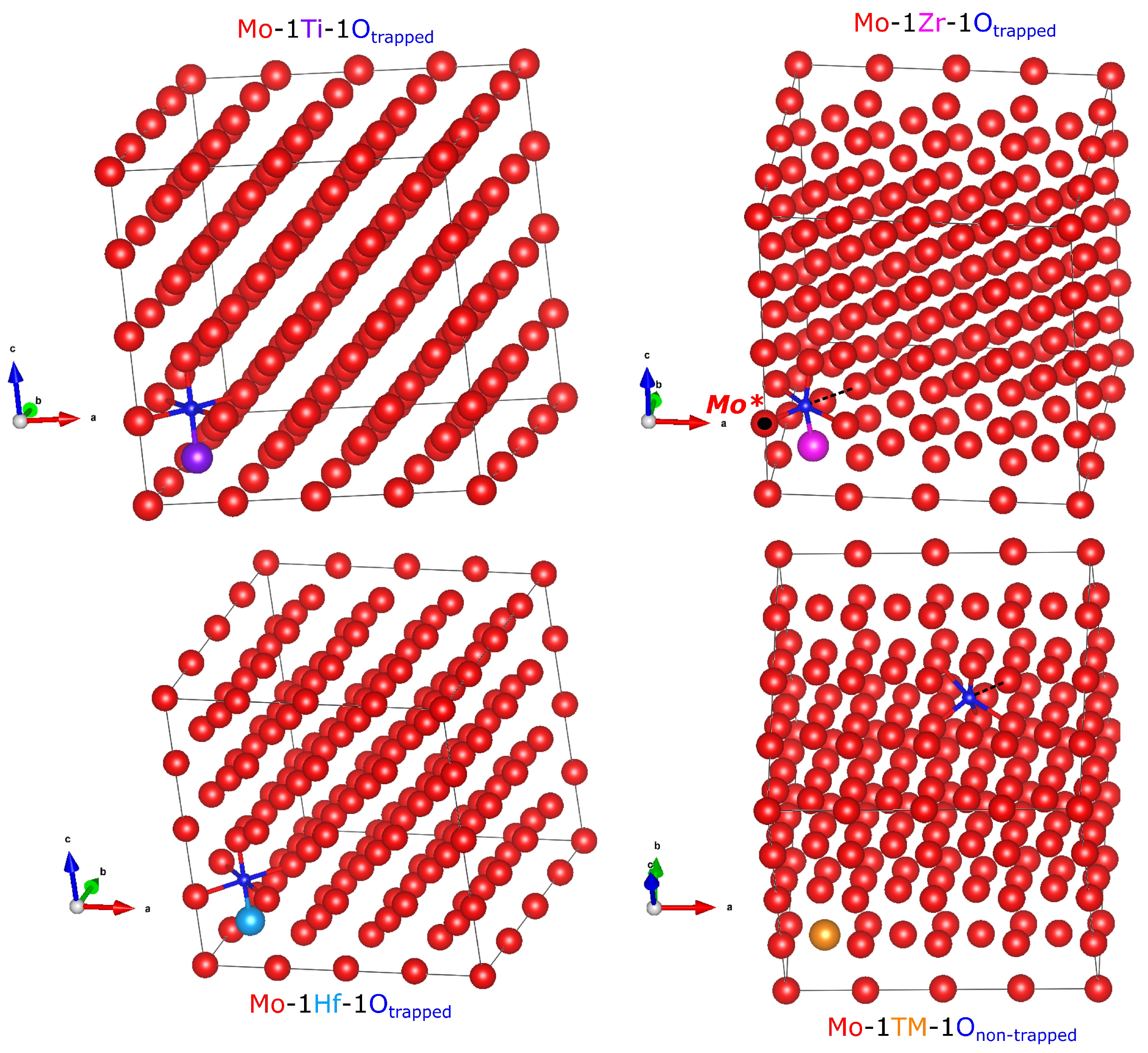
3.3. Relaxed Crystal Structures and Bond Lengths
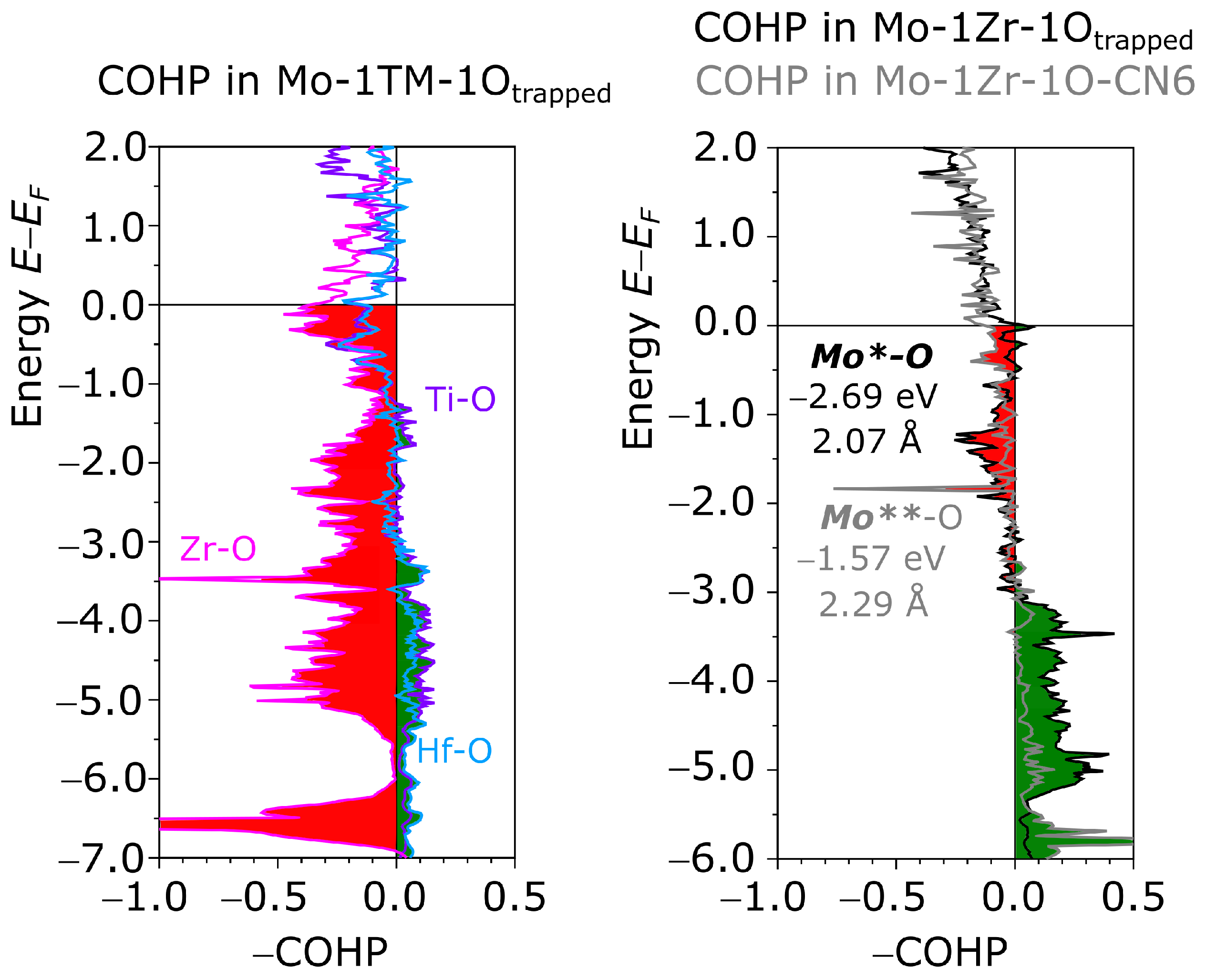
3.4. Chemical Bonding Analysis
3.4.1. Ionic Bonding
3.4.2. Covalent Bonding
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soboyejo, W.; Srivatsan, T. Advanced Structural Materials: Properties, Design Optimization, and Applications; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Schliephake, D.; Kauffmann, A.; Cong, X.; Gombola, C.; Azim, M.; Gorr, B.; Christ, H.J.; Heilmaier, M. Constitution, oxidation and creep of eutectic and eutectoid Mo-Si-Ti alloys. Intermetallics 2019, 104, 133–142. [Google Scholar] [CrossRef]
- Perepezko, J.; Krüger, M.; Heilmaier, M. Mo-Silicide Alloys for High-Temperature Structural Applications. Mater. Perform. Charact. 2021, 10, 122–145. [Google Scholar] [CrossRef]
- Brosse, J.; Fillit, R.; Biscondi, M. Intrinsic Intergranular Brittleness of Molybdenum. Scr. Metall. 1981, 15, 619–623. [Google Scholar] [CrossRef]
- Tsurekawa, S.; Tanaka, T.; Yoshinaga, H. Grain Boundary Structure, Energy and Strength in Molybdenum. Mater. Sci. Eng. A 1994, 176, 341–348. [Google Scholar] [CrossRef]
- Geller, C.B.; Smith, R.W.; Hack, J.E.; Saxe, P.; Wimmer, E. A computational search for ductilizing additives to Mo. Scr. Mater. 2005, 52, 205–210. [Google Scholar] [CrossRef]
- Saage, H.; Krüger, M.; Sturm, D.; Heilmaier, M.; Schneibel, J.; George, E.; Heatherly, L.; Somsen, C.; Eggeler, G.; Yang, Y. Ductilization of Mo–Si solid solutions manufactured by powder metallurgy. Acta Mater. 2009, 57, 3895–3901. [Google Scholar] [CrossRef]
- Kumar, A.; Eyre, B.L. Grain Boundary Segregation and Intergranular Fracture in Molybdenum. Proc. R. Soc. A Math. Phys. Eng. Sci. 1980, 370, 431–458. [Google Scholar] [CrossRef]
- Fichtner, D.; Schmelzer, J.; Yang, W.; Heinze, C.; Krüger, M. Additive Manufacturing of a Near-Eutectic Mo–Si–B Alloy: Processing and Resulting Properties. Intermetallics 2021, 128, 107025. [Google Scholar] [CrossRef]
