Abstract
Roseolumiflavin is a deep red microcrystalline derivative of isoalloxazine that exhibits a weak photophysical activity in the solid state. In aqueous as well as in acidic solution of formic or acetic acid, respectively, it tends to form solvates. Herein, we present a set of binary and ternary roseolumiflavin solvates including one hydrate and a solvate hydrate. The impact of the solvent on solvate formation along with an in-depth structural analysis was investigated. Calculations of the lattice energies provide insight into the phase stability of the evaluated systems showing an energetic benefit for all solvates with values up to −395.82 kJ/mol. The total interaction energies between molecules calculated via Crystal Explorer further identified cofacial π···π stacks to be the most strongly bonding fragments in the crystal lattices for all systems except the formic acid solvate, followed by remarkably weaker hydrogen-bonded arrangements. The energetic contributions of single intermolecular interactions within the fragments are evaluated by an atoms-in-molecules approach. It is shown that physicochemical properties, such as thermal stability, can be tuned depending on the incorporated solvent molecules despite a high decomposition temperature of the chromophore.
1. Introduction
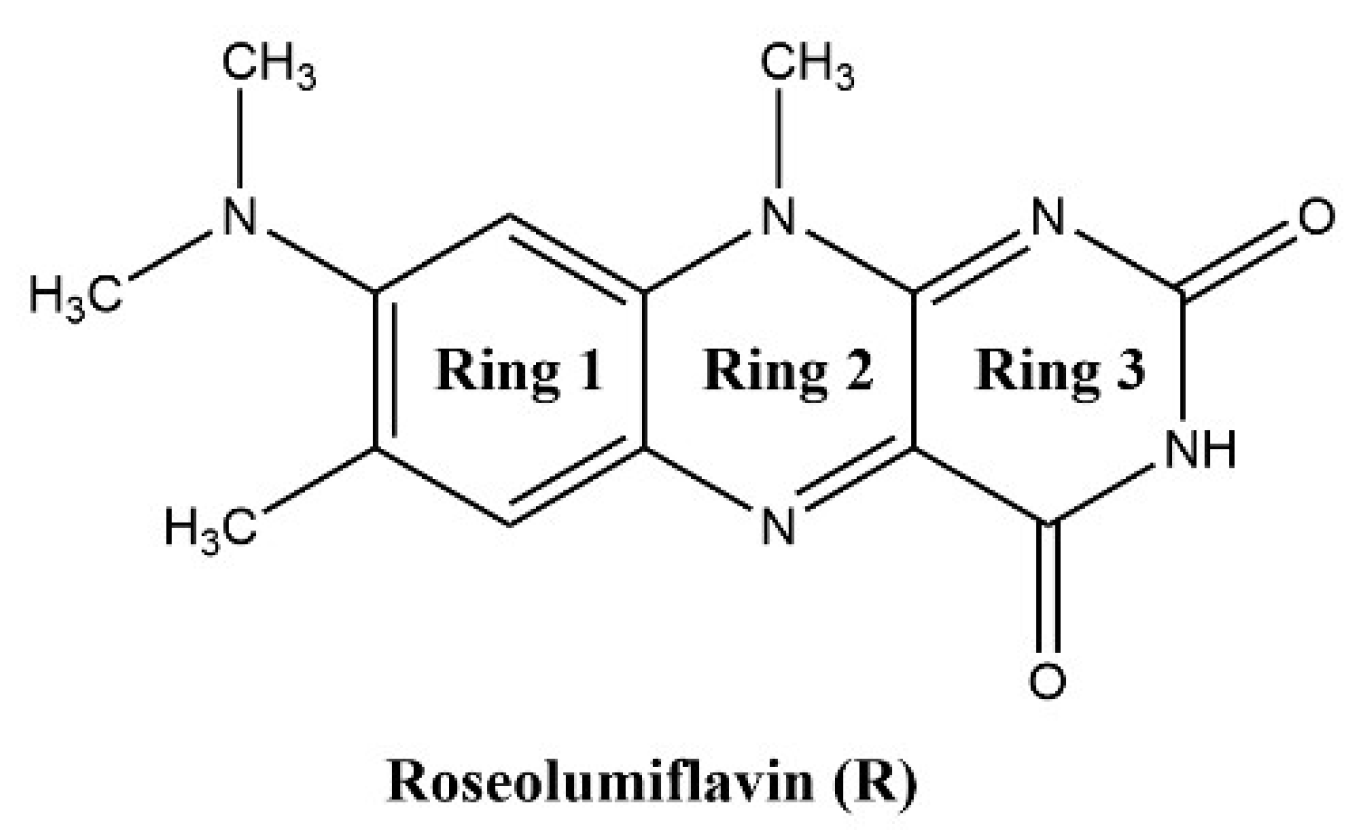
8-(Dimethylamino)-7,10-dimethylbenzo[g]pteridine-2,4(3H,10H)-dione, also known as roseolumiflavin (R), is an organic chromophore emitting deep red-light which is derived from the class of flavin compounds. Prior research on R has shown its capability to co-crystalize with a set of miscellaneous coformers, providing either hydrogen-bonded or halogen-bonded interaction motifs, which led to altered photophysical properties [1]. These findings display the versatility of such compounds with regard to its propensity to build up novel structures. Therefore, different rational design strategies could be adopted on solid state organic chromophores to enhance their applicability in a wide range of fields [2,3]. The aforementioned crystal engineering approach can be stressed out even further when solvate systems are considered. Solvates are solid phases of a compound that form in the respective environment, typically in solution or by exposition to solvent vapor, where solvent components are incorporated into the crystal lattice by weak interactions [4,5]. If the solvent is water, these systems are referred to as hydrates. Just like their co-crystalline counterparts, these multicomponent systems can alter physicochemical properties of their respective host molecule as well such as melting point, solubility, mechanical properties, stability, or processability, amongst others [5,6]. Setting a clear cut between terminologies for crystalline phases has been a topic of discussion. Reminiscent of the scientific debate regarding the justification of the term pseudopolymorphism [7,8,9,10], for example, we may also point to a recent proposal of Grothe et al. in 2016 to employ a set of seven classifications to differentiate between salts, co-crystals, and solvates to avoid ambiguity [11]. In the following, solvates and hydrates are merely used as a general term for a multicomponent system of R including either solvent molecules or, in the case of hydrates, water molecules. Knowledge about the occurrence and the circumstances resulting in solvate formation is of industrial importance, especially for pharmaceuticals and dyes, considering the effect that solvent inclusion solely based on intermolecular interactions can have on the compound properties in the crystalline state [4,12]. Granted, weak intermolecular interactions, the most prominent being the hydrogen bond, induce molecular recognition and provide stability for the crystalline state of organic compounds [13,14]. For example, the hydrate of the pharmaceutical drug phenibut, although thermodynamically not preferred, may form as a metastable intermediate in the transformation of phenibut and its salt phase, which is solely stabilized by weak intermolecular hydrogen bonds [15]. Likewise, in the case of the drug 5-fluorouracil, Heinen et al. faced challenges when producing solvent-free co-crystals due to the propensity of the corresponding hydrate to form highly stabilized hydrogen-bonded networks [16]. These examples show how solvates can compete with and hinder the desired outcome, e.g., co-crystal formation, generally in an unpredictable and uncontrolled manner. Indeed, solvates often appear as unwanted by-products during crystallizations or in synthesis processes. Thus, not much attention is paid to extensive investigations of the driving factors for the solvate formation. However, a combination of the solvates’ crystal structure interpretation and topological analysis of the involved intermolecular interactions on the one hand, alongside the evaluation of energetic contributions for their formation on the other hand, can provide important information, which can help to improve the prediction and to gain control over the desired phase crystallisation. With this work, we aim to encourage researchers not to disregard investigations of solvate and hydrate solid-state systems, as this set of multicomponent crystals in many cases contains useful information to add to a better understanding of properties, favoured mechanisms, and structural characteristics of its underlying component.
Herein, we report the discovery and structural characterisation of two new binary and one ternary solvates of R (Scheme 1) along with its hydrate: roseolumiflavin:formic acid solvate (1:1, R:HCOOH), roseolumiflavin:acetic acid solvate (1:1, R:AcOH), roseolumiflavin:water:acetic acid solvate hydrate (1:1:1, R:H2O:AcOH), and roseolumiflavin:trihydrate (1:3, R:3H2O).
