Abstract
This study investigated the crystallographic and electronic properties of 1,5-benzodiazepine compounds, namely: cis-(3S,4S)-3-hydroxy-7,8-dimethyl-4-phenyl-1,3,4,5-tetrahydro-1,5-benzodiazepin-2-one 3b, trans-(3R,4R)-1-ethyl-3-hydroxy-7,8-dimethyl-4-phenyl-1,3,4,5-tetrahydro-2H-1,5-benzodiazepin-2-one 4, and trans-(3S,4S) 1-ethyl-3-ethoxy-7,8-dimethyl-4-phenyl-1,3,4,5-tetrahydro-1,5-benzodiazepin-2-one 5. Hirshfeld surface analysis was also applied to discern the intermolecular interactions, highlighting the significance of hydrogen bonding, van der Waals forces, and the influence of specific substituents. Furthermore, the MESP maps created using the density functional theory revealed the electrostatic nature of these molecules. The absence of dark blue regions on the MESP maps and variations due to different functional groups and substitutions were noteworthy findings. Collectively, this research offers crucial insights into the behaviour, interactions, and potential applications of new compounds. Finally, the anticancer effects of compounds 3b, 4, and 5 were evaluated against three cancer cell lines and one normal cell line, and the results showed that 3b and 4 had potent antiproliferative effects against all three cancer cell lines.
1. Introduction
1,5-benzodiazepine derivatives are known as powerful scaffolds and building blocks for the construction of a wide variety of heterocyclic compounds that play efficient roles in medicinal and industrial chemistry [1,2,3].
Many synthetic routes have been developed to prepare 1,5-benzodiazepine derivatives with potent biological and pharmacological activities [4,5,6,7]. In continuation of our research on the synthesis and crystal structure of 1,5-benzodiazepin-2-one derivatives [8,9,10,11,12,13,14,15], we report in this current work the crystal structures of 3-hydroxy-7,8-dimethyl-4-phenyl-1,5-benzodiazepin-2-one derivatives.
The compounds in question are derivatives of the 1,3,4,5-tetrahydro-2H-1,5-benzodiazepin-2-one structure. All three compounds share a common core skeleton but are distinguished by their unique substituents. The core of these compounds is a seven-membered diazepine ring, which contains two nitrogen atoms and is fused to a benzene ring. Additionally, the 2-position of the diazepine ring contains a carbonyl group (C=O). The compound, designated 3b “(3R,4R)-3-hydroxy-7,8-dimethyl-4-phenyl-1,3,4,5-tetrahydro-2H-1,5-benzodiazepin-2-one”, is characterized by the presence of a hydroxy group at the 3-position, methyl groups at the 7- and 8-positions, and a phenyl group at the 4-position. The second compound (4), “(3R,4R)-1-ethyl-3-hydroxy-7,8-dimethyl-4-phenyl-1,3,4,5-tetrahydro-2H-1,5-benzodiazepin-2-one”, introduces an ethyl group at the 1-position, with keeping the hydroxy group at the 3-position, the methyl groups at the 7- and 8-positions, and the phenyl group at the 4-position. Finally, compound 5, “(3R,4R)-3-ethoxy-1-ethyl-7,8-dimethyl-4-phenyl-1,3,4,5-tetrahydro-2H-1,5-benzodiazepin-2-one”, has an ethoxy group at the 3-position, an ethyl group at the 1-position, methyl groups at the 7- and 8-positions, and a phenyl group at the 4-position. These textual descriptions aim to provide a comprehensive overview of the molecular structures; however, a chemical drawing or diagram would offer a clearer and more detailed representation.
To obtain a comprehensive understanding of the structural attributes, we have initiated our study using single-crystal X-ray diffraction (SCXRD) analysis, complemented by Hirshfeld–Becke surfaces (BSs) [16,17,18,19]. Using the conceptual DFT (CDFT), we predict global reactivity descriptors along with local Parr functions with both nucleophilic and electrophilic characteristics [20,21,22,23], which would provide insight into possible chemical reactivities. Our current research delineates the structures and properties of the three discussed compounds and guides synthetic organic chemists in designing and crafting new materials within the 1,3,4,5-tetrahydro-2H-1,5-benzodiazepin-2-one framework.
2. Materials and Methods
2.1. General Procedures
2.1.1. Synthesis of 3-Hydroxy-4-phenyl tetrahydro-1,5-benzodiazepin-2-one (3)
1,5-benzodiazepine diastereoisomers, 3a and 3b, were prepared according to the procedure described by our team through the condensation of 4,5-dimethyl-o-phenylenediamine with ethyl phenyl glycidate in refluxing xylene for 48 h (Scheme 1) [24,25].
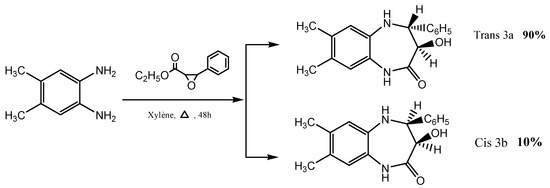
Scheme 1.
Synthesis of diastereoisomeric 1,5-benzodiazepin-2-ones trans 3a and cis 3b.
2.1.2. Synthesis of the Alkylated Compounds (4) and (5)
These compounds have been synthesized according to the procedure previously described in reference [24].
2.2. Anticancer Activity
The cytotoxicity assay of 3b, 4, and 5 was investigated using the colorimetric MTT assay against three cancer cell lines and one normal cell line according to previously reported procedures [26,27,28]. Human lung fibroblasts (WI38), hepatocellular carcinoma (HEPG-2), mammary gland breast cancer (MCF-7), breast cancer (MDA-MB-231) and epithelioid carcinoma cervical cancer (HeLa) cell lines were obtained from ATCC (Manassas, VA, USA). Briefly, the cell lines were cultured in 96-well plates in triplicate in the presence of 10% foetal bovine serum (FBS), an antibiotic cocktail of 100 µL/mL of streptomycin and 100 units/mL of penicillin, and RPMI 11640 (Sigma-Aldrich, St. Louis, MO, USA) as a medium with a density of 1.0 × 104 for 48 h at 37 °C, 5% CO2, and 100% relative humidity. The cells were then exposed to five concentrations of each compound and the reference compound as a positive control or left untreated. Afterwards, the cells were then incubated with 20 mL of MTT solution. To dissolve the solid formazan that formed, 100 mL of dimethyl sulfoxide (DMSO) was added before the absorbance was read at 570 nm using the BioTek EXL 800 plate reader (Agilent Technologies, Inc., Santa Clara, CA, USA). The relative cell viability percentage was calculated as follows (A570 of treated samples/A570 of untreated sample) × 100.
2.3. Computational Methodology
The density functional theory (DFT) calculations used input geometries obtained from the experimental X-ray structure determinations [29,30].
Geometry optimizations for various isolated monomer and dimer models were performed in the gas phase using the Gaussian 09, Rev D.01 software package (Gaussian Inc., Wallingford, CT, USA). The GaussView 6.0 program (Gaussian Inc., Wallingford, CT, USA) was employed to visualize, analyse, modify, and export the resulting data.
