

Effect of Mn+2 Doping and Vacancy on the Ferromagnetic Cubic 3C-SiC Structure Using First Principles Calculations


Abstract
1. Introduction
2. Computational Method
3. Results and Discussion
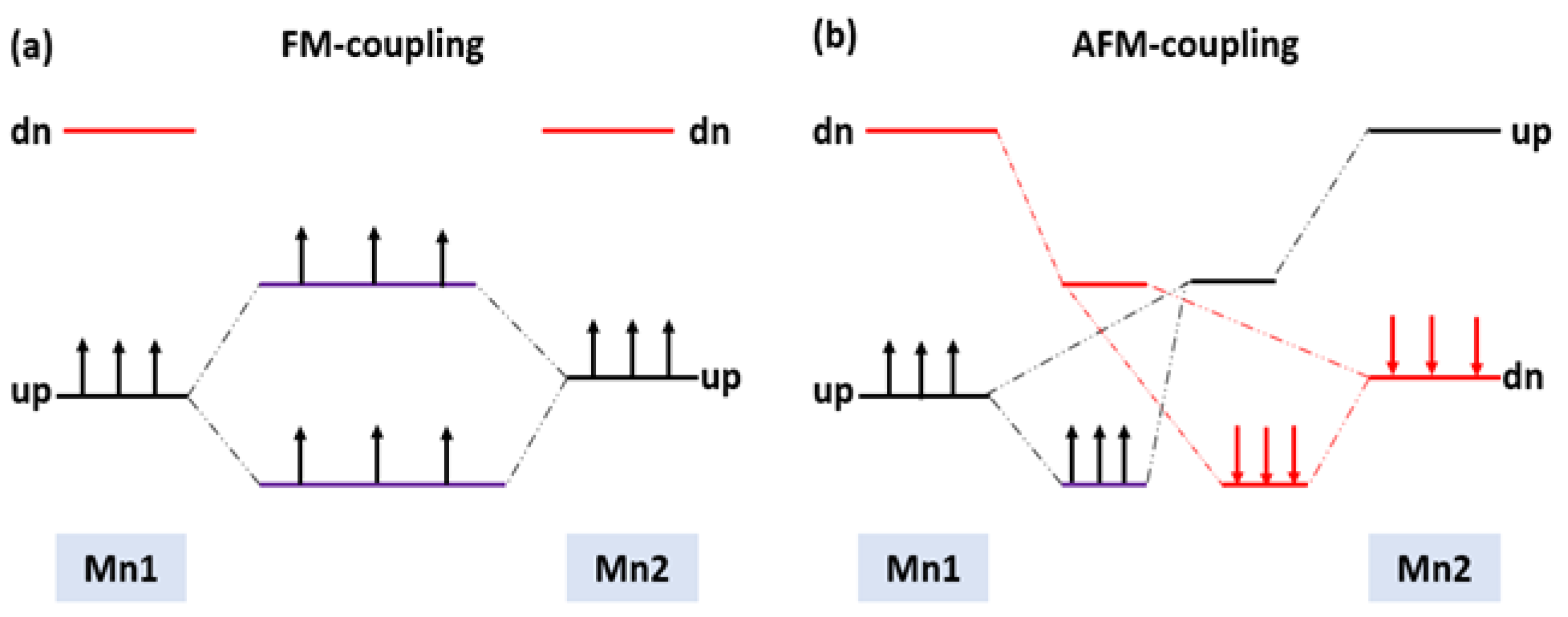
3.1. Spins Coupling of Pure (Mn+2)-Doped 3C-SiC
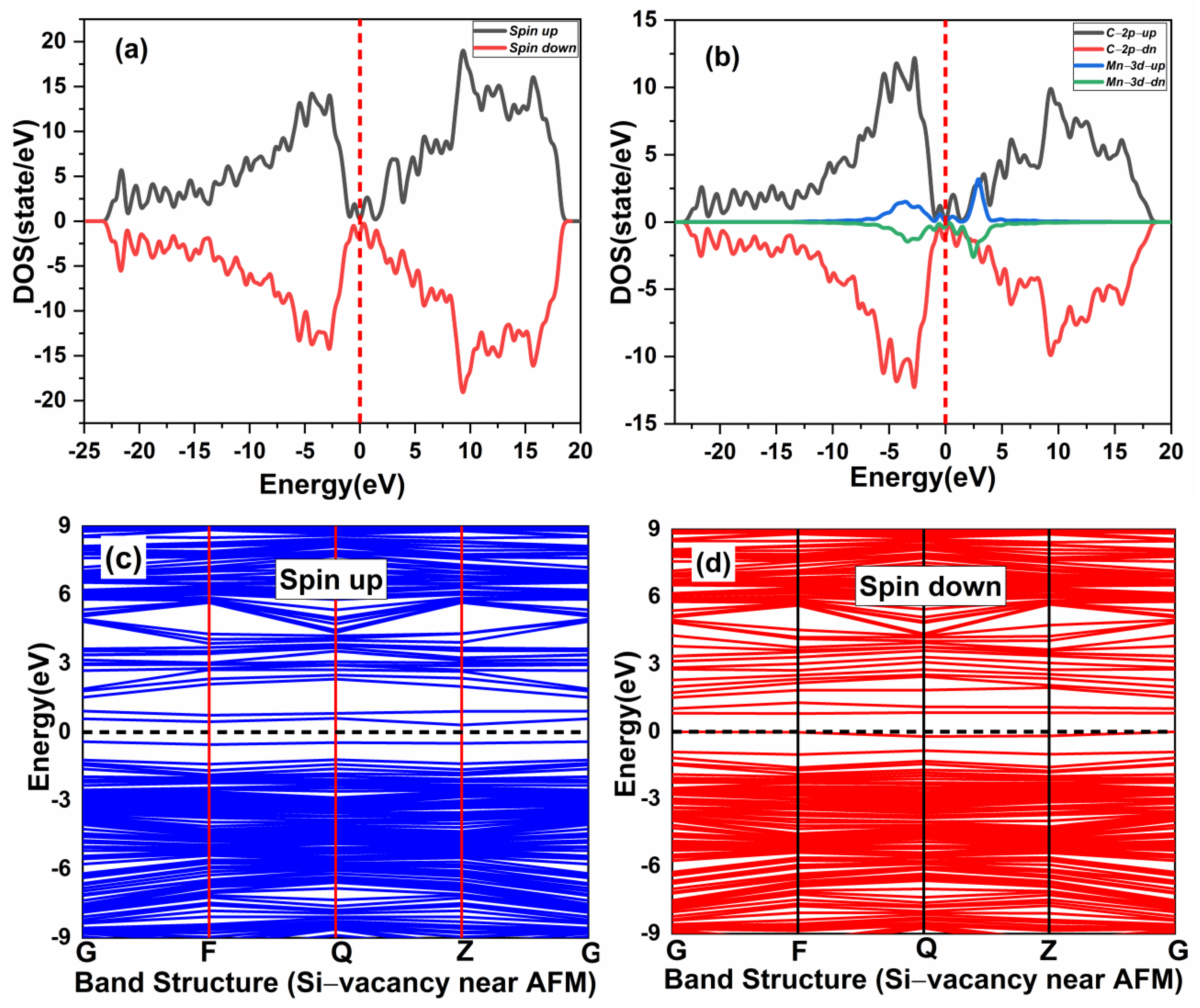
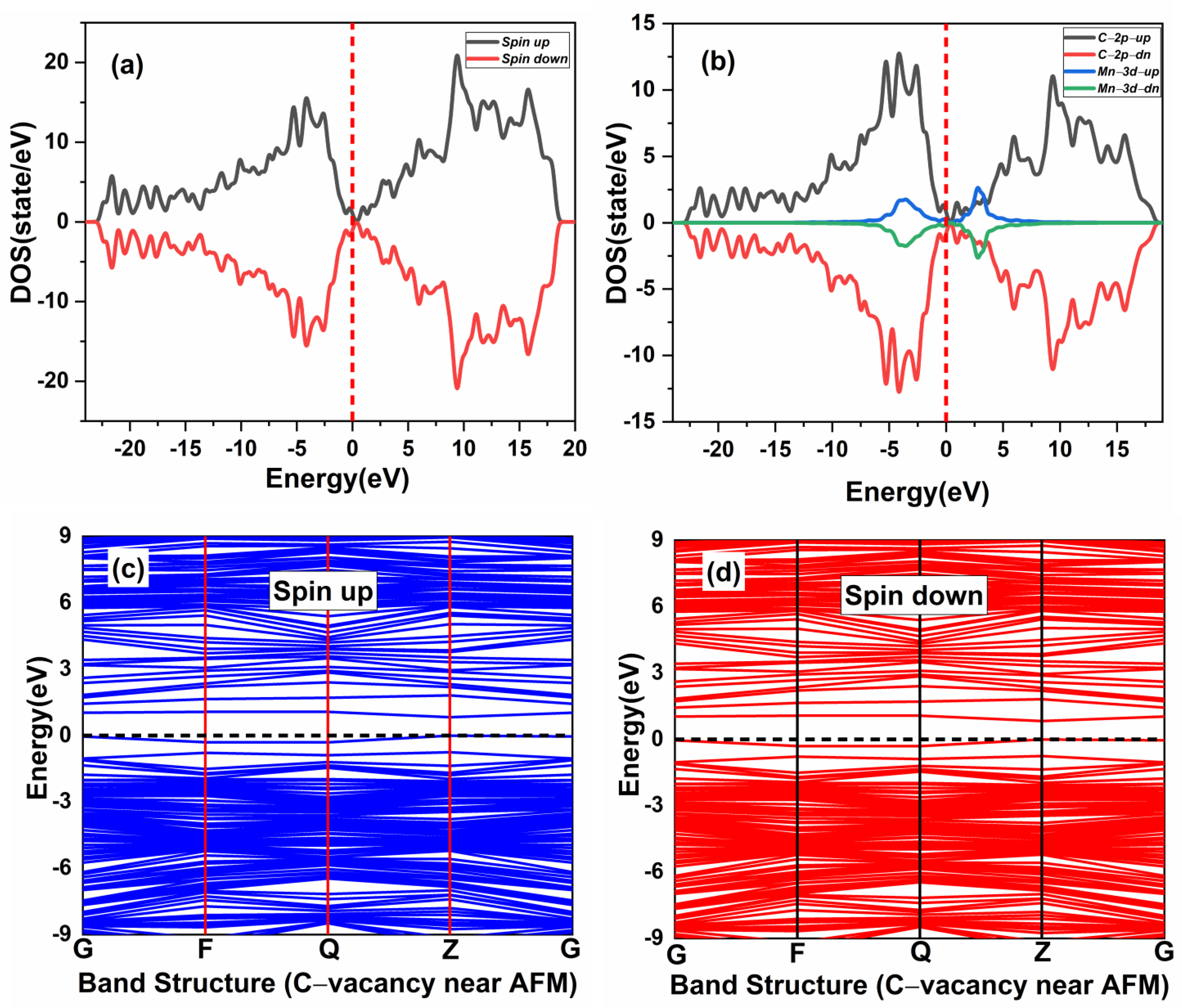
3.2. Magnetic Coupling of Mn Doped 3C-SiC with Native Defect
3.3. Curie Temperature Estimation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wolf, S.A.; Awschalom, D.D.; Buhrman, R.A.; Daughton, J.M.; von Molnár, S.; Roukes, M.L.; Chtchelkanova, A.Y.; Treger, D.M. Spintronics: A Spin-Based Electronics Vision for the Future. Science 2001, 294, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Ohno, H. Making Nonmagnetic Semiconductors Ferromagnetic. Science 1998, 281, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Dietl, T.; Ohno, H.; Matsukura, F.; Cibert, J.; Ferrand, D. Zener Model Description of Ferromagnetism in Zinc-Blende Magnetic Semiconductors. Science 2000, 287, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, T.; Sinova, J.; Mašek, J.; Kučera, J.; MacDonald, A.H. Theory of ferromagnetic (III,Mn)V semiconductors. Rev. Mod. Phys. 2006, 78, 809–864. [Google Scholar] [CrossRef]
- Griffin, K.A.; Pakhomov, A.B.; Wang, C.M.; Heald, S.M.; Krishnan, K.M. Intrinsic Ferromagnetism in Insulating Cobalt Doped Anatase TiO2. Phys. Rev. Lett. 2005, 94, 157204. [Google Scholar] [CrossRef]
- Hong, N.H.; Sakai, J.; Prellier, W.; Hassini, A.; Ruyter, A.; Gervais, F. Ferromagnetism in transition-metal-doped TiO2 thin films. Phys. Rev. B 2004, 70, 195204. [Google Scholar] [CrossRef]
- Liu, X.; Lin, F.; Sun, L.; Cheng, W.; Ma, X.; Shi, W. Doping concentration dependence of room-temperature ferromagnetism for Ni-doped ZnO thin films prepared by pulsed-laser deposition. Appl. Phys. Lett. 2006, 88, 062508. [Google Scholar] [CrossRef]
- Javid, H.; Aldaghfag, S.A.; Butt, M.K.; Mubashir, S.; Yaseen, M.; Ishfaq, M.; Saleem, S.; Ali, H.E.; Hegazy, H.H. Physical characteristics of LaCrxAl1−xO3: DFT approach. J. Ovonic Res. 2022, 18, 481–489. [Google Scholar] [CrossRef]