Scheme 1.
Roseolumiflavin (R).
Intermolecular interactions present in the obtained crystal lattice of R and its multicomponent systems are further investigated in terms of their energetic impact on the phase formation, since this biologically relevant organic chromophore is still scarcely examined, especially on a molecular level. Besides our initial experimental article [1], to the best of our knowledge, only one publication exists investigating the photophysical activity of R in different virtual environments based on theoretical models [17]. In that regard, it is of importance to gain an in-depth understanding of the intricate interaction patterns in the crystal structures that ultimately construct the solid phases. Previous research indicated the strength of the hydrogen-bonded dimeric motif in the crystal structure of pure R, which has been a primary target of a co-crystallization strategy to functionalize [1]. This is also in agreement with prior publications of related flavin co-crystals [18,19,20,21], which determine hydrogen-bonded interactions to be the driving force for structural arrangement. Based on the crystalline structure of R, we proceed to scrutinize the strongest interactions present in the crystalline phase via both the atoms-in-molecules (AIM) method implemented in the MultiWFN package [22] and the model interaction energies approach [23] provided by CrystalExplorer21 [24]. These results are compared to those of the herein reported crystal structures of the roseolumiflavin solvates to gain better insight in the crucial determining factors for the assembly of multicomponent systems of the flavin.
2. Materials and Methods
2.1. Synthesis
1:1 Roseolumiflavin:formic acid solvate (R:HCOOH): Red needles of R:HCOOH were obtained from a solution of 3 mg of R in 1 mL formic acid and slow evaporation of the solvent at ambient temperature. R:HCOOH could be reproduced confidently via a solvent evaporation method. Bragg reflections [°2θ]: 6.4, 10.32, 14.7, 25.12, 27.0; IR [cm−1]: 3341, 3159, 3015, 2800, 1694, 1668, 1643; TGA [°C] (mass loss [%]): 119 (5), 371 (58).
1:1 Roseolumiflavin:acetic acid solvate (R:AcOH): Clear, red rectangular-shaped plates of R:AcOH were obtained from a solution of 3 mg of R in 2 mL acetic acid and slow evaporation of the solvent at ambient temperature. Reproduction of the structure was possible in arbitrary amounts of solvent via a solvent evaporation method. Bragg reflections [°2θ]: 6.44, 11.93, 14.4, 15.3; IR [cm−1]: 3428, 3280, 3207, 3065, 3040, 2811, 1688, 1644; TGA [°C] (mass loss [%]): 103 (16), 353 (52).
1:3 Roseolumiflavin:trihydrate (R:3H2O): Thin, light-red needles of R:3H2O were obtained from dissolution of 1 mg of R in 1 mL water and slow evaporation of the solvent. The hydrate precipitates immediately after R comes into contact with the solvent at ambient temperature. R:3H2O could be reproduced confidently via a solvent evaporation method. Bragg reflections [°2θ]: 7.26, 9.62, 10.94, 13.11, 15.97, 18.94, 30.22, 31.45, 33.36, 35.24, 41.42, 44.79; IR [cm−1]: 3379, 3249, 3131, 3070, 2995, 2804, 1695, 1637; TGA [°C] (mass loss [%]): 48 (15), 288 (51).
1:1:1 Roseolumiflavin:water:acetic acid solvate hydrate (R:H2O:AcOH): Red, block-shaped crystals of R:H2O:AcOH were noticed as a side product in vessels of R:AcOH samples due to their distinct morphology and were picked for X-ray measurements. Numerous further attempts for a targeted reproduction of the structure under variation of conditions such as solvent amount, additives, and temperature have failed; incremental addition of water resulted in precipitation of the hydrate. Thus, besides the theoretical calculations based on the crystal structure obtained from single crystal X-ray measurements, no further experimental analyses could be carried out for this particular compound.
2.2. X-ray Measurements
A Rigaku Miniflex diffractometer (Rigaku, Tokyo, Japan) was used for powder X-ray diffraction (PXRD) measurements in θ/2θ geometry at ambient temperature (20 °C) using Cu-Kα radiation (λ = 1.54182 Å). Single-crystal X-ray diffraction (SCXRD) measurements were carried out on a Bruker APEX Duo diffractometer (Bruker, Billerica, MA, USA) with a CCD detector, micro-focus X-ray tube, and Mo-Kα radiation (λ = 0.71073 Å) at 140(2) K for R:AcOH, R:3H2O and R:AcOH:H2O and a Rigaku XtaLAB Synergy-S diffraction system (Rigaku, Tokyo, Japan) with a HiPyx 6000 photon detector, and micro-focus X-ray tube with Cu-Kα radiation (λ = 1.54182 Å) measured at 100(2) K for R:HCOOH. Cell refinement, data collection, and data reduction on the Rigaku Synergy-S system were performed with CrysAlisPro [25]. On the Bruker APEX Duo, data collection and cell refinement were achieved with APEX2 [26] and data reduction was performed via SAINT [27]. Structure solution was conducted by SHELXT 2014/5 [28] for R:AcOH, R:3H2O, and R:AcOH:H2O and SHELXT 2018/2 [28] for R:HCOOH. R:AcOH:H2O was refined with SHELXL-2014/7 [28], R:AcOH and R:3H2O were refined with SHELXL-2017/1 [29] and R:HCOOH was refined with SHELXL-2018/3 [29]. R:HCOOH was refined with the Olex2 software package (OlexSys Ltd., Durham, England, v.1.5) [30]. All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were experimentally refined. All structures are deposited in the CCDC with following numbers: 2294770-2294773.
2.3. Thermogravimetric Analysis (TGA)
TGA measurements were performed on a Netzsch TG 209 (NETZSCH, Selb, Germany) in the range between 30 °C and 600 °C with a 10 °C min−1 heating rate under a nitrogen atmosphere. In total, 5 mg of each compound was analysed.
2.4. FTIR Measurements
The FTIR spectra were recorded on a Bruker Tensor 37 (Bruker, Billerica, MA, USA) with an ATR-unit at ambient temperature (25 °C) in the range between 4000 cm−1 and 400 cm−1.
2.5. Chemicals
Roseolumiflavin was synthesized as reported previously [1]. Formic acid (≥98%) and acetic acid (≥99.8%) were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA) and used without further purification. Deionized water was used for the hydrate crystallisation.
2.6. Software
The software package MultiWFN [22] was used for AIM calculations based on .wfn files of molecular geometries based on crystal structure data. The .wfn files were generated by a density function theory (DFT) method using Gaussian16 [31] at a B3LYP/6-31G** level of theory for comparability with calculated model interaction energies [23] derived from CrystalExplorer21 [24], which are parametrized for the same combination of functional and basis set. Quantum Espresso (QE) PWSCF (Quantum ESPRESSO Foundation, Cambridge, UK, v. 6.6) was utilized for calculations of lattice energies with the atomic pseudopotentials originating from the QE pslibrary, following the approach described by Komisarek et al. in 2022 [32], with the exception that initial geometry optimization was done via the “relax” command. Hydrogen bond interaction energies based on bond critical points were predicted via the model proposed by Emamian et al. in 2019 [33]. Mercury 2022.3.0 [34] was used for structural figure preparation, and verbose crystal structure information was derived from PLATON for Windows Taskbar (A.L.Spek, Utrecht University, Utrecht, The Netherlands, Version 1.19) calculations [35].
3. Results
3.1. Single Crystal X-ray Diffraction (SCXRD)
Four new solvate structures, including a hydrate, were yielded from single crystal X-ray measurements. In the following section, a structural analysis of each structure is provided. Table 1 displays the overview of the important crystallographic data for R:HCOOH, R:AcOH, R:3H2O, and R:AcOH:H2O. In all the presented structures, no (de)protonation is observed, verified additionally by IR-analysis (see Figure S1 in ESI).

Table 1.
Crystallographic data of R:HCOOH, R:AcOH, R:3H2O and R:AcOH:H2O.