The calculations employed the B3LYP functional, a well-known hybrid density functional in computational chemistry and quantum mechanics. This functional combines Becke’s three-parameter exchange (B3) functional [29] with Lee, Yang, and Parr’s correlation (LYP) function [30]. The 6-311(d,p) basis set has emerged as a highly cost-efficient choice for accurately predicting a range of properties, including geometry optimization, thermochemical behaviour, crystal structures, spectroscopic analyses, and noncovalent interactions. Employing the B3LYP/6-311(d,p) level of theory has been shown to strike an optimal balance between minimal error margins and manageable computational demands, as is evident in [31,32]. Further validation of the DFT/B3LYP method, using the 6-311G++(d,p) basis set, was conducted by comparing the calculated bond lengths and angles with those reported in the literature [32,33].
Frequency calculations of the optimized geometries confirmed that all stationary points were true minima (with zero imaginary frequencies) on the potential energy surface [29,30,34].
The stable geometries obtained from the optimizations were used to calculate the reactivity descriptors and the molecular electrostatic nature of the interactions at the same level of theory. Three-dimensional (3D) Hirshfeld surfaces (HSs) and corresponding two-dimensional (2D) fingerprint maps were generated using the CrystalExplorer17.5 program [35]. The dnorm surfaces for 3b, 4, and 5 were mapped using a fixed colour scale ranging from −0.4449, −0.4296, and −0.3318 a.u. (red) to 1.5467, 1.4336, and 1.3823 a.u. (blue), respectively. The shape index and curvedness mapping ranges for the three compounds were set to −1.0 to 1.0 and −4.0 to 0.4 Å, respectively.
Global chemical reactivity descriptors were computed based on concepts from a conceptual DFT, also known as the chemical reactivity theory (CRT) [36,37]. A detailed theoretical background and computational information are found in the Supplementary Materials.
For 3b, 4, and 5, the electrophilic P+k and nucleophilic P−k regions were identified using the Parr functions. These were derived by examining the Mulliken atomic spin densities (ASD) of both radical anions and cations. This examination was carried out through single-point energy calculations of their optimized neutral geometries, leveraging the unrestricted UB3LYP approach tailored for radical species [20,35,36,37,38].
2.4. X-ray Crystallography
Suitable crystals of 3b, 4, and 5 were mounted on polymer loops with a drop of heavy oil and placed in a cold nitrogen stream using the Bruker D8 Venture diffractometer. The intensity data were collected under the control of the APEX4 software (APEX4, Bruker AXS LLC, Madison, WI, USA) and reduced to F2 values with SAINT (“Saint. Data Reduction and Correction Program. Version 8.34A. Bruker AXS Inc., Madison, WI, USA, 2014”, n.d.), which also performed a least-squares refinement of unit cell parameters with ca. 9500 reflections chosen from the full data set. The application of empirical absorption corrections and merging of equivalent reflections employed SADABS ((Version 2014/4). Bruker AXS Inc., Madison, WI, USA) [39], and the structures were solved with a dual space method using SHELXT (Bruker. APEX3, SADABS, SAINT and SHELXTL. Bruker AXS Inc., Madison, WI, USA, 2016) [40]. The structural models were refined by full-matrix, least-squares procedures with SHELXL (Sheldrick, 2015) [40] with hydrogen atoms attached to carbon included as riding contributions with isotropic displacement parameters tied to those of the attached atoms. For 3b and 4, hydrogen atoms attached to nitrogen and oxygen were refined with the appropriate DFIX instructions. For 5, the atoms attached to nitrogen were refined independently. The crystal structures and refinement data are presented in Table 1.

Table 1.
Crystal and refinement data for 4, 3b, and 5.
2.5. In Silico Physicochemical Properties
The in silico pkCSM descriptors algorithm program was utilized to analyse the physicochemical characteristics and pharmacokinetics of compounds 3b, 4, and 5 following a previously reported methodology [41].
3. Results and Discussion
3.1. Structural Elucidation of Compounds 3a, 3b, 4, and 5
The synthesis and structures of the two diastereoisomeric 3-hydroxy-7,8-dimethyl-1,5-benzodiazepin-2-ones (3a and 3b) are illustrated in Scheme 1.
The structure of the two diastereoisomeric compounds, 3a and 3b, were first characterized using 1H NMR spectroscopy [24,25], and the configuration of each diastereoisomer was established through the coupling constant between the proton atoms at positions 3 and 4 of the seven-membered ring. The 1H NMR spectrum of compound 3a showed two doublets with a coupling constant of 10 Hz corresponding to the trans configuration. Moreover, the 1H NMR spectra of 3b presented two doublets with a coupling constant of 5 Hz, related to the cis configuration of 3b. To confirm the results of the 1H NMR spectroscopy, we performed a single-crystal X-ray diffraction study of compound 3b. Furthermore, 1-ethyl-3-hydroxy-7,8-dimethyl-4-phenyl-1,3,4,5-tetrahydro-2H-1,5-benzodiazepin-2-one 4 and trans dialkylated -1,5- benzodiazepin-2-one 5 were synthesized according to the procedure described by Rida et al. [24] (Scheme 2).
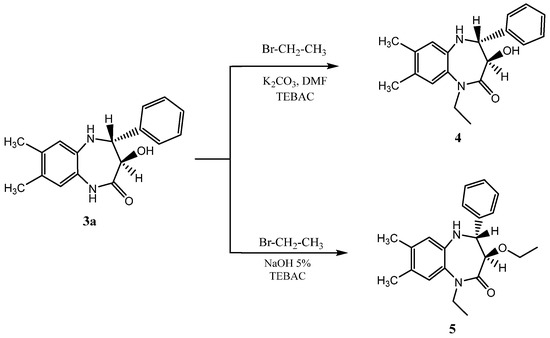
Scheme 2.
Alkylation of trans-3-hydroxy-7,8-dimethyl-4-phenyl-1,5-benzodiazepin-2-one under phase transfer catalysis (PTC) conditions [31].
The structures of 4 and 5 were elucidated from the spectral data (1H NMR, 13C NMR, mass spectrometry) and confirmed by a single-crystal X-ray diffraction study.
3.2. Plausible Mechanisms for the Formation of the Synthesized Compounds
It is worth noting that Mamedov et al. [42] described in 2020 the condensation of substituted o-phenylenediamines with methyl aryl glycidate in boiling acetic acid led to two series of heterocyclic systems: 1,5-benzodiazepine and quinoxaline compounds. The authors proposed possible mechanisms explaining their formation under an acidic medium. On our part, we proposed a plausible mechanism explaining the formation of the diastereisomers, 3a and 3b, in neutral conditions. The initial step starts with a nucleophilic attack of the amino group of 4,5-dimethyl-o-phenylenediamine 1 on the carbon atom at the 3-position of ethyl glycidate 2, leading to N-substituted-o-phenylenediamine [A], which undergoes an intramolecular cyclization involving the loss of an ethanol molecule from the intermediate [B] to afford the two diastereoisomers, 3a and 3b (Scheme 3).