- Arshad, M.; Yaseen, M.; Aldaghfag, S.A.; Saleem, S.; Ishfaq, M.; Nazar, M.; Yousef, E.; Hegazy, H.H. Physical characteristics of Pb1−xAxSe (A = Fe, Mn, V) for spintronic applications. Chalcogenide Lett. 2022, 19, 553–563. [Google Scholar] [CrossRef]
- Katayama-Yoshida, H.; Sato, K. Spin and charge control method of ternary II–VI and III–V magnetic semiconductors for spintronics: Theory vs. experiment. J. Phys. Chem. Solids 2003, 64, 1447–1452. [Google Scholar] [CrossRef]
- Saito, H.; Zayets, V.; Yamagata, S.; Ando, K. Room-Temperature Ferromagnetism in a II–VI Diluted Magnetic Semiconductor Zn1−xCrxTe. Phys. Rev. Lett. 2003, 90, 207202. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.W. Antiferromagnetism. Theory of Superexchange Interaction. Phys. Rev. 1950, 79, 350. [Google Scholar] [CrossRef]
- Ysl, P.; Ew, C.R.E. Interaction Between the d SheHs in the Transition Metals. Phys. Rev. 1951, 81, 440. [Google Scholar]
- Tian, Z.; Yuan, S.; He, J.; Li, P.; Zhang, S.; Wang, C.; Wang, Y.; Yin, S.; Liu, L. Structure and magnetic properties in Mn doped SnO2 nanoparticles synthesized by chemical co-precipitation method. J. Alloy. Compd. 2008, 466, 26–30. [Google Scholar] [CrossRef]
- Kaminski, A.; Das Sarma, S. Polaron Percolation in Diluted Magnetic Semiconductors. Phys. Rev. Lett. 2002, 88, 247202. [Google Scholar] [CrossRef]
- Xun, Q.; Xun, B.; Li, Z.; Wang, P.; Cai, Z. Application of SiC power electronic devices in secondary power source for aircraft. Renew. Sustain. Energy Rev. 2017, 70, 1336–1342. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, X.; Tolbert, L.M.; Wang, F.; Liang, Z.; Costinett, D.; Blalock, B.J. A high temperature silicon carbide mosfet power module with integrated silicon-on-insulator-based gate drive. IEEE Trans. Power Electron. 2014, 30, 1432–1445. [Google Scholar] [CrossRef]
- Goela, J.S.; Brese, N.E.; Burns, L.E.; Pickering, M.A. High-Thermal-Conductivity SiC and Applications. In High Thermal Conductivity Materials; Springer-Verlag: Berlin, Germany, 2006; pp. 167–198. [Google Scholar] [CrossRef]