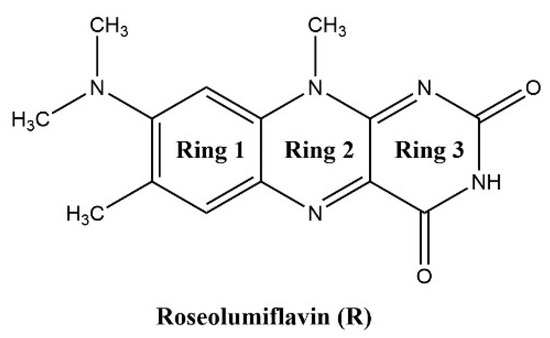
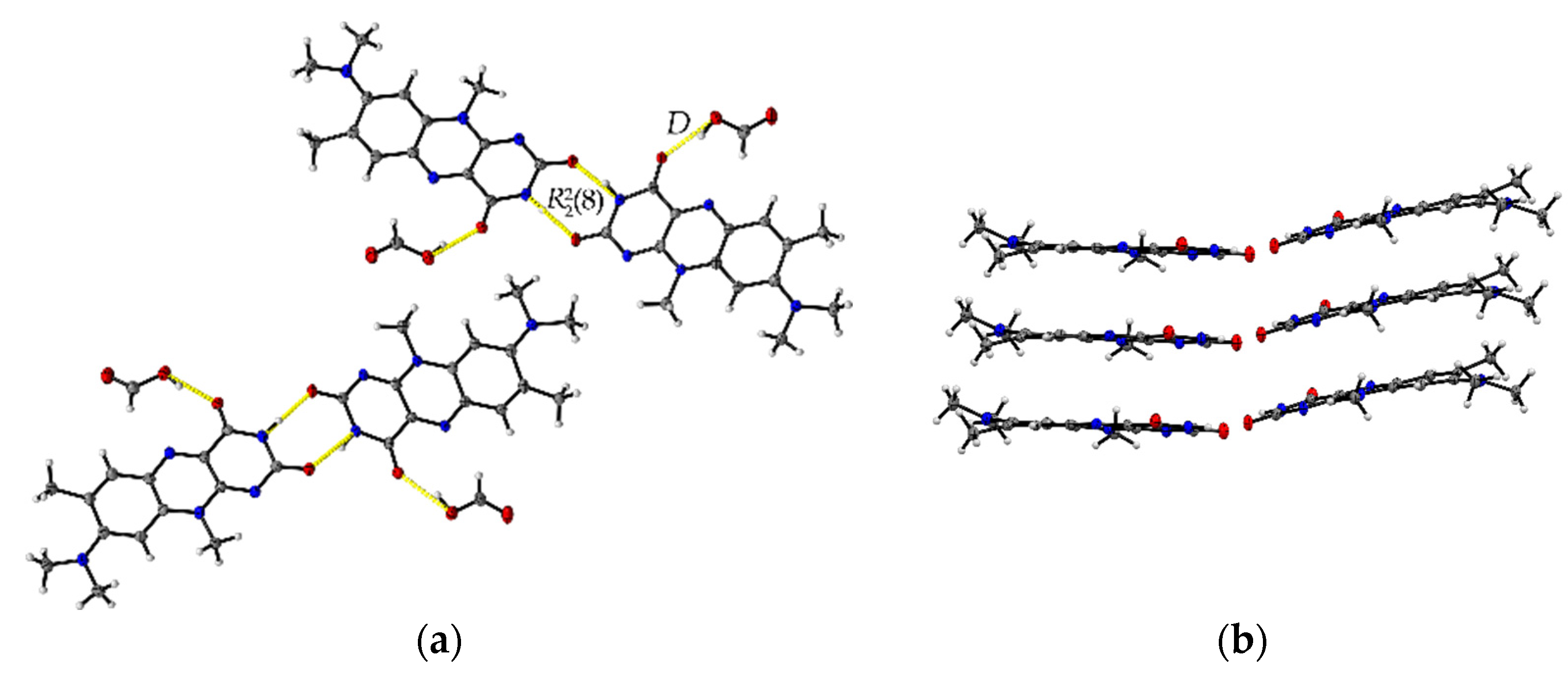
The formic acid solvate R:HCOOH (1:1) crystallizes in the monoclinic space group P21. The asymmetric unit contains two units of each molecule (Z = 4, Z’ = 2). Each R molecule is connected to an adjacent formic acid molecule via hydrogen-bonded (HB) interactions (graph set notation D [36] O3-H3···O1: 2.577(5) Å and O7-H7A···O5: 2.598(6) Å). Amide-amide homodimeric interactions ((8), N2-H2···O6: 2.828(6) Å and N7-H7···O2: 2.768(6) Å) between the imide groups of each two neighbouring R units propagate along the crystallographic b-axis, constructing tetrameric motifs of two R and two acid molecules, see Figure 1a. The same amide-amide dimer is present in the single-component R structure, as described previously [1]. Along the c-axis, these tetrameric motifs organize themselves in a herringbone arrangement mode. The R molecules are further arranged along the a-axis in a cofacial π···π stacking orientation (centroidout···centroidout: 3.796(3) Å, centroidmiddle···centroidmiddle: 3.796(3) Å) parallel towards each other, see Figure 1b.
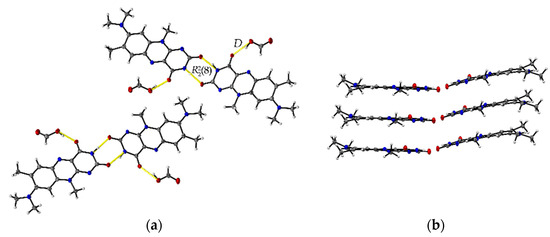
Figure 1.
(a) Herringbone arrangement of tetrameric substructures along the c-axis in R:HCOOH. View along the a-axis. (b) Propagation of π···π interacting flavin stacks along the a-axis. View along the c-axis.
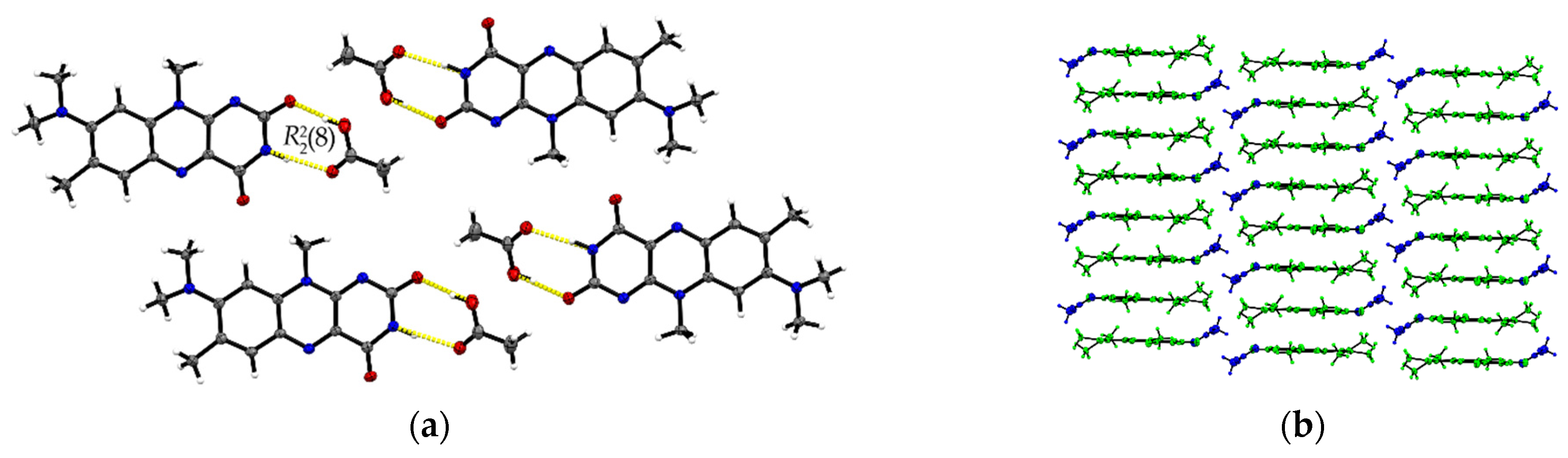
The R:AcOH (1:1) solvates crystallize in the triclinic P space group with one acetic acid molecule and one R molecule in an asymmetric unit cell (Z = 2, Z’ = 1). Along the crystallographic a-axis, two R molecules align via strong inversely oriented cofacial π···π stacks (centroidout···centroidout: 3.5008(14) Å, centroidmiddle···centroidmiddle: 3.4567(13) Å). The stacks build up brick layer structures in the crystal packing connected by offset π···π interactions (C2···C10 3.323(4) Å, see Figure 2b).