- Schneibel, J.H.; Brady, M.; Kruzic, J.; Ritchie, R.O. On the Improvement of the Ductility of Molybdenum by Spinel (MgAl2O4) Particles. Int. J. Mater. Res. 2022, 96, 632–637. [Google Scholar] [CrossRef]
- Cockeram, B.V.; Ohriner, E.K.; Byun, T.S.; Miller, M.K.; Snead, L.L. Weldable Ductile Molybdenum Alloy Development. J. Nucl. Mater. 2008, 382, 229–241. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, T.; Jiang, S.; Zhang, B.; Wang, Y.; Feng, J. Microstructure Evolution and Embrittlement of Electron Beam Welded TZM Alloy Joint. Mater. Sci. Eng. A 2017, 700, 512–518. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, Y.; Jiang, S.; Li, X.; Zhang, B.; Feng, J. Stress Relief and Purification Mechanisms for Grain Boundaries of Electron Beam Welded TZM Alloy Joint with Zirconium Addition. J. Mater. Process. Technol. 2018, 251, 168–174. [Google Scholar] [CrossRef]
- Wang, T.; Li, N.; Zhang, Y.; Jiang, S.; Zhang, B.; Wang, Y.; Feng, J. Influence of Welding Speed on Microstructures and Mechanical Properties of Vacuum Electron Beam Welded TZM Alloy Joints. Vacuum 2018, 149, 29–35. [Google Scholar] [CrossRef]
- Morito, F. Effect of Impurities on the Weldability of Powder Metallurgy, Electron-Beam Melted and Arc-Melted Molybdenum and Its Alloys. J. Mater. Sci. 1989, 24, 3403–3410. [Google Scholar] [CrossRef]
- Babinsky, K.; Weidow, J.; Knabl, W.; Lorich, A.; Leitner, H.; Primig, S. Atom Probe Study of Grain Boundary Segregation in Technically Pure Molybdenum. Mater. Charact. 2014, 87, 95–103. [Google Scholar] [CrossRef]
- Seah, M.P.; Hondros, E.D. Grain Boundary Segregation. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1973, 335, 191–212. [Google Scholar] [CrossRef]
- Srivastava, S.C.; Seigle, L.L. Solubility and Thermodynamic Properties of Oxygen in Solid Molybdenum. Metall. Trans. 1974, 5, 49–52. [Google Scholar] [CrossRef]
- Azim, M.A.; Gorr, B.; Christ, H.J.; Heilmaier, M.; Koch, U.; Engelhard, M. Characterization of Oxidation Kinetics of Mo–Si–B-Based Materials. Oxid. Met. 2017, 87, 89–108. [Google Scholar] [CrossRef]
- Ono, K.; Moriyama, J. Deoxidation of High-Melting-Point Metals and Alloys in Vacuum. Metall. Trans. B 1982, 13, 241–249. [Google Scholar] [CrossRef]