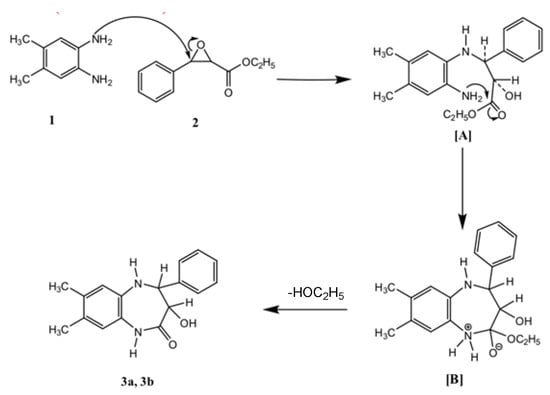
Scheme 3.
Plausible mechanism for the formation of the two diastereoisomers 3a and 3b.
3.3. Description of the Structures of 3b, 4, and 5
In both 3b and 4, the seven-membered ring adopts a slightly flattened boat conformation, while in 5, the conformation is a slightly twisted boat. In 3b, the phenyl substituent is in an axial position directed away from the “bottom” of the boat, while in 4 and 5, the three substituents occupy approximately equatorial sites (Figure 1). The orientation of the hydroxyl group in 3b is determined by the intramolecular O1—H1⋯O2 hydrogen bond (Table 2) and the sum of angles about N2 is 360° within experimental error. This implicates the involvement of its lone pair in N→C π bonding, and this is primarily with C9 as indicated by the short N2—C9 distance of 1.339(2) Å. Similar situations were obtained for N1 in 4 and in 5 where the N1—C7 distances are 1.3517(9) and 1.3577(15) Å, respectively. In the crystal of 3b, N2—H2A⋯O1 hydrogen bonds generate chains of molecules extending along the a-axis direction. These are connected across inversion centres by C2—H2⋯O2 hydrogen bonds and N1—H1A⋯Cg2 interactions (Table 2) with the formation of layers parallel to the ac plane. The layers pack with normal van der Waals contacts. For 4, inversion dimers are formed by O2—H2A⋯O1 and C15—H15⋯O1 hydrogen bonds (Table 2). These are joined by inversion-related C11—H11B⋯O2 hydrogen bonds (Table 2) into ribbons of molecules extending along the b-axis direction. The ribbons are connected by N2—H2B⋯Cg2 interactions (Table 2) into layers parallel to (101), and the layers are joined by C12—H12B⋯Cg2 interactions (Table 2) to form the full 3D structure. In the supramolecular structure of 5, N2—H2A⋯O1 hydrogen bonds (Table 2) form zigzag chains of molecules extending along the c-axis direction. The chains pack with normal van der Waals contacts.
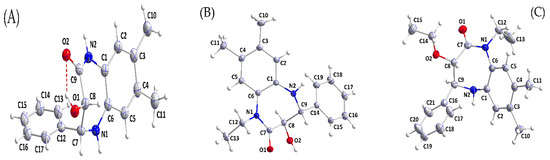
Figure 1.
Perspective views of (A) 3b, (B) 4, and (C) 5, with labelling schemes and 50% probability ellipsoids. The intramolecular hydrogen bond in (A) is depicted by a dashed line.
Table 2.
Hydrogen bond geometry (Å,°) for 3b, 4, and 5.
3.4. Hirshfeld Surface Analysis
Hirshfeld surface analysis helps to understand molecular interactions and design molecules with targeted properties by studying their noncovalent bonds.
3.5. dnorm Mapping
The comparison of the Hirshfeld surfaces for 3b, 4, and 5 provides valuable insights into the effects of derivatization or substitutions on their intermolecular interactions (Figure 2). All three compounds exhibit similar molecular contours, which is indicative of their closely related structural frameworks. Nevertheless, specific substituent groups, such as the 3-ethoxy in 5, might introduce steric and electronic variations, influencing the molecular behaviour. For both 3b and 4, the O—H⋯O and N—H⋯O hydrogen bonds (Table 2) show up as dark red spots, while the C—H⋯O hydrogen bonds are indicated by the lighter red spots. For 5, the only significant intermolecular interactions are the N—H⋯O hydrogen bonds, which are depicted by the dark red spots.
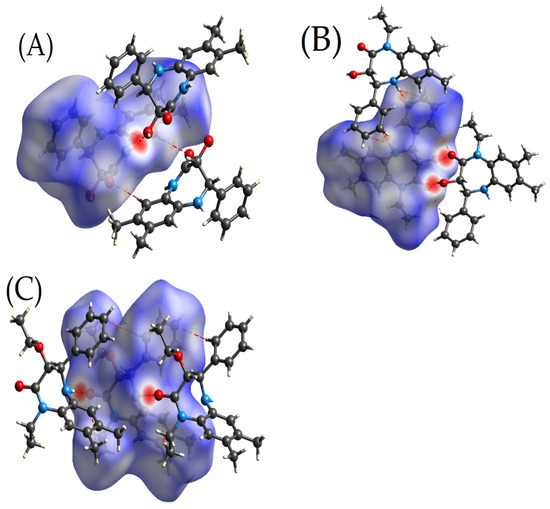
Figure 2.
Three-dimensional Hirshfeld surfaces showing the intermolecular interactions within the crystalline structures: Panel (A) for 3b, Panel (B) for 4, and Panel (C) for 5.
Furthermore, the hydroxy group present in 4 might offer enhanced solubility and can act as a potential hydrogen bond donor. This structural feature could influence its biological activity. Meanwhile, the ethoxy substituent in 5 might affect the compound’s lipophilicity, possibly altering its permeability and distribution in biological systems. The presence or absence of the hydroxyl group and other substituents between the three molecules will likely influence their binding affinities, reactivities, and overall pharmacokinetic and pharmacodynamic profiles.
3.5.1. Curvedness and Shape Index
In our investigation of the molecular packing properties of 3b, 4, and 5, the measures of curvature, notably the curvedness and shape index as delineated by Koenderink, prove indispensable. Koenderink’s [43] seminal works have shed light on the pertinence of these metrics in offering an in-depth understanding of intricate molecular frameworks [44].
In the molecular assembly of compound 5, the Hirshfeld surface analysis reveals distinctive regions of marked curvature, differentiating it from its counterparts. Conversely, the Hirshfeld surfaces of compounds 3b and 4 are distinguished by the presence of planar regions. Specifically, the planarity is apparent over the centroid of the C12…C17 ring (Cg2) in compound 3b and the C14…C18 ring (Cg2) in compound 4, located at the symmetry operations 2 – x, 1 – y, 1 – z and 2 – x, 2 – y, −z, respectively. These planar domains are indicative of the potential for π–hydrogen bonding interactions, which are known to contribute significantly to the structural coherence in various molecular frameworks, as illustrated in Figure 3A–C. In contrast, compound 5 is characterized by discernible regions with dark blue perimeters on the Hirshfeld surface, suggesting the lack of π–π stacking interactions within its crystal structure [44].