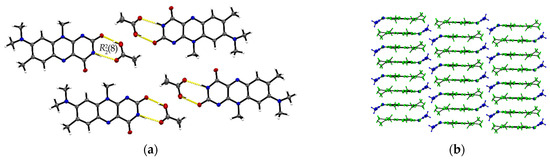
Figure 2.
(a) Hydrogen-bonded acid-amide dimers of R and acetic acid in R:AcOH. View along the crystallographic b-axis. (b) Stacking motifs formed along the a-axis. Coloured by symmetry equivalent entities (R: green, acetic acid: blue). View perpendicular to bc-plane.
Opposite to the R and R:HCOOH structures, the amide-amide homodimers in R:AcOH are replaced by an acid-amide heterodimeric motif. An acetic acid molecule interacts with the flavin’s imide group ((8), O3–H···O2: 2.596(2) Å, N2–H···O3: 2.879(2) Å). Adjacent acetic acid molecules support the dimeric synthons via short C16–H···O2 contacts (3.420(3) Å) to give tetrameric units. Further, tetramers are connected via multiple R molecules stabilizing the pattern by C–H···O interactions of average 3.2–3.5 Å, in particular facilitated by the convenient orientation of the R methyl substituents to form narrow hydrogen-bonded layers along the ab-plane (Figure 2a).
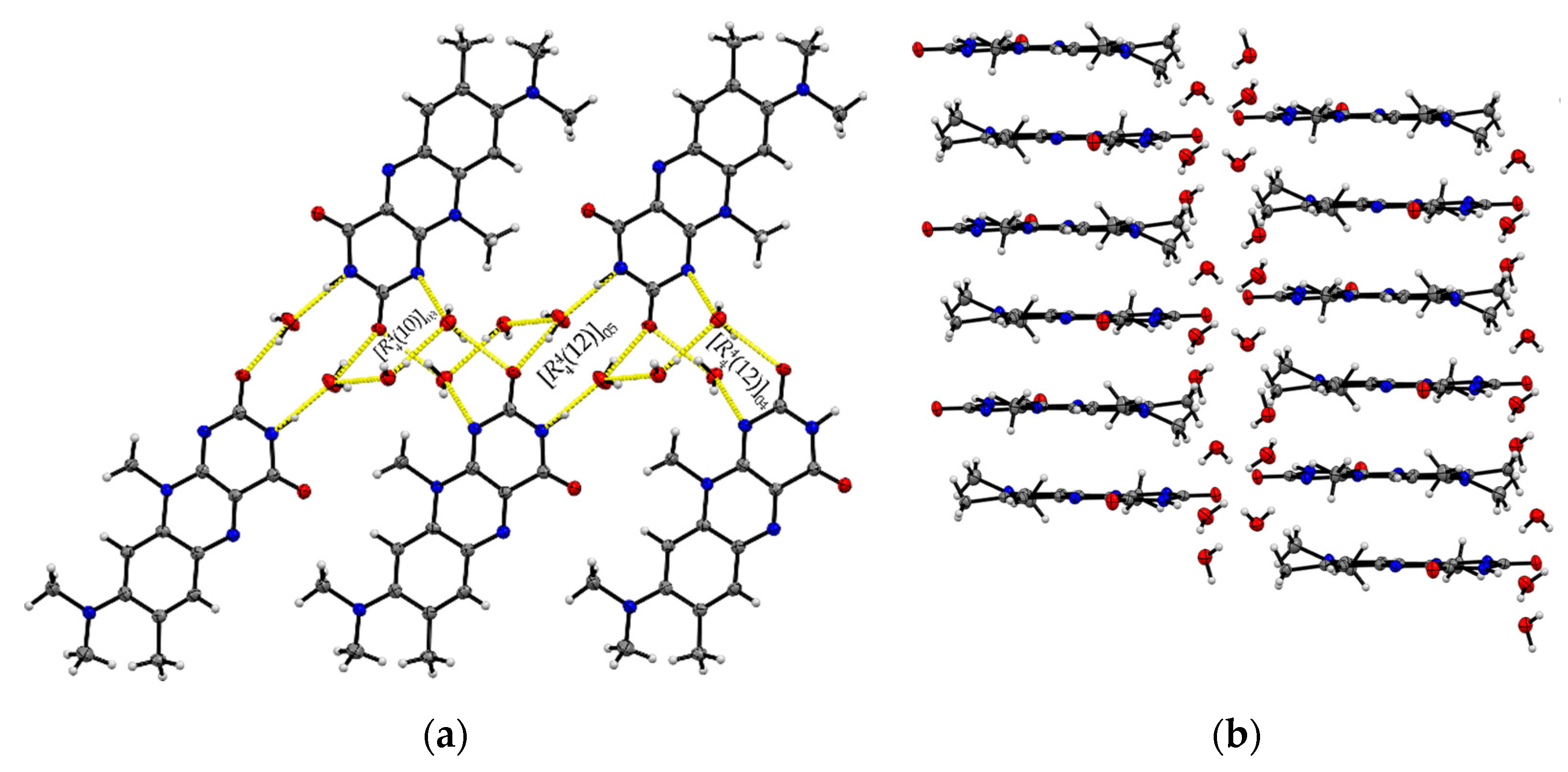
Single crystals of R:3H2O (1:3) are of a remarkably thin, fine needle-like shape in bright red colour, which tend to agglomerate immediately. The needles are sized between 0.01 mm and 0.05 mm in width and up to 0.5 mm in length. The resolved structure belongs to the triclinic space group with one R and three water molecules in an asymmetric unit cell (Z = 2, Z’ = 1). R molecules align via π···π interactions along the crystallographic a-axis and construct slip-stacked interaction geometries (centroidout…centroidout: 3.678(4) Å and 3.597(4) Å, centroidmiddle…centroidmiddle: 3.697(4) Å and 3.678(4) Å). These stacking motifs are separated along the c-axis by water molecules that replace the typical amide-amide synthon and instead provide several O–H···O and N–H···O interactions between the flavin stacks (N2–H···O5: 2.774(5) Å, N3–H···O4: 2.981(5) Å, O1–H···O5: 2.752(4) Å, O1–H···O4: 2.759(4) Å) as well as O–H···O interactions to neighbouring water molecules (O3–H5···O4: 2.833(5) Å, O3–H4···O4: 2.887(4) Å, O3–H···O5: 2.805(5) Å). This results in a much more complex crystal architecture when compared to all other reported structures, which consist of alternating R stacks and hydrogen-bonded sequences of water molecules (Figure 3b), propagating along the b-axis. Among the complexity of the present HB interactions, we can identify three main ring-motives, each built up by four molecules. Hereby, two similar tetrameric subsequences of the type [(12)], constructed of two R and two water molecules each, form a zig-zag chain (10)[[(12)]O5, [(12)]O4] (Figure 3a). The third ring system [(10)]O3 encloses one R and three water molecules which allows a connection to the next stacked layer. The short contacts between neighbouring R molecules provided by the C11-methyl side chain and the amide group (C11–H···O2: 2.66 Å) allow for the expansion of the flavins along the b-axis.
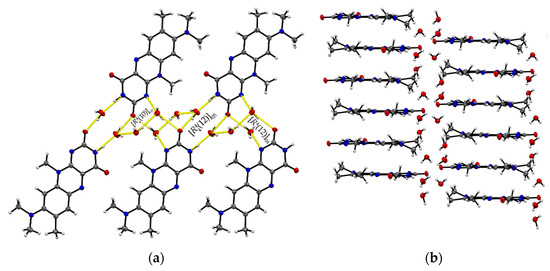
Figure 3.
(a) The imide group in R:3H2O interacts with water molecules giving different tetrameric ring motifs; those build up hydrogen-bonded chains ( (10)[[(12)]O5, [(12)]O4]) along the crystallographic b-axis. View along the a-axis. (b) R stacks are connected via complex O–H···O interaction patterns involving water entities.