- Sultan, N.M.; Albarody, T.M.B.; Al-Jothery, H.K.M.; Abdullah, M.A.; Mohammed, H.G.; Obodo, K.O. Thermal Expansion of 3C-SiC Obtained from In-Situ X-ray Diffraction at High Temperature and First-Principal Calculations. Materials 2022, 15, 6229. [Google Scholar] [CrossRef]
- Amy, F.; Douillard, L.; Aristov, V.Y.; Soukiassian, P. Oxynitridation of cubic silicon carbide (100) surfaces. J. Vac. Sci. Technol. A 1999, 17, 2629–2633. [Google Scholar] [CrossRef]
- Fraga, M.A.; Bosi, M.; Negri, M. Silicon Carbide in Microsystem Technology—Thin Film Versus Bulk Material. In Advanced Silicon Carbide Devices and Processing; Saddow, S.E., Via, F.L., Eds.; InTech: London, UK, 2015. [Google Scholar] [CrossRef]
- Gorin, S.N.; Ivanova, L.M. Cubic Silicon Carbide (3C-SiC): Structure and Properties of Single Crystals Grown by Thermal Decomposition of Methyl Trichlorosilane in Hydrogen. Phys. Status Solidi B 1997, 202, 221–245. [Google Scholar] [CrossRef]
- Zheng, H.W.; Wang, Z.Q.; Liu, X.Y.; Diao, C.L.; Zhang, H.R.; Gu, Y.Z. Local structure and magnetic properties of Mn-doped 3C-SiC nanoparticles. Appl. Phys. Lett. 2011, 99, 222512. [Google Scholar] [CrossRef]
- Ma, S.; Sun, Y.; Zhao, B.; Tong, P.; Zhu, X.; Song, W. Magnetic properties of Mn-doped cubic silicon carbide. Phys. B Condens. Matter 2007, 394, 122–126. [Google Scholar] [CrossRef]
- Sun, X.; Guo, R.; An, Y.; Liu, J. Investigation of structure, magnetic, and transport properties of Mn-doped SiC films. J. Vac. Sci. Technol. A 2013, 31, 41507. [Google Scholar] [CrossRef]
- Bouziane, K.; Al Azri, M.; Elzain, M.; Chérif, S.; Mamor, M.; Declémy, A.; Thomé, L. Mn fraction substitutional site and defects induced magnetism in Mn-implanted 6H-SiC. J. Alloy. Compd. 2015, 632, 760–765. [Google Scholar] [CrossRef]
- Al Azri, M.; Elzain, M.; Bouziane, K.; Chérif, S.M.; Roussigné, Y.; Declemy, A.; Drouet, M.; Thomé, L. Magnetic properties of Mn-implanted 6H-SiC single crystal. J. Appl. Phys. 2012, 111, 07C315. [Google Scholar] [CrossRef]