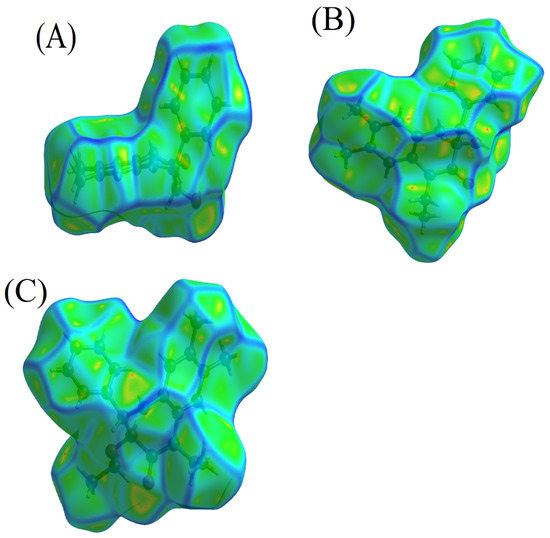
Figure 3.
Curvature analyses of compounds: (A) 3b, (B) 4, and (C) 5.
The shape index serves as another layer of insight, being a qualitative measure of the differences in surface shapes. Its sensitivity is particularly pronounced in detecting even subtle alterations, especially in areas that manifest as relatively flat. The dichotomous nature of the shape index is evident when studying the complementary facets, namely ‘bumps and hollows’. In molecular interactions, a blue bump shape marked by a shape index exceeding one indicates the donor entity. Conversely, a shape index falling below one, visualized as a red hollow, pinpoints the acceptor in the interaction (Figure 4A–C) [44].
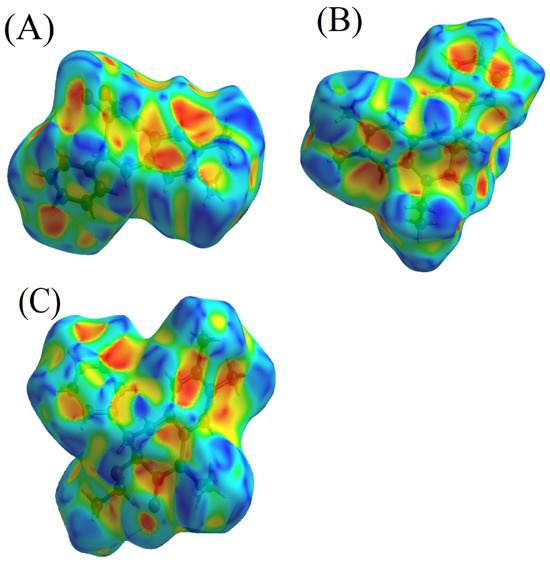
Figure 4.
Shape index visualizations for 3b (A), 4 (B), and 5 (C).
The detailed understanding of the molecular interactions in compounds 3b and 4, particularly the apparent π–hydrogen bonding, enriches our discourse on their chemical behaviours and intermolecular relationships. Such knowledge broadens the scope of exploring their functionalities and prospective utilizations under diverse conditions, a contrast to compound 5, where such π–hydrogen bonding interactions are absent.
3.5.2. Distribution of Individual Intermolecular Interactions
The distribution of individual intermolecular interactions in 4, 3b, and 5, was determined using Hirshfeld surface (HS) analysis. The results, presented in Table 3 and Figure 5, reveal insights into the nature and strength of these interactions.
Table 3.
The distribution of individual intermolecular interactions in 4, 3b, and 5 using Hirshfeld surface (HS) analysis.
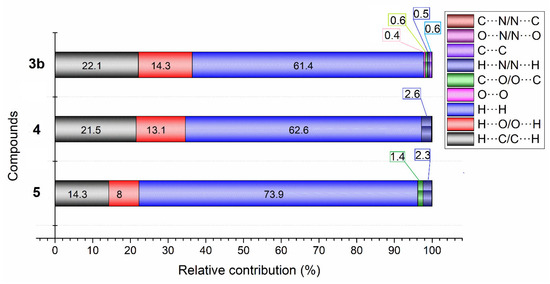
Figure 5.
Percentage distribution of different intermolecular forces contributing to the Hirshfeld surface for 3b, 4, and 5.
An analysis of Table 3 and Figure 6 highlights that in compounds 4, 3b, and 5, a significant majority of the intermolecular interactions are hydrogen-based, specifically H···H (62.6% in 4, 61.5% in 3b, and 73.8% in 5), H···C/C···H (21.5% in 4, 22.1% in 3b, and 14.3% in 5), and H···O/O···H (13.1% in 4, 14.3% in 3b, and 8.0% in 5), suggesting a pivotal role of hydrogen bonding and van der Waals forces in their structural stability. The symmetrical 2D fingerprint plots further corroborate that these interactions mainly occur between identical monomers, with H···H contacts dominating due to the minimal van der Waals radius of hydrogen (Figure S1).
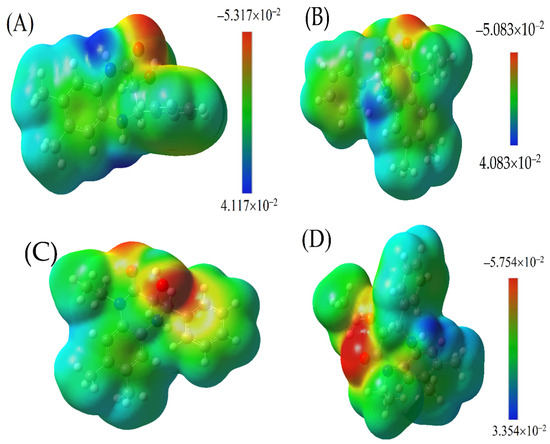
Figure 6.
Molecular electrostatic potential (MESP) maps of: (A) 3b, (B,C) 4, and (D) 5. The colour spectrum ranges from red (indicating highly negative regions) to dark blue (denoting highly positive regions).
Compounds 3b and 4 exhibit more pronounced H···C/C···H interactions, constituting over 22% of the total, compared to just 14.3% in compound 5, implying a denser molecular packing and more robust intermolecular forces that could enhance the stability and influence the crystal lattice structure. Compound 5 is distinguished by its exceptionally high H···H interactions at 73.8%, which is indicative of potent van der Waals forces shaped by its unique molecular framework. In contrast, 3b and 4 have a higher incidence of H···O/O···H interactions, 14.3% and 13.1%, respectively, compared to 8% in compound 5, highlighting the significance of hydrogen bonds involving oxygen atoms in determining the compounds’ stability and modifying properties, such as the solubility and boiling points. The ethoxy substitutions in compound 5 reduce the potential for such oxygen-involved hydrogen bonding, thus differentiating its intermolecular interactions and resultant properties from those of 3b and 4 [44].
In 4 and 5, the H···N/N···H interactions (2.6% and 2.3%, respectively,) are more significant than in 3b, while 5 has a higher percentage of C···O (1.4%) interactions compared to 4 and 3b, which may be due to the presence of the ethoxy group in its structure [45,46,47,48].