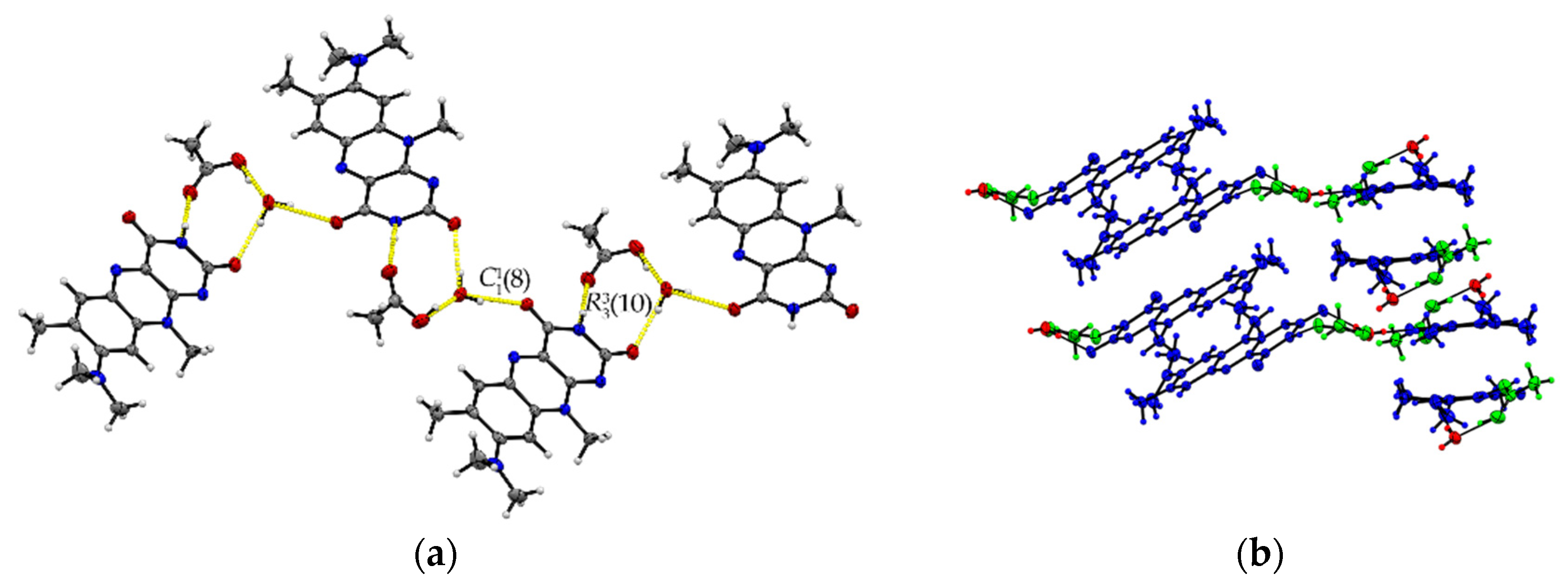
The ternary R:AcOH:H2O (1:1:1) solvate hydrate crystallizes in the monoclinic P21/c space group and the red block-shaped crystals appear in sizes between 0.04 mm to 0.20 mm. One molecule of each individual component is found in the asymmetric unit (Z = 4, Z’ = 1). Remarkably, similar to R:AcOH, an amide-amide dimeric motif typical for flavins is replaced by a supramolecular acid-amide synthon (Figure 4). However, not only does one acetic acid molecule serve as a substitute but is supported by a water molecule to give strongly bound trimeric motifs with donor-acceptor distances O5–H···O4 (2.7077(18) Å) and N3–H···O2 (2.952(2) Å). Such an arrangement enables tilted R stacks at 34.93° angle along the b-axis due to the flexible positioning of the small solvate molecules. The slip-stacked π···π connections (centroidout···centroidout: 3.5732(10) Å, centroidmiddle···centroidmiddle: 3.5545(9) Å) are formed with one flavin molecule oriented inversely to the other one along the a-axis. Each isolated, nearly cofacial stack is alternating with a neighbouring offset stack (C2···N4: 3.429(2) Å and C7···N2: 3.4108(19) Å). The tilted stacks are stabilized by hydrogen-bonded zig-zag chains built up of trimers along the b-axis (graph set notation (8)(10)). Those are mainly induced by water molecules serving as a mediator between the carbonyl groups of two flavin molecules (O5–H···O3: 2.8608(17) Å) and one acetic acid component, itself stabilized by the O1–H···O5 (2.5483(19) Å) interaction.
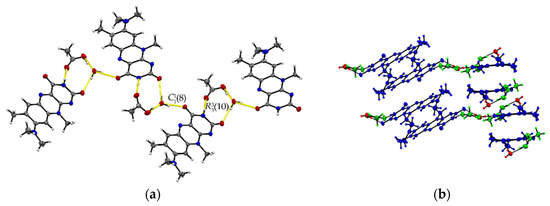
Figure 4.
(a) Hydrogen-bonded chains between solvate O–H groups and R amide groups connect trimeric motifs along the b-axis in R:AcOH:H2O. View along the a-axis. (b) Quasi-cofacial π···π stacks connected by offset stacking interaction alternate along the a-axis. View along the c-axis. Coloured by symmetry inequivalence for clarity (R: blue, acetic acid: green, water: red).
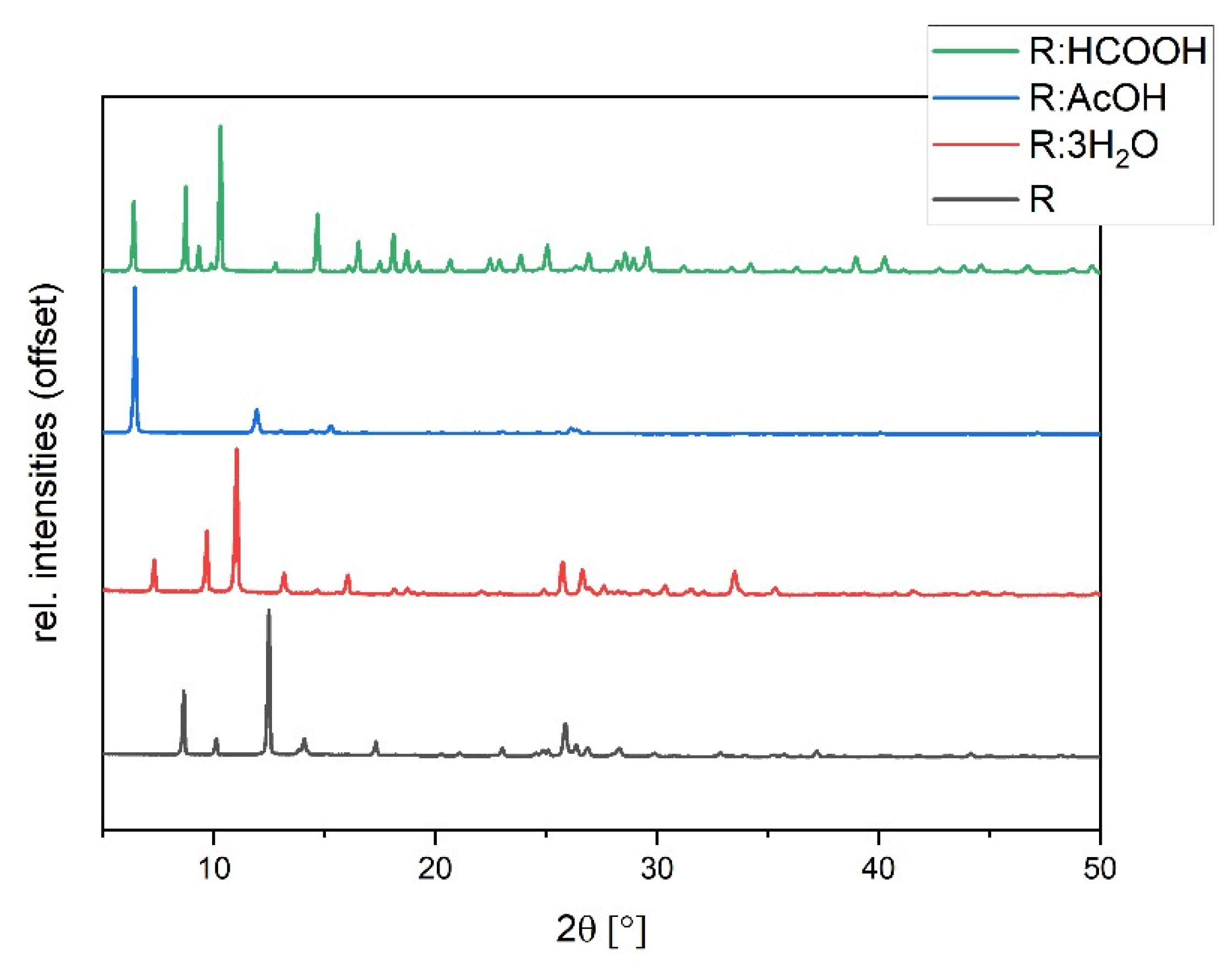
3.2. Powder X-ray Diffraction (PXRD)
The synthesized solvates were characterized via powder diffraction pattern analysis. In comparison to pure R, clearly distinct new diffraction patterns can be observed, verifying the synthesis of new phases (Figure 5). Experimental powder patterns are reproducible in agreement with the simulated patterns obtained from SCXRD data (see Figures S2–S4 in ESI), which confirms the overall phase purity for R:3H2O, R:HCOOH, and R:AcOH. Hereby, R:AcOH shows a strong preferred orientation of the (0 0 1)-plane in the experimental powder pattern. The simulated PXRD was therefore adapted with a March–Dollase factor of 0.6 for a better comparison (Figure S2 in ESI).