- Kurishita, H.; Kitsunai, Y.; Hiraoka, Y.; Shibayama, T.; Kayano, H. Development of Molybdenum Alloy with High Toughness at Low Temperatures. Mater. Trans. Jim 1996, 37, 89–97. [Google Scholar] [CrossRef]
- Hasemann, G.; Bogomol, I.; Schliephake, D.; Loboda, P.; Krüger, M. Microstructure and creep properties of a near-eutectic directionally solidified multiphase Mo–Si–B alloy. Intermetallics 2014, 48, 28–33. [Google Scholar] [CrossRef]
- Stephens, J.R.; Witzke, W.R. Alloy softening in group via metals alloyed with rhenium. J. Less Common Met. 1971, 23, 325–342. [Google Scholar] [CrossRef]
- Stephens, J.R.; Witzke, W.R. Alloy Hardening and Softening in Binary Molybdenum Alloys as Related to Electron Concentration. J. Less Common Met. 1972, 29, 371–388. [Google Scholar] [CrossRef]
- Schneibel, J.H.; Liu, C.T.; Heatherly, L.; Kramer, M.J. Assessment of Processing Routes and Strength of a 3-Phase Molybdenum Boron Silicide (Mo5Si3-Mo5SiB2-Mo3Si). Scr. Mater. 1998, 38, 1169–1176. [Google Scholar] [CrossRef]
- Majumdar, S.; Kapoor, R.; Raveendra, S.; Sinha, H.; Samajdar, I.; Bhargava, P.; Chakravartty, J.K.; Sharma, I.G.; Suri, A.K. A Study of Hot Deformation Behavior and Microstructural Characterization of Mo–TZM Alloy. J. Nucl. Mater. 2009, 385, 545–551. [Google Scholar] [CrossRef]
- Obert, S.; Kauffmann, A.; Heilmaier, M. Characterisation of the Oxidation and Creep Behaviour of Novel Mo-Si-Ti Alloys. Acta Mater. 2020, 184, 132–142. [Google Scholar] [CrossRef]
- Zhou, W.; Tsunoda, K.; Nomura, N.; Yoshimi, K. Effect of Hot Isostatic Pressing on the Microstructure and Fracture Toughness of Laser Additive Manufactured Mosibtic Multiphase Alloy. Mater. Des. 2020, 196, 109132. [Google Scholar] [CrossRef]
- Hinrichs, F.; Kauffmann, A.; Schliephake, D.; Seils, S.; Obert, S.; Ratschbacher, K.; Allen, M.; Pundt, A.; Heilmaier, M. Flexible Powder Production for Additive Manufacturing of Refractory Metal-Based Alloys. Metals 2021, 11, 1723. [Google Scholar] [CrossRef]
- Jéhanno, P.; Heilmaier, M.; Kestler, H. Characterization of an Industrially Processed Mo-based Silicide Alloy. Intermetallics 2004, 12, 1005–1009. [Google Scholar] [CrossRef]