- Lu, X.; Yang, P.; Luo, J.; Guo, X.; Ren, J.; La, P. Density functional theory capture of electronic structures and optical properties of vacancy doped 3C-SiC systems. Mater. Res. Express 2019, 6, 115905. [Google Scholar] [CrossRef]
- Al Azri, M.; Elzain, M.; Bouziane, K.; Chérif, S.M. First principal calculation of the electronic and magnetic properties of Mn-doped 6H-SiC. Eur. Phys. J. B 2013, 86, 402. [Google Scholar] [CrossRef]
- Takano, F.; Wang, W.; Ofuchi, H.; Ohshima, T.; Akinaga, H.; Caldas, M.; Studart, N. Structural and Magnetic Properties of Mn-Doped SiC. AIP Conf. Proc. 2010, 1199, 443. [Google Scholar] [CrossRef]
- Tang, S.-A.; Mao, F.; Zhao, X.-D.; Zhang, C. First-principles study of the ferromagnetism of Mn-doped 3C-SiC. Solid State Commun. 2020, 321, 114039. [Google Scholar] [CrossRef]
- Houmad, M.; Dakir, O.; Benzidi, H.; Mounkachi, O.; El Kenz, A.; Benyoussef, A. Magnetic behavior of Mn-doped silicon carbide nanosheet. Int. J. Mod. Phys. B 2017, 31, 1750163. [Google Scholar] [CrossRef]
- Sato, K.; Katayama-Yoshida, H. Ab initio Study on the Magnetism in ZnO-, ZnS-, ZnSe-and ZnTe-Based Diluted Magnetic Semiconductors. Phys. Stat. Sol. 2002, 229, 673–680. [Google Scholar] [CrossRef]
- Iuşan, D.; Sanyal, B.; Eriksson, O. Role of defects on the magnetic interactions in Mn-doped ZnO. Phys. Status Solidi A 2007, 204, 53–60. [Google Scholar] [CrossRef]
- Kittilstved, K.; Liu, W.K.; Gamelin, D.R. Electronic structure origins of polarity-dependent high-TC ferromagnetism in oxide-diluted magnetic semiconductors. Nat. Mater. 2006, 5, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Shi, L.-J.; Abdalla, A.; Zou, B.; Ikram, M. Computational insights into electronic, magnetic and optical properties of Mn(II)-doped ZnTe with and without vacancy defects. Mater. Sci. Semicond. Process. 2022, 150, 106965. [Google Scholar] [CrossRef]
- Lin, X.-L.; Zhang, H.-X.; Yang, W.-X.; Chen, H.-M.; Pan, F.-C. The high Curie temperature and long-range ferromagnetism in Mn-doped 3C-SiC: A study using first-principles calculation. J. Korean Phys. Soc. 2021, 79, 546–551. [Google Scholar] [CrossRef]