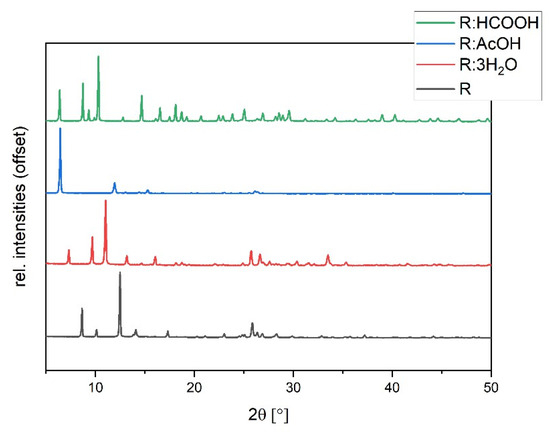
Figure 5.
Experimental powder diffraction patterns of R:AcOH, R:3H2O, R:HCOOH in comparison to R.
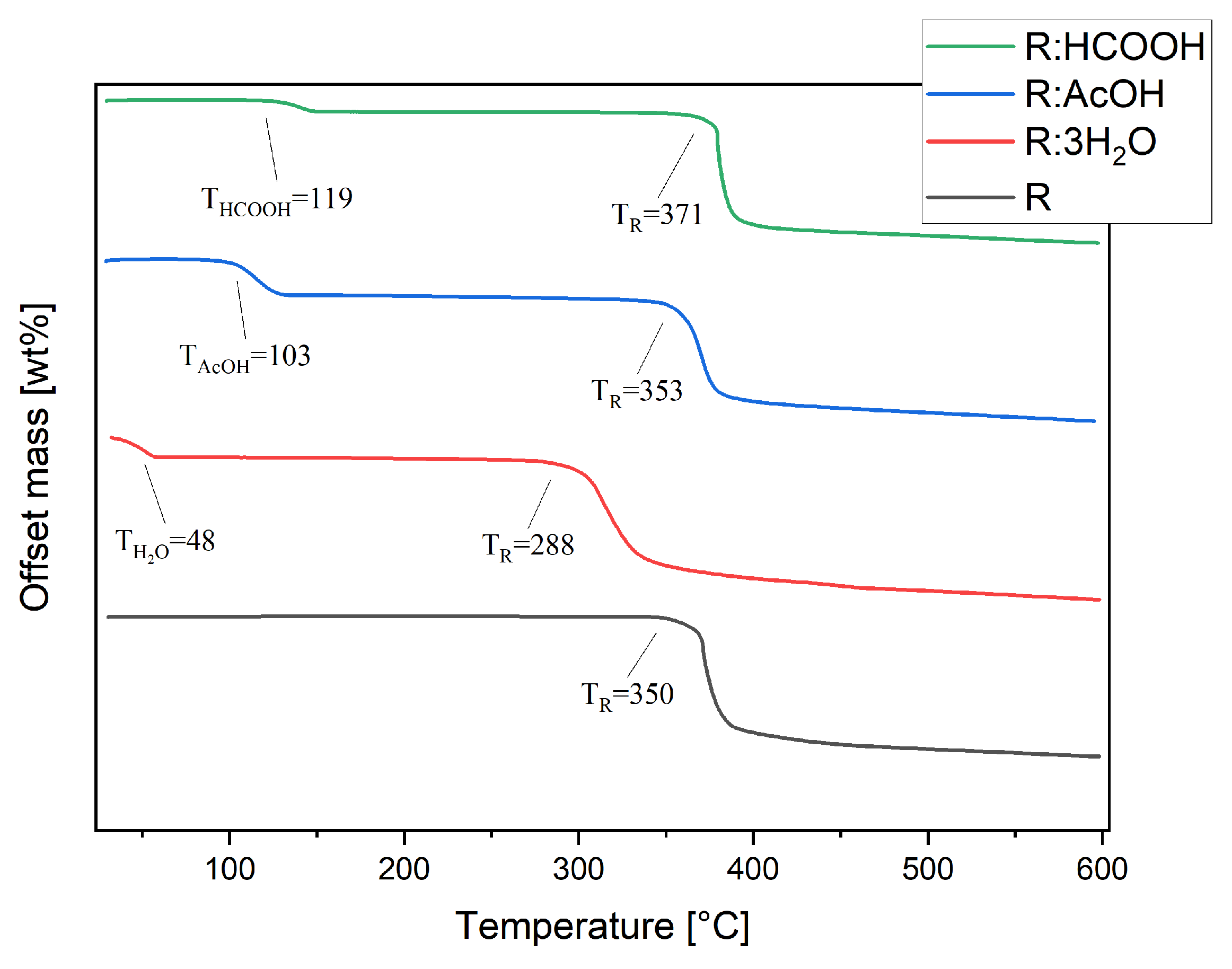
3.3. Thermogravimetric Analysis (TGA)
Thermal stability studies were conducted by thermogravimetric analysis of each compound, including R. Pure R decomposes in one step at TR of 350 °C (Figure 6). The thermogram of R:AcOH features two mass losses. The first one, with an onset of 103 °C and a mass loss of 16%, is apparently due to the solvent release from the crystal lattice. This equates to 0.93 moles of solvent molecules. The second one at 353 °C is indistinctively attributed to the decomposition point of R with a mass loss of 52%. R:3H2O also degrades in two steps. The first with an onset at 48 °C indicates an early evaporation of water, causing 15% of a mass decrease (2.82 moles of water molecules). The second step at 288 °C (mass loss of 51%) shows a remarkably strong shift in decomposition temperature of R to lower temperatures.
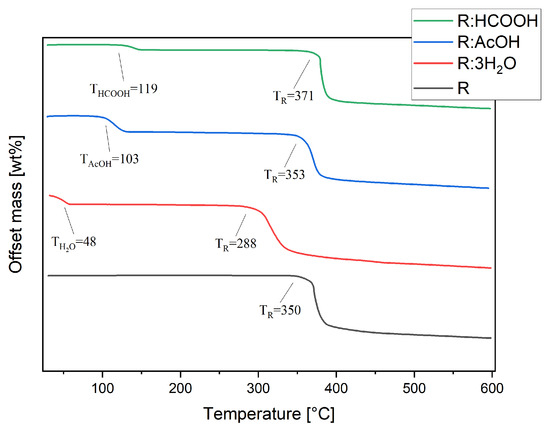
Figure 6.
Thermogravimetric analyses of R, R:AcOH, R:3H2O and R:HCOOH taken from 30 to 600 °C, under nitrogen atmosphere at 10 K/min heating rate. Extrapolated onset decomposition temperatures are given.