3.6. DFT Density Functional Theory (DFT) Examination
3.6.1. Refinement of Structural Configuration
The molecular geometries of the triad of compounds were meticulously refined utilizing the density functional theory (DFT) with the B3LYP functional and the 6-311G (d,p) basis set. Depicted in Figure 1 are the resultant optimized conformations. Subsequent quantification of internuclear distances and interatomic angles facilitated through this theoretical framework afforded a comprehensive dataset, which was subsequently aligned with empirical findings procured from X-ray crystallographic analyses, as enumerated in Supplementary Tables S1 and S2. Scrutinizing these tables furnished a rigorous correlative assessment between the calculated theoretical bond lengths and the experimentally determined parameters.
In the study, the mean absolute error (MAE) values were calculated and are presented in Tables S1 and S2 for selected bond distances and bond angles of three compounds (4, 3b, and 5). The MAE values indicate the average magnitude of the errors between the predicted values and the actual values.
For the bond distances, the MAE values for 4, 3b, and 5 are reported as 0.00583, 0.0475, and 6.66667 × 10−5, respectively. These values are considered satisfactory, taking into account that the average actual bond distances are 1.4041, 1.4507, and 1.4250 Å for 4, 3b, and 5, respectively. The MAE values for bond distances provide an indication of how accurately the predicted bond distances match the real bond distances. The fact that the MAE values are relatively small compared to the average actual bond distances suggests that the predictions are reasonably accurate (Table S1).
Similarly, for the bond angles, the MAE values for 4, 3b, and 5 are reported as 2.630, 2.067, and 0.00051, respectively. These MAE values were obtained using the B3LYP method. Comparing these values with the average real bond angles of 119.546, 117.121, and 117.929 degrees for 4, 3b, and 5, respectively, it can be concluded that the MAE values are within an acceptable range. The smaller the MAE values, the closer the predicted bond angles are to the actual bond angles (Table S2).
3.6.2. Molecular Electrostatic Nature of Interactions—MESP for 3b, 4, and 5
The calculations pertaining to the molecular electrostatic surface potential (MESP) for the derivatives of 1,3,4,5-tetrahydro-2H-benzo[b][1,4]diazepin-2-one, namely 3b, 4, and 5, were conducted using the advanced DFT level by the B3LYP/6-311G(d,p) method. The objective of these calculations was to derive an in-depth understanding of the electrostatic nature of these molecules and how potential interactions are manifested on their surfaces.
The MESP maps generated for 3b, 4, and 5, Figure 6, are coloured to indicate the range of electrostatic potentials. A palette from red to dark blue represents the shift from extremely negative to extremely positive sites on the molecular surface. The sequence of colour gradation—red, yellow, green, light blue, and dark blue—mirrors the progression from the most negative to the most positive potential. Red zones depict regions of high negative potentials, while dark blue indicates areas of large positive potentials. Areas coloured in yellow are less negative than the red zones, and light blue signifies regions less positive than the darker blue counterparts. The green zones highlight nearly neutral potentials.
The electronegativity of the bonded atoms strongly influences this colour distribution. Regions around highly electronegative atoms exhibit a red hue when these atoms are bonded to less electronegative ones. Conversely, when atoms of comparable electronegativity values are bonded, the colour distribution spectrum tends to narrow.
The MESP maps of 3b, 4, and 5 do not feature dark blue regions, implying an absence of highly positive sites. This observation can be rationalized by the dominance of C and H atoms in these structures. Two primary characteristics are evident across the MESP maps of these compounds:
- A pronounced electron cloud localized at the oxygen atoms in both the carbonyl and the hydroxide groups, appearing as a red ellipse, most likely results from the lone pairs of electrons present in these functional groups.
- Surrounding these structures is a light blue hue, suggestive of slightly positive regions. This can largely be attributed to the hydrogen atoms enveloping the structure. However, for those derivatives with nitrogen substitutions, the light blue distribution varies.
Further inspection reveals a transition from the core diazepin-2-one structure towards its terminal end, marked by a blend of green and light blue. This indicates the presence of phenyl rings, which play a pivotal role in dictating the electrostatic potential in these zones.
3.6.3. The Global Descriptors
The global descriptors for 3b, 4, and 5 show their inherent electronic properties. The data in Table 4 offer an analysis of these three compounds based on the CDFT calculations conducted using multiwfn and the DFT.
Table 4.
The global descriptors of compounds 4, 3b, and 5.
The highest occupied molecular orbital (HOMO) energies, represented as EHOMO, provide a significant understanding of the electron donating tendencies of molecules. Compound 5, with the EHOMO value of −6.198 eV, seems to have the most prominent inclination towards electron donation compared with 3b and 4. The latter two have almost equal HOMO energies, with values of −5.4646 eV and −5.4602 eV, respectively.
Vertical ionization potentials (IP) and electron affinities (EA) further extend our comprehension of the electronic behaviour of these compounds. Compound 3b exhibits the largest at 7.0644 eV, indicating it would be the most difficult of the three to ionize. However, all compounds have comparable vertical EA values of ca. −1 eV, with 3b having the lowest value (−1.0934 eV).
The softness and hardness provide a way to view the reactivity of molecules. Compound 5, with a softness of 0.1272 eV−1, emerges as the most reactive, signifying its susceptibility to potential chemical interactions, implying a higher propensity for undergoing electron-related chemical processes, such as redox reactions, nucleophilic or electrophilic substitutions, or forming complexes with metals. Conversely, the highest hardness value is attributed to 3b (8.1577 eV), which indicates its relatively stable nature in chemical environments.
Upon comparative analysis, compounds 3b, 4, and 5 each display a marked tendency for electron donation, as indicated by their chemical potentials of −2.9855 eV, −3.0456 eV, and −3.009 eV, respectively. This aligns them with the behaviour of moderate nucleophiles. Their Mulliken electronegativities (2.9855 eV for 3b, 3.0456 eV for 4, and 3.009 eV for 5) suggest that all three have electropositive characters, with the ability to release electrons during chemical reactions. Notably, 3b and 4, with nucleophilicity indices of 3.6566 eV and 3.661 eV, respectively, are classified as strong nucleophiles, indicating a high degree of reactivity and a pronounced inclination to donate electrons. Compound 5, with a nucleophilicity index of 2.9232 eV, is characterized as a moderate nucleophile, showing a slightly lower, yet significant, reactivity. Despite their nucleophilic strengths, all three compounds exhibit marginal electrophilic behaviours, with electrophilicity indices below 0.6 eV, pointing to a lessened tendency to accept electrons. In essence, while compounds 3b and 4 demonstrate substantial nucleophilic characters with strong reactivity, compound 5, though still nucleophilic, offers a more moderate reactivity profile. This indicates that compounds 3b and 4 are more likely to participate in reactions involving electron pair donations to electrophiles, whereas compound 5, while still reactive, may engage in such interactions with slightly less fervour.
3.6.4. The Local Descriptors
The nucleophilicity (Parr functions ()) provides information on the electronic properties of 3b, 4, and 5. These functions categorize regions as electrophilic when they have positive Parr functions and nucleophilic when they exhibit negative Parr functions () (Table 5).
Table 5.
Electrophilic and Nucleophilic Parr Function Values for 3b, 4, and 5.