- Moharana, G.P.; Singh, S.K.; Narayanan, H.K. Ferromagnetism in Mn doped 3C-SiC: A variable temperature ESR study. J. Magn Magn Mater. 2021, 527, 167707. [Google Scholar] [CrossRef]
- Izadifard, M.; Ghazi, M.E.; Hosaini, S. Effects of Mn Substitution on Magnetic and Electronic Properties of β-SiC Semiconductor. Appl. Phys. Res. 2010, 2, 2. [Google Scholar] [CrossRef]
- Patnaik, P.; Mukhopadhyay, G.; Singh, P.P.; Garg, A.B.; Mittal, R.; Mukhopadhyay, R. Magnetism in Transition metal doped Cubic SiC. AIP Conf. Proc. 2011, 1349, 1087. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Ouma, C.N.M.; Singh, S.; Obodo, K.O.; Amolo, G.O.; Romero, A.H. Controlling the magnetic and optical responses of a MoS2 monolayer by lanthanide substitutional doping: A first-principles study. Phys. Chem. Chem. Phys. 2017, 19, 25555–25563. [Google Scholar] [CrossRef]
- Mulwa, W.M.; Ouma, C.N.; Onani, M.O.; Dejene, F.B. Energetic, electronic and optical properties of lanthanide doped TiO2: An ab initio LDA+U study. J. Solid State Chem. 2016, 237, 129–137. [Google Scholar] [CrossRef]
- Obodo, K.; Chetty, N. First principles LDA +Uand GGA +Ustudy of protactinium and protactinium oxides: Dependence on the effectiveUparameter. J. Phys. Condens. Matter 2013, 25, 145603. [Google Scholar] [CrossRef]
- Obodo, K.; Chetty, N. GGA+U studies of the early actinide mononitrides and dinitrides. J. Nucl. Mater. 2013, 442, 235–244. [Google Scholar] [CrossRef]
- Lin, L.; Yao, L.; Li, S.; Shi, Z.; Xie, K.; Huang, J.; Tao, H.; Zhang, Z. Influence of Mn and Co doping on optical and magnetic properties in 3C–SiC. J. Phys. Chem. Solids 2021, 153, 110002. [Google Scholar] [CrossRef]
- Cockayne, E.; Levin, I.; Wu, H.; Llobet, A. Magnetic structure of bixbyite α-Mn2O3: A combined DFT+U and neutron diffraction study. Phys. Rev. B 2013, 87, 184413. [Google Scholar] [CrossRef]
- Fletcher, R. Practical Methods of Optimization; Wiley: New York, NY, USA, 1981; Volume 1. [Google Scholar]
- Aradi, B.; Gali, A.; Deák, P.; Lowther, J.E.; Son, N.T.; Janzén, E.; Choyke, W.J. Ab initio density-functional supercell calculations of hydrogen defects in cubic SiC. Phys. Rev. B 2001, 63, 245202. [Google Scholar] [CrossRef]