For R:HCOOH, the first step at 119 °C with a mass loss of 5% indicates the release of the formic acid. This corresponds to only 0.35 mole of the solvent, which is probably caused by a partial loss of solvent molecules from the crystal lattice prior to the TG analysis during the sample storage. The second step, again representing the start of decomposition of the chromophore, is set at 371 °C with a mass loss of 58%, reaching a significantly higher decomposition point of the chromophore.
3.4. Computational Studies
An evaluation of the phase stability of the new structures is investigated by comparison of lattice energies and topology analyses via the atoms-in-molecules (AIM) approach, as well as intermolecular interaction energies. Based on the method described and validated by Komisarek et al. [32], lattice energies (Elat) for each solvate structure and R are calculated. The results demonstrate that the formation of all reported solvate structures is driven by a considerable gain in energy when compared to pure R with its Elat of −181.08 kJ/mol (Table 2) This explains why the formation of the solvate phases is preferred in the respective solvent environment. Hereby, R:HCOOH shows just a slight energetic benefit with −197.58 kJ/mol. A noteworthy observation is the relative positioning of energies for R:AcOH:H2O with Elat of −352.46 kJ/mol, which is set between both R:AcOH and R:3H2O, showing lattice energies of −283.55 kJ/mol and −395.82 kJ/mol, respectively. Since in all our attempts to systematically reproduce said system, the hydrate formation was favoured, these findings indicate that the ternary system is an intermediate product when both solvents are present, thus a targeted crystallization of that intermediary stage is challenging. The formation of R:3H2O is the most beneficial, which goes along with our experimental observation of its immediate precipitation once R is treated with water.
Table 2.
Overview of the calculated lattice energies (Elat), strongest pairwise interaction energies (Etot) of a central flavin molecule in a 3.8 Å cluster, and estimated binding energies (BE) for HB interactions for R, R:HCOOH, R:AcOH, R:AcOH:H2O, and R:3H2O.
To gather a profound understanding of the driving forces that lead to the formation of the respective crystal lattice, the specific molecular surroundings of the chromophore weres investigated for each structure. For this, we utilized the model interaction energies method implemented in CrystalExplorer21 [23]. Here, a 3.8 Å cluster of molecules encompassing one flavin molecule is generated, approximating the first interaction environment of the selected central molecule. The energy of interaction acting upon the central flavin molecule with each of the neighbour molecules within the cluster is calculated via a DFT approach at a B3LYP/6-31G** level of theory. For each two interacting molecules, the total interaction energy Etot, which encloses the sum of electrostatic (Eele), polarization (Epol), dispersion (Edis), and exchange-repulsion (Erep) energies, each multiplied with respective scaling factors, is calculated and can be quantified. This approach provided us a first overall understanding of the most strongly bonded molecular fragments in each crystalline phase, which dominate and drive their architectures. The highest impact within the total interaction energy between the molecules is provided by the intermolecular interactions involved. Following this, we approximate the strongest contributors. For each structure, the four highest calculated total energies of molecular interaction are given in Table 2, along with the assigned contributing intermolecular interactions. A complete table is provided in the ESI (Table S5). For R:3H2O, additional interactions are given, generally of a weaker total energy but of a high binding energy, which will be discussed further.
Evaluation of the pairwise interactions for R via CrystalExplorer shows the highest values being supplied by units that provide π···π stacks as well as the aforementioned amide-amide hydrogen-bonded dimer between two flavins. Noteworthy, a flavin moiety adjacent to the dimethylamine residue provides some strong molecular interaction energies in close energetic range as well, which can be found in nearly all structures except R:HCOOH, with Etot values around −30 kJ/mol. The highest contribution in R:AcOH, R:AcOH:H2O, and R:3H2O again is given by cofacial π···π stacked units with total interaction energies around −110 kJ/mol. Still, cofacially stacked flavin moieties provide the highest total molecular energetic gain in R with up to −127.1 kJ/mol. The contribution energies drop down to −64 kJ/mol the more the stacks are offset in the arrangements present in R:AcOH, R:AcOH:H2O. Solely in R:AcOH, a pairwise interaction of a solvate molecule and the flavin’s imide group, which corresponds to HB (−62.0 kJ/mol), provides an energetic benefit at a similar range compared to the stacks. Otherwise, all hydrogen-bonded fragments corresponding to amide-amide, acid-amide dimers, or interactions with water molecules in R, R:AcOH, R:AcOH:H2O, and R:3H2O exhibit highest Etot values between −30 to −43 kJ/mol, being clearly outnumbered by the π···π stacking. The R:HCOOH structure stands out of all investigated systems with an absolute value of the π···π interactions contributing fragment being significantly lower than in the other structures. Here, the strongest total energy is directly induced by the formic acid unit, which interacts via hydrogen bonds with the flavin.
Finally, based on these initial observations and results, an AIM analysis [37] shall elucidate whether bond critical points (BCPs), in line with the underlying theoretical concept, do exist along the respective pathways, aiding in the identification of the respective interactions. Additionally, by applying a fitted equation provided by Emamian et al. [33] we estimate the binding energies (BE) of the located HBs. In this way, an energetic contribution of the single intermolecular bond can be verified. To the best of our knowledge, no equation for the estimation of π···π interactions is available yet. The AIM analysis confirms bond critical points with stabilizing interaction energies located between the dimeric units of two R molecules in the pure flavin structure. The estimated binding energies are summarized in Table 2. These interactions can be identified as moderately strong hydrogen bonds at −19.3 kJ/mol, particularly in relative comparison to the further identified hydrogen-bonding interactions between the dimethylamine residue and the imide group of two adjacent R moieties, ranging between −2.3 and −5.1 kJ/mol. The structural positioning of the units allows further C6-H···O1 and C11-H···O1 HBs with similar energetic gain. Comparable stabilizing interactions are observed in R:AcOH, with binding energies ranging between −2.4 and −4.2 kJ/mol, which is slightly lower than those in R. In R:AcOH, the dimeric flavin unit is replaced by hydrogen-bonded motifs with the acid unit. In absolute values, the binding energies are significantly stronger when compared to the hydrogen bonds provided in the amide-amide synthon in the R crystal lattice. In R:AcOH:H2O, moderately strong interaction motifs are provided not only by the water molecules but the acetic acid unit as well. The water molecules interact with the imide group over O4···H-O5 (−23.4 kJ/mol) and O3···H-O5 (15.4 kJ/mol) HBs. The acetic acid units provide stabilizing HB via N3-H···O2 at 14.7 kJ/mol and even weak interactions with the solvate methyl group via O3···H-C1 at −3.6 kJ/mol. In R:3H2O, the dimeric motif has also been replaced by water molecules. Comparing the binding energies, we observe interactions around 1.5 times higher than the amide-amide synthon in pure R. The strongest interaction is observed between O5···H-N2 at −30.1 kJ/mol, followed by an adjacent water molecule interacting with the carbonyl group via O1···H-O5 with an energetic benefit of −22.94 kJ/mol and further stabilizing interactions with energetic values around −10 kJ/mol. On the other hand, the aforementioned methyl group HB interactions to neighbouring flavin units are weaker ranging from −0.92 to −2.69 kJ/mol. Lastly, in R:HCOOH we identified strong HBs between acid units and the flavin with −31.15 (O3-H···O1) and −27.75 kJ/mol (O7-H···O5), while the dimeric hydrogen-bonded flavin units also contribute to the stability with −25.88 and −22.88 kJ/mol, respectively. Moreover, according to our AIM analyses, the structural peculiarity of tilted adjacent flavin molecules with their dimethylamine residue directed towards the nearby carbonyl group provide even further stabilizing effects with −8.95 kJ/mol between C28-H···O6 and −4.88 kJ/mol between C13-H···O5.