Analysing the electrophilic and nucleophilic behaviours of compounds 3b, 4, and 5 via Parr functions provides valuable insights into their reactive sites. For compound 3b, the N1 nitrogen exhibits the highest electrophilic activation with a Parr function value () of 8.15 eV, surpassing the electrophilic activations of the C3 and C1 carbons, which have values of 5.49 eV and 4.70 eV, respectively. This positions the N1 nitrogen as the primary electrophilic centre in compound 3b. Conversely, the C4 carbon is identified as the primary nucleophilic site with a Parr function value () of 6.18 eV, indicating it is more nucleophilically activated than the C1, C9, and C2 carbons.
For compound 4, the N2 nitrogen stands out with an electrophilic Parr function value of 8.51 eV, which is notably higher than those of the C4 and C6 carbons, marked at 5.72 eV and 4.69 eV, respectively, making it the most electrophilic site. Nucleophilically, the C3 carbon exhibits a higher activation with a Parr function value of 5.92 eV, compared to the C6 and C7 carbons, which have lower values of 4.23 eV and 3.93 eV.
In the case of compound 5, the electrophilic character is dominated by the N2 nitrogen with a Parr function value of 7.60 eV, greater than that of the C4 and C6 carbons, which are 4.86 eV and 4.70 eV. Thus, the N2 nitrogen is the most electrophilic centre. The nucleophilic Parr functions suggest the C3 carbon as the most activated site with a value of 5.95 eV, exceeding the activations of the C6 and C19 carbons, with values of 4.40 eV and 3.80 eV, respectively [20].
Collectively, these theoretical Parr function analyses reveal that nitrogen centres in all three compounds exhibit the highest electrophilic activation, suggesting they are the preferred sites for electrophilic attacks. Moreover, certain carbon atoms in each compound are identified as key sites for nucleophilic reactivity, with compound 3b’s C4, compound 4’s C3, and compound 5’s C3 carbons showing the greatest potential for nucleophilic interactions. These insights are crucial for predicting the behaviour of these compounds in various chemical environments, guiding synthetic strategies, and understanding their interaction with other molecules.
3.7. Anticancer Effect
The antiproliferative activity of 4, 3b, and 5 as well as sunitinib as a reference standard was investigated against three cancer cell lines: a colorectal carcinoma cell line (HCT 116), human hepatocellular carcinoma cell line (HepG2), and human breast cancer cell line (MCF-7) and one normal cell line (WI38) using the MTT assay. The results in Table 6 show that 5 has very weak activity on all tested cell lines and, unexpectedly, was more cytotoxic on the normal cells. Compound 4 demonstrated good antiproliferative activity, similar to sunitinib, on the three cell lines. Remarkably, 3b exhibited a significant antiproliferative effect on all cell lines, ranging from 6.13 to 9.18 µM. Finally, 4 and 3b displayed lower cytotoxicity on the normal cells compared to the cancer cells. In summary, 4 and 3b revealed promising anticancer activities, which is worth further investigating for the mechanism of their anticancer effects.
Table 6.
In vitro cytotoxicity of compounds 3b, 4, and 5 against cancer and normal cell lines.
3.8. In Silico Studies of the Pharmacokinetics and Pharmacodynamics Properties
The in silico properties of compounds 3b, 4, and 5, including absorption, distribution, metabolism, elimination, and toxicity (ADMET), were evaluated using the pkCSM protocol. The results in Table 7 demonstrate that the three compounds follow Lipinski’s Rule of Five, suggesting a good oral bioavailability and potentially useful drug-like properties. All compounds seem to cross the blood–brain barrier which indicates possible CNS side effects. Compounds 4 and 5 could be a substrate for CYP3A4, and, therefore, they may need to be used with caution if patients coadminister other drugs affecting CYP3A4. Finally, compound 3b has a higher maximum tolerated dose than compounds 4 and 5. All compounds are not supposed to induce hepatotoxicity or a skin allergy.
Table 7.
In silico physical and chemical properties of compounds 3b, 4, and 5.
4. Conclusions
In this current work, we report the crystal structures and the electronic properties of three 3-hydroxy- 4-phenyl- 1,5-benzodiazepine derivatives, namely cis-(3S,4S)-3-hydroxy-7,8-dimethyl-4-phenyl-1,3,4,5-tetrahydro-1,5-benzodiazepin-2-one 3b, trans-(3R,4R)-1-ethyl-3-hydroxy-7,8-dimethyl-4-phenyl-1,3,4,5-tetrahydro-2H-1,5-benzodiazepin-2-one 4, and trans-(3S,4S) 1-ethyl-3-ethoxy-3,7,8-dimethyl-4-phenyl-1,3,4,5-tetrahydro-1,5-benzodiazepin-2-one 5. Compound 3b crystallized uniquely in the triclinic system, while 4 and 5 predominantly settled in the monoclinic system, with 4 showing subtle triclinic attributes. Metrics, such as cell parameters and unit cell volume, revealed both shared attributes and subtle distinctions among the compounds. The Hirshfeld surface analysis further highlighted specific intermolecular interactions, with hydrogen bonding and van der Waals forces emerging as dominant influences, modulated by specific substituents. The density functional theory, in creating MESP maps, provided deeper insights into their electrostatic characteristics, with the absence of dark blue regions and variations due to diverse functional groups being noteworthy. The hydrogen bonding profiles, influenced by the unique substituents, played a critical role in determining the molecular structures, stabilities, and potential reactivities of these compounds. Furthermore, the Hirshfeld surfaces, mapped with the dnorm, shape index, and curvedness, offer insights into factors like π–π stacking, packing configurations, and other defining crystallographic features, all contributing to their stability and properties. Table 1 encapsulated these interactions, underscoring the importance of hydrogen bonds in these benzodiazepinone derivatives. This research amplifies our understanding of these compounds and sets a foundation for future studies, emphasizing the impact of substituent groups on their properties. Finally, 3b and 4 demonstrated a potent anticancer effect comparable to the reference standard, sunitinib, against the HCT 116, HepG2 and MCF-7 cancer cell lines.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/2073-4352/13/12/1693/s1, Figure S1: Full two-dimensional fingerprint plots depicting the interactions of the three compounds (3b, 4, and 5); Table S1: Comparison of Selected Bond Lengths (Å) of compounds 3b, 4, and 5) by SC-XRD and DFT; Table S2: Comparison of Selected Bond Angles (°) of compounds (3b, 4, and 5) by SC-XRD and DFT.
Author Contributions
Conceptualization, E.M.E. and S.L.; methodology, S.L. and J.T.M.; software, J.T.M. and A.H.B.; validation, M.M.A. and S.L.; formal analysis, A.H.B.; investigation, M.M.A.; resources, M.M.A.; data curation, J.T.M. and A.H.B.; writing—original draft preparation, A.H.B. and S.L.; writing—review and editing, M.M.A.; visualization, E.M.E.; supervision, M.M.A. and E.M.E.; project administration, M.M.A. and S.L.; funding acquisition, M.M.A.; All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Researchers Supporting Project number (RSPD2023R628), King Saud University, Riyadh, Saudi Arabia.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2023R628), King Saud University, Riyadh, Saudi Arabia for funding this research.