- Madelung, O.; Schulz, M.; Weiss, H. Numerical Data and Functional Relationships in Science and Technology, 2 nd ed; Springer: New York, NY, USA, 1982. [Google Scholar]
- Dou, Y.; Jin, H.-B.; Cao, M.; Fang, X.; Hou, Z.; Li, D.; Agathopoulos, S. Structural stability, electronic and optical properties of Ni-doped 3C–SiC by first principles calculation. J. Alloy. Compd. 2011, 509, 6117–6122. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, Z.; Li, Y.; Ouyang, X. The mechanical and thermodynamic properties of β-Si1−xC. RSC Adv. 2017, 7, 28499–28505. [Google Scholar] [CrossRef]
- Gholami, M.; Golsanamlou, Z.; Soleimani, H.R. Effects of 3D transition metal impurities and vacancy defects on electronic and magnetic properties of pentagonal Pd2S4: Competition between exchange splitting and crystal fields. Sci. Rep. 2022, 12, 10838. [Google Scholar] [CrossRef] [PubMed]
- Schultz, P.A.; Van Ginhoven, R.M.; Edwards, A.H. Theoretical study of intrinsic defects in cubic silicon carbide 3C-SiC. Phys. Rev. B 2021, 103, 195202. [Google Scholar] [CrossRef]
- Yang, J.; Song, W.H.; Ma, Y.Q.; Zhang, R.L.; Zhao, B.C.; Sheng, Z.G.; Zheng, G.H.; Dai, J.M.; Sun, Y.P. Structural, magnetic, and transport properties in thePr-doped manganites La0.9−xPrxTe0.1MnO3 (0 ≤ x ≤ 0.9). Phys. Rev. B 2004, 70, 144421. [Google Scholar] [CrossRef]
- Song, B.; Bao, H.; Li, H.; Lei, M.; Jian, J.; Han, J.; Zhang, X.; Meng, S.; Wang, W.; Chen, X. Magnetic properties of Mn-doped 6H-SiC. Appl. Phys. Lett. 2009, 94, 102508. [Google Scholar] [CrossRef]
- Bolduc, M.; Awo-Affouda, C.; Stollenwerk, A.; Huang, M.B.; Ramos, F.G.; Agnello, G.; LaBella, V.P. Above room temperature ferromagnetism in Mn-ion implanted Si. Phys. Rev. B 2005, 71, 033302. [Google Scholar] [CrossRef]
- Moharana, G.P.; Singh, S.; Babu, P.; Narayanan, H.K. Investigation of magnetic order and spin dynamics in Mn doped 3C-SiC. J. Alloy. Compd. 2018, 765, 314–323. [Google Scholar] [CrossRef]