- Suzuki, M.; Nutt, S.R.; Aikin, R.M., Jr. Creep Behavior of an SiC-reinforced XDTM MoSi2 Composite. Mater. Sci. Eng. A-Struct. Mater. Prop. Microstruct. Process. 1993, 162, 73–82. [Google Scholar] [CrossRef]
- Schmelzer, J.; Rittinghaus, S.K.; Weisheit, A.; Stobik, M.; Paulus, J.; Gruber, K.; Wessel, E.; Heinze, C.; Krüger, M. Printability of Gas Atomized Mo-Si-B Powders by Laser Metal Deposition. Int. J. Refract. Met. Hard Mater. 2019, 78, 123–126. [Google Scholar] [CrossRef]
- Floquet, N.; Bertrand, O.; Heizmann, J.J. Structural and Morphological Studies of the Growth of MoO3 Scales during High-Temperature Oxidation of Molybdenum. Oxid. Met. 1992, 37, 253–280. [Google Scholar] [CrossRef]
- Bolbut, V.; Bogomol, I.; Bauer, C.; Krüger, M. Gerichtet erstarrte Mo-Zr-B-Legierungen/Directionally solidified Mo-Zr-B alloys. Mater. Werkst. 2017, 48, 1113–1124. [Google Scholar] [CrossRef]
- Bolbut, V.; Bogomol, I.; Loboda, P.; Krüger, M. Microstructure and mechanical properties of a directionally solidified Mo-12Hf-24B alloy. J. Alloys Compd. 2018, 735, 2324–2330. [Google Scholar] [CrossRef]
- Krüger, M.; Schliephake, D.; Jain, P.; Kumar, K.; Schumacher, G.; Heilmaier, M. Effects of Zr additions on the microstructure and the mechanical behavior of PM Mo-Si-B alloys. JOM 2013, 65, 301–306. [Google Scholar] [CrossRef]
- Krüger, M.; Kauss, O.; Naumenko, K.; Burmeister, C.; Wessel, E.; Schmelzer, J. The potential of mechanical alloying to improve the strength and ductility of Mo–9Si–8B–1Zr alloys—Experiments and simulation. Intermetallics 2019, 113, 106558. [Google Scholar] [CrossRef]
- Scheiber, D.; Pippan, R.; Puschnig, P.; Ruban, A.; Romaner, L. Ab-Initio Search for Cohesion-Enhancing Solute Elements at Grain Boundaries in Molybdenum and Tungsten. Int. J. Refract. Met. Hard Mater. 2016, 60, 75–81. [Google Scholar] [CrossRef]
- Scheiber, D.; Romaner, L.; Pippan, R.; Puschnig, P. Impact of Solute-Solute Interactions on Grain Boundary Segregation and Cohesion in Molybdenum. Phys. Rev. Mater. 2018, 2, 093609. [Google Scholar] [CrossRef]