Conflicts of Interest
The authors declare no conflict of interest.
Correction Statement
This article has been republished with a minor correction to the title. This change does not affect the scientific content of the article.
References
- Arora, N.; Dhiman, P.; Kumar, S.; Singh, G.; Monga, V. Recent Advances in Synthesis and Medicinal Chemistry of Benzodiazepines. Bioorg. Chem. 2020, 97, 103668. [Google Scholar] [CrossRef]
- Verma, R.; Bhatia, R.; Singh, G.; Kumar, B.; Mehan, S.; Monga, V. Design, Synthesis and Neuropharmacological Evaluation of New 2,4-Disubstituted-1,5-Benzodiazepines as CNS Active Agents. Bioorg. Chem. 2020, 101, 104010. [Google Scholar] [CrossRef] [PubMed]
- Gawandi, S.J.; Desai, V.G.; Joshi, S.; Shingade, S.; Pissurlenkar, R.R. Assessment of Elementary Derivatives of 1,5-Benzodiazepine as Anticancer Agents with Synergy Potential. Bioorg. Chem. 2021, 117, 105331. [Google Scholar] [CrossRef] [PubMed]
- Zellou, A.; Cherrah, Y.; Hassar, M.; Essassi, E.M. Synthese et Etude Pharmacologique Des 1,5-Benzodiazepine-2,4-Diones et de Leurs Derives Alkyles. Ann. Pharm. Fr. 1998, 56, 169–174. [Google Scholar] [PubMed]
- Di Braccio, M.; Grossi, G.; Roma, G.; Vargiu, L.; Mura, M.; Marongiu, M.E. 1,5-Benzodiazepines. Part XII. Synthesis and Biological Evaluation of Tricyclic and Tetracyclic 1,5-Benzodiazepine Derivatives as Nevirapine Analogues. Eur. J. Med. Chem. 2001, 36, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Mazimba, O.; Molefe, T.C. 1, 5-Benzodiazepines: A Review Update. Int. J. Chem. Stud. 2015, 3, 46–52. [Google Scholar]
- Tolu-Bolaji, O.O.; Sojinu, S.O.; Okedere, A.P.; Ajani, O.O. A Review on the Chemistry and Pharmacological Properties of Benzodiazepine Motifs in Drug Design. Arab. J. Basic. Appl. Sci. 2022, 29, 287–306. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A.; Pinto, J.S.; Kumbar, S.A.; Bhat, N.; Nayak, P. Synthesis and Antimicrobial Evaluation of Some Novel 1,5 Benzodiazepine Derivatives Derived from Pyrrolyl Chalcones. Res. J. Pharm. Technol. 2022, 15, 1811–1814. [Google Scholar] [CrossRef]
- Ongone, T.N.; El Ouasif, L.; El Ghoul, M.; Achour, R.; Chakchak, H.; El Jemli, M.; Cherrah, Y.; Alaoui, K.; Zellou, A. Synthesis of Surfactants Derived from 2-Mercaptobenzimidazole and Study of Their Acute Toxicity and Analgesic and Psychotropic Activities. Biochem. Res. Int. 2019, 2019, 9615728. [Google Scholar] [CrossRef]
- El Abbassi, M.; Essassi, E.M.; Fifani, J. Nouvelle Synthese Des Benzodiazepines-1,5 a Partir de La γ-Pyrone. Tetrahedron Lett. 1987, 28, 1389–1392. [Google Scholar] [CrossRef]
- Essassi, E.M.; Salem, M. Synthèse Des Pyrazolyl-1 Benzimidazoles A Partir Des Dihydro-1,3(2H) Benzo[2,3-b] Diazepine-1,5 Ones-2. Bull. des Sociétés Chim. Belges 1985, 94, 755–758. [Google Scholar] [CrossRef]
- Sebhaoui, J.; El Bakri, Y.; El Aoufir, Y.; Anouar, E.H.; Guenbour, A.; Nasser, A.A.; Mokhtar Essassi, E. Synthesis, NMR Characterization, DFT and Anti-Corrosion on Carbon Steel in 1M HCl of Two Novel 1,5-Benzodiazepines. J. Mol. Struct. 2019, 1182, 123–130. [Google Scholar] [CrossRef]
- Saber, A.; Zouihri, H.; Essassi, E.M.; Ng, S.W. 4-Methyl-2,3-Dihydro-1H-1,5-Benzodiazepin-2-One Monohydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, o1408. [Google Scholar] [CrossRef] [PubMed]
- El Bakri, Y.; Anouar, E.H.; Ahmad, S.; Nassar, A.A.; Taha, M.L.; Mague, J.T.; El Ghayati, L.; Essassi, E.M. Synthesis and Identification of Novel Potential Molecules Against COVID-19 Main Protease Through Structure-Guided Virtual Screening Approach. Appl. Biochem. Biotechnol. 2021, 193, 3602–3623. [Google Scholar] [CrossRef] [PubMed]
- Chkirate, K.; Akachar, J.; Hni, B.; Hökelek, T.; Anouar, E.H.; Talbaoui, A.; Mague, J.T.; Sebbar, N.K.; Ibrahimi, A.; Essassi, E.M. Synthesis, Spectroscopic Characterization, Crystal Structure, DFT, ESI-MS Studies, Molecular Docking and in Vitro Antibacterial Activity of 1,5-Benzodiazepin-2-One Derivatives. J. Mol. Struct. 2022, 1247, 131188. [Google Scholar] [CrossRef]
- Altowyan, M.S.; Haukka, M.; Soliman, S.M.; Barakat, A.; Alaswad, S.O.; Boraei, A.T.A.; Gad, E.M.; Youssef, M.F. Synthesis, Characterization and Single Crystal X-Ray Diffraction Analysis of Fused Triazolo/Thiadiazole Clubbed with Indole Scaffold. Crystals 2023, 13, 423. [Google Scholar] [CrossRef]
- Bakheit, A.H.; Al-Salahi, R.; Ghabbour, H.A.; Ali, E.A.; AlRuqi, O.S.; Mostafa, G.A.E. Synthesis, X-Ray Crystal Structure, and Computational Characterization of Tetraphenylborate, 3-(5H-Dibenzo[a,d] Cyclohepten-5-Ylidene)-N, N-Dimethyl-1-Propanamine. Crystals 2023, 13, 1088. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards Quantitative Analysis of Intermolecular Interactions with Hirshfeld Surfaces. Chem. Commun. 2007, 37, 3814–3816. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Bakheit, A.H.; Al-Salahi, R.; Al-Majed, A.A. Thermodynamic and Computational (DFT) Study of Non-Covalent Interaction Mechanisms of Charge Transfer Complex of Linagliptin with 2,3-Dichloro-5,6-Dicyano-1,4-Benzoquinone (DDQ) and Chloranilic Acid (CHA). Molecules 2022, 27, 6320. [Google Scholar] [CrossRef]
- Bakheit, A.H.; Abuelizz, H.A.; Al-Salahi, R. Hirshfeld Surface Analysis and Density Functional Theory Calculations of 2-Benzyloxy-1,2,4-Triazolo[1,5-a] Quinazolin-5(4H)-One: A Comprehensive Study on Crystal Structure, Intermolecular Interactions, and Electronic Properties. Crystals 2023, 13, 1410. [Google Scholar] [CrossRef]