- Chattopadhyay, M.K.; Roy, S.B.; Chaddah, P. Kinetic arrest of the first-order ferromagnetic-to-antiferromagnetic transition in Ce(Fe0.96Ru0.04)2: Formation of a magnetic glass. Phys. Rev. B 2005, 72, 180401. [Google Scholar] [CrossRef]
- Sokhey, K.; Chattopadhyay, M.; Nigam, A.; Roy, S.; Chaddah, P. Signatures of phase separation across the disorder broadened first order ferromagnetic to antiferromagnetic transition in doped-CeFe2 alloys. Solid State Commun. 2004, 129, 19–23. [Google Scholar] [CrossRef]
- Roy, S.B.; Perkins, G.K.; Chattopadhyay, M.K.; Nigam, A.K.; Sokhey, K.J.S.; Chaddah, P.; Caplin, A.D.; Cohen, L.F. First Order Magnetic Transition in Doped CeFe2 Alloys: Phase Coexistence and Metastability. Phys. Rev. Lett. 2004, 92, 147203. [Google Scholar] [CrossRef]
- Chattopadhyay, M.K.; Roy, S.B.; Nigam, A.K.; Sokhey, K.J.S.; Chaddah, P. Metastability and giant relaxation across the ferromagnetic to antiferromagnetic transition in Ce(Fe0.96Ru0.04)2. Phys. Rev. B 2003, 68, 174404. [Google Scholar] [CrossRef]
- Galanakis, I. Slater-Pauling Behavior in Half-Metallic Magnets. J. Surfaces Interfaces Mater. 2014, 2, 74–78. [Google Scholar] [CrossRef]
- Tan, Z.; Xiao, W.; Wang, L.; Yang, Y. Magnetic properties of ZnS doped with noble metals (X = Ru, Rh, Pd, and Ag). J. Appl. Phys. 2012, 112, 123920. [Google Scholar] [CrossRef]
- Turek, I.; Kudrnovsky, J.; Diviš, M.; Franek, P.; Bihlmayer, G.; Blügel, S. First-principles study of the electronic structure and exchange interactions in bcc europium. Phys. Rev. B 2003, 68, 224431. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Chung, Y.-C.; Yi, S.-C. Electronic structure and half-metallic property of Mn-doped β-SiC diluted magnetic semiconductor. Mater. Sci. Eng. B 2006, 126, 194–196. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
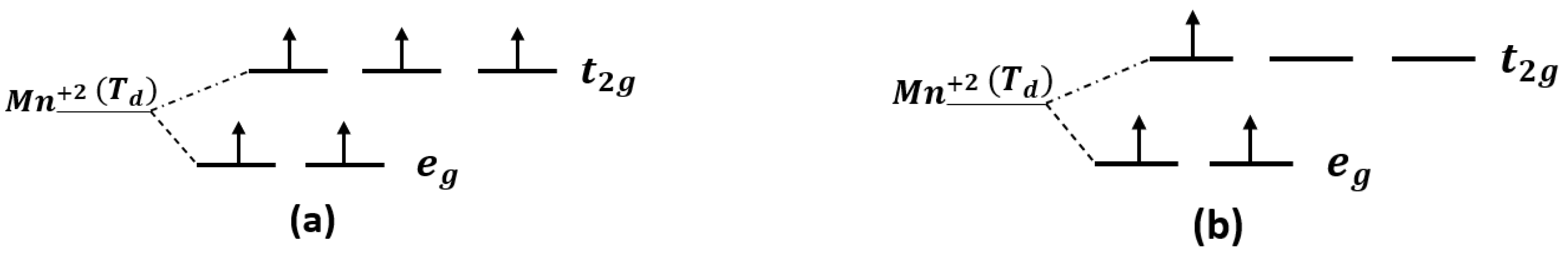{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
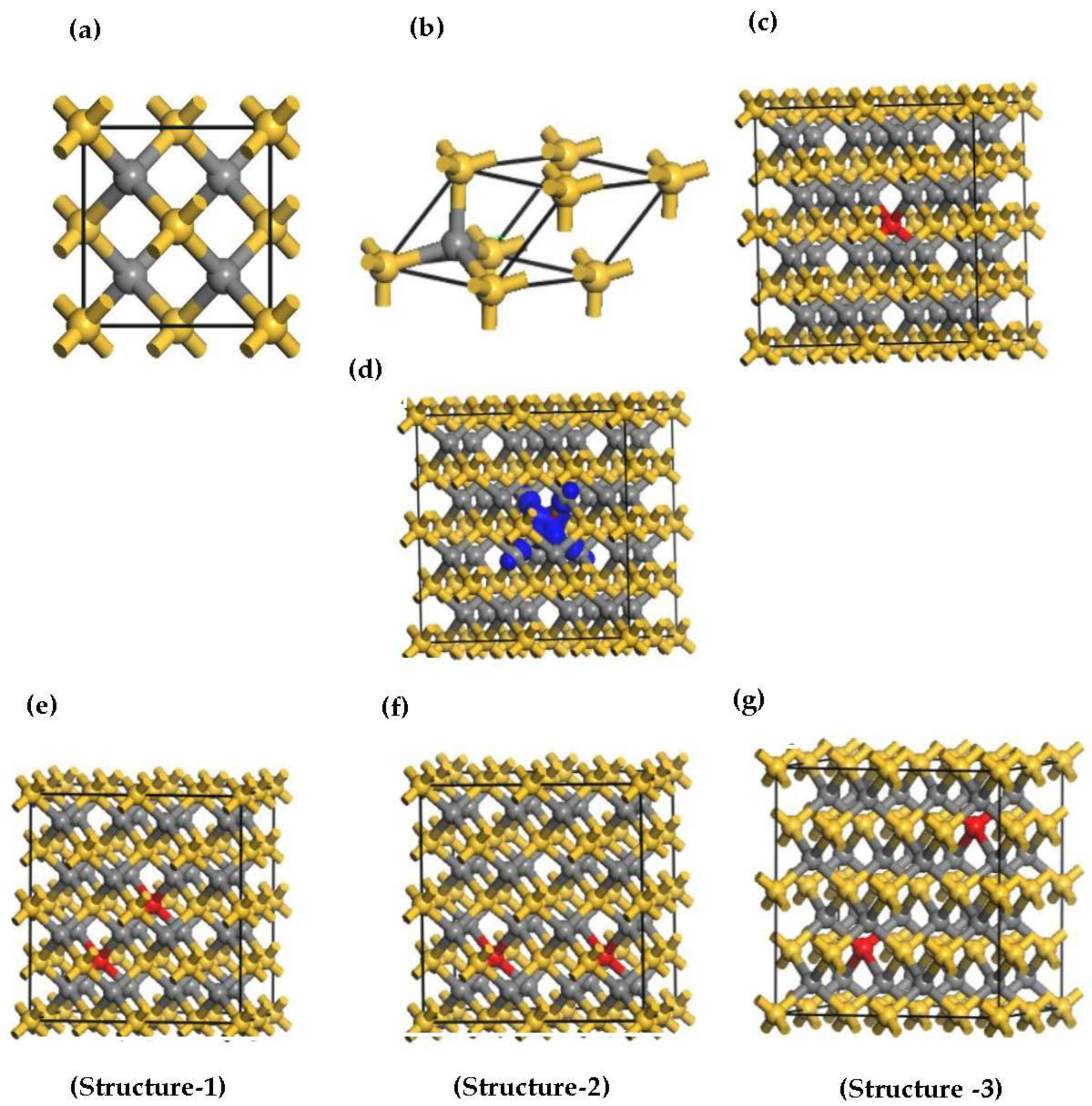
| Configuration | (Å) | (meV) | (meV) | Coupling | |
|---|---|---|---|---|---|
| Before | After | ||||
| Structure -1 | 4.348 | 4.364 | 2.88 | −218 | FM |