- Ma, H.; Ding, X.; Zhang, L.; Sun, Y.; Liu, T.; Ren, Q.; Liao, Y. Segregation of Interstitial Light Elements at Grain Boundaries in Molybdenum. Mater. Today Commun. 2020, 25, 101388. [Google Scholar] [CrossRef]
- Reynolds, C.; Pollock, T.M.; Van Der Ven, A. Solute-Solute Interactions in Dilute Nb-X-O Alloys from First Principles. Acta Mater. 2024, 266, 119621. [Google Scholar] [CrossRef]
- Yu, X.; Li, Z.; Jain, P.; Gao, H.; Kumar, S. Effect of Si content on the uniaxial tensile behavior of Mo-Si solid solution alloys. Acta Mater. 2021, 207, 116654. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Physics Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Physics Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Dal Corso, A. Pseudopotentials periodic table: From H to Pu. Comput. Mater. Sci. 2014, 95, 337–350. [Google Scholar] [CrossRef]
- Marzari, N.; Vanderbilt, D.; De Vita, A.; Payne, M.C. Thermal Contraction and Disordering of the Al(110) Surface. Phys. Rev. Lett. 1999, 82, 3296–3299. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Yu, M.; Trinkle, D.R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 2011, 134, 064111. [Google Scholar] [CrossRef]
- de-la Roza, A.O.; Blanco, M.; Pendás, A.M.; Luaña, V. Critic: A new program for the topological analysis of solid-state electron densities. Comput. Phys. Commun. 2009, 180, 157–166. [Google Scholar] [CrossRef]
- de-la Roza, A.O.; Johnson, E.R.; Luaña, V. Critic2: A program for real-space analysis of quantum chemical interactions in solids. Comput. Phys. Commun. 2014, 185, 1007–1018. [Google Scholar] [CrossRef]
- Andersen, O.; Skriver, H.; Nohl, H.; Johansson, B. Electronic structure of transition metal compounds; Ground-state properties of the 3d-monoxides in the atomic sphere approximation. Pure Appl. Chem. VI 1980, 52, 93–118. [Google Scholar] [CrossRef]
- Andersen, O.K.; Jepsen, O. Explicit, First-Principles Tight-Binding Theory. Phys. Rev. Lett. 1984, 53, 2571–2574. [Google Scholar] [CrossRef]
- Jepsen, O.; Andersen, O. TB-LMTO-ASA (version 47), 2000.