- Bakheit, A.H.; Attwa, M.W.; Kadi, A.A.; Ghabbour, H.A.; Alkahtani, H.M. Exploring the Chemical Reactivity, Molecular Docking, Molecular Dynamic Simulation and ADMET Properties of a Tetrahydrothienopyridine Derivative Using Computational Methods. Crystals 2023, 13, 1020. [Google Scholar] [CrossRef]
- Rida, M.; Meslouhi, H.E.; Ahabchane, N.H.; Garrigues, B.; Es-Safi, N.; Essassi, E.M. A Convenient Method for the Synthesis of 1,5-Benzodiazepin-2-One. Open Org. Chem. J. 2014, 2, 83–87. [Google Scholar] [CrossRef]
- Meslouhi, H.E.; Essassi, E.M.; Zerzouf, A.; Salem, M.; PETRUS, C.; Viallefont, P. ChemInform Abstract: Synthesis of New 1,5-Benzodiazepin-2-Ones from Ethyl Glycidate. ChemInform 1997, 28. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Alanazi, A.S. Novel 7-Deazapurine Incorporating Isatin Hybrid Compounds as Protein Kinase Inhibitors: Design, Synthesis, In Silico Studies, and Antiproliferative Evaluation. Molecules 2023, 28, 5869. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, A.S.; Mirgany, T.O.; Alsaif, N.A.; Alsfouk, A.A.; Alanazi, M.M. Design, Synthesis, Antitumor Evaluation, and Molecular Docking of Novel Pyrrolo[2,3-d]Pyrimidine as Multi-Kinase Inhibitors. Saudi Pharm. J. 2023, 31, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, A.S.; Mirgany, T.O.; Alsfouk, A.A.; Alsaif, N.A.; Alanazi, M.M. Antiproliferative Activity, Multikinase Inhibition, Apoptosis- Inducing Effects and Molecular Docking of Novel Isatin–Purine Hybrids. Medicina 2023, 59, 610. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; Taie, H.A.A.; Bakheit, A.H.; Mostafa, G.A.E.; Marzouk, M.; Rashid, H.; Al-Salahi, R. Investigation of 4-Hydrazinobenzoic Acid Derivatives for Their Antioxidant Activity: In Vitro Screening and DFT Study. ACS Omega 2021, 6, 31993–32004. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Zeng, W.; Wang, X.; Zhang, Y. Crystal Structure, Thermodynamic Properties and DFT Studies of 5,6-Dimethyl-1H-Benzo[d]Imidazol-3-Ium 3-((2,4-Dioxo-1,5-Dioxaspiro[5.5]Undecan-3-Ylidene)Methyl) -2,4-Dioxo-1,5-Dioxaspiro[5.5]Undecane Hydrate. Crystals 2021, 11, 1393. [Google Scholar] [CrossRef]
- Priyambada Biswal, S.; Ranjan Dash, M.; Misra, P.K. Synthesis and Characterization of Tailor-Made o-Hydroxysubstituted Anils through 1H NMR, 13C NMR, SC-XRD and DFT Studies for Possible Optoelectronic Applications. Mol. Phys. 2023, 121, 2225651. [Google Scholar] [CrossRef]
- Dash, M.R.; Rajakumar, B. Theoretical Investigations of the Gas Phase Reaction of Limonene (C10H16) with OH Radical. Mol. Phys. 2015, 113, 3202–3215. [Google Scholar] [CrossRef]
- Lardy, S.W.; Luong, K.C.; Schmidt, V.A. Formal Aniline Synthesis from Phenols through Deoxygenative N-Centered Radical Substitution. Chem. Eur. J. 2019, 25, 15267–15271. [Google Scholar] [CrossRef]
- Zahariev, F.; Levy, M. Density- and Spin-Density-Functional Theories through Spin-Free Wave Functions. Phys. Rev. A 2019, 100, 062507. [Google Scholar] [CrossRef]
- Bakheit, A.H.; Ghabbour, H.A.; Hussain, H.; Al-Salahi, R.; Ali, E.A.; Mostafa, G.A.E. Synthesis and Computational and X-Ray Structure of 2, 3, 5-Triphenyl Tetrazolium, 5-Ethyl-5-Phenylbarbituric Acid Salt. Crystals 2022, 12, 1706. [Google Scholar] [CrossRef]
- Hossen, J.; Pal, T.K.; Hasan, T. Theoretical Investigations on the Antioxidant Potential of 2,4,5-Trihydroxybutyrophenone in Different Solvents: A DFT Approach. Results Chem. 2022, 4, 100515. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Spackman, M.A. CrystalExplorer: A Tool for Displaying Hirshfeld Surfaces and Visualising Intermolecular Interactions in Molecular Crystals. Acta Crystallogr. Sect. A Found. Crystallogr. 2006, 62, s90. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. IUCr SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Mamedov, V.A.; Mamedova, V.L.; Syakaev, V.V.; Voronina, J.K.; Mahrous, E.M.; Korshin, D.E.; Latypov, S.K.; Sinyashin, O.G. Regioselective Syntheses of 3-Hydroxy-4-Aryl-3,4,5-Trihydro-2H-Benzo[b][1,4]Diazepin-2(1H)-Ones and 3-Benzylquinoxalin-2(1H)-Ones from Arylglycidates When Exposed to 1,2-Diaminobenzenes. Tetrahedron 2020, 76, 131478. [Google Scholar] [CrossRef]
- Koenderink, J.J.; van Doorn, A.J. Surface Shape and Curvature Scales. Image Vis. Comput. 1992, 10, 557–564. [Google Scholar] [CrossRef]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld Surface Calculations, Non-Covalent Inter action (NCI) Plots and the Calculation of Interaction Energies in the Analysis of Molecular Packing. Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, 75, 308. [Google Scholar] [CrossRef] [PubMed]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Hydrogen Atomic Positions of O-H···O Hydrogen Bonds in Solution and in the Solid State: The Synergy of Quantum Chemical Calculations with 1H-NMR Chemical Shifts and X-Ray Diffraction Methods. Molecules 2017, 22, 415. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.; Andres, J.; Safont, V.S.; Campodonico, P.; Santos, J.G. A Theoretical Study on the Relationship between Nucleophilicity and Ionization Potentials in Solution Phase. J. Phys. Chem. A 2003, 107, 5588–5593. [Google Scholar] [CrossRef]
- Mukta, S.; Rahul, K.; Shekhar, S. Synthesis, Characterisation and Hirshfeld Surface Analysis of Some Mercury(II) Molecular Adducts with Macrocyclic Ligands. Res. J. Chem. Environ. 2023, 27, 46–57. [Google Scholar] [CrossRef]
- Stondus, J.; Kant, R. Crystallographic, DFT, Lattice Energy and Hirshfeld Surface Analysis of Some CSD-Based 6-Chloropurines. Chem. Pharm. Res. 2022, 4, 1–14. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).