| Structure -2 | 5.325 | 5.345 | 3.75 | −259 | FM |
| Structure -3 | 6.149 | 6.171 | 4.83 | −290 | FM |
| Type of Doping | E (Nearby Vacancy) | E (Far Vacancy) |
|---|---|---|
| (Mn+2)-doped 3C-SiC + | 398 meV | 239 meV |
| (Mn+2)-doped 3C-SiC + | 227 meV | −405 meV |
| Type of Doping | Phase Type | Band Structure Type | |
|---|---|---|---|
| Pure 3C-SiC | Non-magnetic (NM) | 0 | 2.379 eV Semiconductor |
| Single (Mn+2)-doped 3C-SiC | FM | 3 | Half-metallic |
| (Mn+2)-(Mn+2)-doped 3C-SiC | FM | 6 | Half metallic |
| (Mn+2)-(Mn+2)-doped 3C-SiC + (Nearby) | AFM | 0 | 0.317 eV Semiconductor |
| (Mn+2)-(Mn+2)-doped 3C-SiC + (Far) | AFM | 0 | Half metallic |
| (Mn+2)-(Mn+2)-doped 3C-SiC + (Nearby) | AFM | 0 | 0.828 eV Semiconductor |
| (Mn+2)-(Mn+2)-doped 3C-SiC + (Far) | FM | 8 | Half metallic |
| Type Configuration | Curie Temperature (k) |
|---|---|
| structure-1 | 843 |
| structure-2 | 1000 |
| Structure-3 | 1121 |
| Nearby Si-vacancy | - |
| Far Si-Vacacny | - |
| Nearby C-vacancy | - |
| Far C-vacancy | 1566 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultan, N.M.; Albarody, T.M.B.; Obodo, K.O.; Baharom, M.B. Effect of Mn+2 Doping and Vacancy on the Ferromagnetic Cubic 3C-SiC Structure Using First Principles Calculations. Crystals 2023, 13, 348. https://doi.org/10.3390/cryst13020348
Sultan NM, Albarody TMB, Obodo KO, Baharom MB. Effect of Mn+2 Doping and Vacancy on the Ferromagnetic Cubic 3C-SiC Structure Using First Principles Calculations. Crystals. 2023; 13(2):348. https://doi.org/10.3390/cryst13020348
Chicago/Turabian StyleSultan, Najib M., Thar M. Badri Albarody, Kingsley Onyebuchi Obodo, and Masri B. Baharom. 2023. "Effect of Mn+2 Doping and Vacancy on the Ferromagnetic Cubic 3C-SiC Structure Using First Principles Calculations" Crystals 13, no. 2: 348. https://doi.org/10.3390/cryst13020348
APA StyleSultan, N. M., Albarody, T. M. B., Obodo, K. O., & Baharom, M. B. (2023). Effect of Mn+2 Doping and Vacancy on the Ferromagnetic Cubic 3C-SiC Structure Using First Principles Calculations. Crystals, 13(2), 348. https://doi.org/10.3390/cryst13020348