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blöchl, P. Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Maroef, I.; Olson, D.L.; Eberhart, M.; Edwards, G.R. Hydrogen trapping in ferritic steel weld metal. Int. Mater. Rev. 2002, 47, 191–223. [Google Scholar] [CrossRef]
- Slater, J.C. Atomic Radii in Crystals. J. Chem. Phys. 2004, 41, 3199–3204. [Google Scholar] [CrossRef]
- Pearson, R. Absolute Electronegativity and Hardness: Application to Inorganic Chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mo-1Ti | Mo-1Zr | Mo-1Hf | |
|---|---|---|---|
| (eV per cell) |
| Mo-1Ti-1O | Mo-1Zr-1O | Mo-1Hf-1O | |
|---|---|---|---|
| (kJ/mol per atom) | |||
| (eV per cell) | |||
| (eV per cell) |
| Mo-1Ti-1O | Mo-1Zr-1O | Mo-1Hf-1O | Mo-1O | |
|---|---|---|---|---|
| 1 | ||||
| 2 | ||||
| 6 | 6 | |||
| Bader Charge Mo (e) 1 | Bader Charge TM (e) | Bader Charge O (e) | |
|---|---|---|---|
| Mo-1Ti-1Otrapped | 0.21 | ||
| Mo-1Ti-1Onontrapped | 0.24 | ||
| Mo-1Ti | +0.05 | +1.06 | |
| Mo-1Zr-1Otrapped | 0.26 | ||
| Mo-1Zr-1Onontrapped | 0.24 | ||
| Mo-1Zr | +0.05 | ||
| Mo-1Hf-1Otrapped | 0.24 | ||
| Mo-1Hf-1Onontrapped | 0.24 | ||
| Mo-1Hf | +0.05 | ||
| Mo-1O | <0.25 |
| Mo-1Ti-1O | Mo-1Zr-1O | Mo-1Hf-1O | |
|---|---|---|---|
| (eV per cell) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Touzani, R.S.; Nizinkovskyi, R.; Krüger, M. Effect of Oxygen in Mo-TM (TM = Ti, Zr, Hf) Solid Solutions as Studied with Density Functional Theory Calculations. Crystals 2024, 14, 213. https://doi.org/10.3390/cryst14030213
Touzani RS, Nizinkovskyi R, Krüger M. Effect of Oxygen in Mo-TM (TM = Ti, Zr, Hf) Solid Solutions as Studied with Density Functional Theory Calculations. Crystals. 2024; 14(3):213. https://doi.org/10.3390/cryst14030213
Chicago/Turabian StyleTouzani, Rachid Stefan, Rostyslav Nizinkovskyi, and Manja Krüger. 2024. "Effect of Oxygen in Mo-TM (TM = Ti, Zr, Hf) Solid Solutions as Studied with Density Functional Theory Calculations" Crystals 14, no. 3: 213. https://doi.org/10.3390/cryst14030213
APA StyleTouzani, R. S., Nizinkovskyi, R., & Krüger, M. (2024). Effect of Oxygen in Mo-TM (TM = Ti, Zr, Hf) Solid Solutions as Studied with Density Functional Theory Calculations. Crystals, 14(3), 213. https://doi.org/10.3390/cryst14030213