4. Discussion
Our findings display the versatility of R with respect to solvation, evident not only in the formation of various compounds but also in their altered crystal structure. While an amide-amide synthon between two flavin molecules is present in both R and R:HCOOH, in the other solvates the solvent molecules replace these hydrogen-bonded interactions with additional stabilizing benefits in their respective crystal lattices. The estimated HBs in total outweigh the already moderate to strong hydrogen bonded amide-amide motif. Considering identified π···π interactions, what stands out is the strength of the fragment contributions being significantly higher in comparison to the HBs, at around −100 to −120 kJ/mol in cofacial systems and around −65 to −70 kJ/mol when offset. The strongest molecular contribution by cofacial stacks is present in the pure R structure followed by R:AcOH:H2O, R:AcOH, and R:3H2O. Noteworthy, the R:HCOOH stacks merely provide energetic benefits at −36.6 kJ/mol, which are around one-third of the observed values in the other structures. Comparing the π⋯π distances with those of pure R, we find overall slightly shorter distances between the flavin units in R:AcOH and R:AcOH:H2O, similar range distances in R:3H2O, and longest distances in R:HCOOH. See Table 3 for the respective values.
Table 3.
π⋯π Distances measured for R, R:AcOH, R:3H2O, R:AcOH:H2O and R:HCOOH.
In the case of R:HCOOH, the solvate molecules add to the stability of the dimeric unit by the formation of HB alongside the imide group. Further, graph set notation analyses show that the presence of water molecules results in the formation of hydrogen-bonded chains in both R:3H2O and R:AcOH:H2O, whereas the two other solvates are limited to finite interactions. These added benefits are represented in the calculated lattice energies as well, considering calculated Elat decreases along the row R > R:HCOOH > R:AcOH > R:AcOH:H2O > R:3H2O. This results in the most stable lattice for the R trihydrate. The ring distances, however, do not vary by large values; the longest and shortest absolute centroid-centroid distances observed in R:HCOOH and R:3H2O, respectively, merely differ by ca. 0.2 Å in comparison to R. Assuming the intermolecular interactions to be of significance, this suggests the aforementioned HBs to be the primary determining factor for the lattice energy alteration. The high energetic benefit of the hydrate further confirms the experimental observations, where the slightest presence of water in solvate samples resulted in an exclusively preferred formation of the hydrate. This is due to the contribution of two strong HB between water and flavin molecules. Furthermore, graph set notation analyses display the complexity of the hydrogen-bonded motifs building up HB chains that enclose two hydrogen-bonded rings and further ring-motifs involving water molecules, which contribute to the close interaction of the flavin layers in the crystal structure. Here, AIM calculations show binding energies of up to −30 kJ/mol, being considerably predominant compared to HBs of −19.34 kJ/mol in pure R. The second lowest lattice energy is attributed to the ternary R:AcOH:H2O system, which in absolute values is to be set between R:AcOH and R:3H2O. Again, this is in agreement with our experimental observations and could offer an explanation for the failed attempts to reproduce the structure. Since the energetic benefit from R:3H2O is significantly higher than in R:AcOH, the hydrate formation is thermodynamically preferred. Nonetheless, the ternary system can be an intermediary product in a reorganization process from a first step acetic acid solvate formation towards the building of the hydrate. As a result, the intermediary product is then accessible in randomized occasions, but solely as a side product.
On the other hand, from TG analyses we observed a strongly shifted decomposition point of the chromophore towards lower temperatures in the hydrate at 288 °C when compared to that of R at 350 °C. While R:AcOH is not significantly altered at this point, R:HCOOH shows a significantly higher decomposition temperature for the R component at 371 °C. The thermal stability of R in the solvate systems with TR(R:HCOOH) > TR(R:AcOH) > TR(R:3H2O) nearly counterpose order derived from Elat. We suggest that the structural alteration in the hydrate may be of such significance that an optimized reorganization of the flavin molecules after solvent vaporization is hindered, resulting in earlier decomposition. In addition, the strongest hydrogen bonds formed between water and flavin molecules are at once missing after the water release. On the other hand, the chromophore may rearrange to an even more stable arrangement when the formic acid components evaporate, resulting in a later decomposition point. This might be due to the initial arrangement of stacked flavin units in the R:HCOOH crystal lattice differing significantly from those of the other compounds. From the one side, R:HCOOH is the only solvate structure in which the R-R amide-amide dimer remained intact, and, on the other side, the only one where flavin molecules are not inversely stacked. While this could be the cause of the higher calculated Etot due to a positive electrostatic component (see Table S5 in ESI for the detailed information of the components of Etot), the molecular rearrangement after the thermal activation may cause altered agglomeration of flavin stacks.
5. Conclusions
Within the scope of crystal engineering, challenges arise when solvate systems are considered. Often solvates and hydrates are an unwanted by-product in the experimental setting, yet these multicomponent systems can provide valuable insight into the investigated compounds. Here, we took a deeper look into the structural and energetic environment of three solvates as well as one hydrate of roseolumiflavin. These compounds display interesting features when compared to the flavin, not only from the crystallographic perspective, but based on observations on thermal properties as well. Previous research assumed that the hydrogen-bonded dimeric motif between two adjacent flavin units in roseolumiflavin provides significant benefits in terms of stability, next to the π⋯π stacking interactions. While the latter is present in all solvate systems, the former is replaced by hydrogen-bonded motifs with the respective solvent molecules in all but the R:HCOOH structure. Energetic calculations of pairwise molecular interactions underline the strength of cofacial π⋯π stacking, followed by the offset stacks. Hence, stacking interactions remain the most important driving force in the formation of R and its solvate structures. HBs are energetically considerably outnumbered in average by −40 to −70 kJ/mol. However, confirmed by AIM calculations, high binding energies of single hydrogen bonds in combination with the large overall number of such interactions seem to be able to promote the formation of new phases. The significance of energetic benefits is also evident in the calculated lattice energies. While R:HCOOH provides a more stable lattice by only ca. −16 kJ/mol when compared to R, the lattice energy of R:3H2O is more than twice as low as the pure flavin. And yet, the experimental observations show that from a thermal behaviour aspect, the hydrate leads to significantly earlier decomposition of the chromophore, while the presence of the formic acid component delays the decomposition of R by around 20 °C. To conclude, calculations on the theoretical level based on single crystal structures along with the careful examination of the interaction modes can provide valuable inputs into the crystal architecture and allows a detailed description of its topologies. A reliable prediction of tuneable physicochemical properties, however, remains challenging not least due to the complexity and versatility of roseolumiflavin systems.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst13101512/s1, Figure S1: Comparison of IR spectra of R:HCOOH, R:AcOH, R:3H2O with R. Characteristic bands are highlighted; Figure S2: Experimental powder pattern of R:AcOH compared to simulated patterns of R:AcOH and adjusted simulated pattern for consideration of preferred orientation from single crystal data; Figure S3: Experimental powder pattern of R:3H2O compared to simulated pattern from single crystal data; Figure S4: Experimental powder pattern of R:HCOOH compared to simulated pattern from single crystal data; Figure S5: Asymmetric unit of R:HCOOH. View along the crystallographic a-axis; Figure S6: Asymmetric unit of R:AcOH. View along the crystallographic a-axis; Figure S7: Asymmetric unit of R:3H2O. View along the crystallographic a-axis; Figure S8: Asymmetric unit of R:AcOH:H2O. View along the crystallographic a-axis; Table S1: Geometries of hydrogen-bonded interactions of R:AcOH; Table S2: Geometries of hydrogen-bonded interactions of R:3H2O; Table S3: Geometries of hydrogen-bonded interactions of R:AcOH:H2O; Table S4: Geometries of hydrogen-bonded interactions of R:HCOOH; Table S5: Crystal Explorer model interaction energies calculations, overview of strongest molecular interaction energy contributions in 3.8 Å cluster on central flavin unit with verbose parameters.
Author Contributions
Conceptualization, T.H.H.S. and V.V.; methodology, T.H.H.S. and F.M.; software, T.H.H.S.; validation, T.H.H.S., F.M. and V.V.; formal analysis, T.H.H.S. and F.M.; investigation, T.H.H.S. and F.M.; resources, C.C. and V.V.; data curation, V.V.; writing—original draft preparation, T.H.H.S.; writing—review and editing, T.H.H.S. and V.V.; visualization, T.H.H.S.; supervision, V.V.; project administration, V.V.; funding acquisition, V.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—440366605 and 396890929/GRK 2482.
Data Availability Statement
All crystal structures are deposited in the Cambridge Structural Database CSD (CCDC numbers: 2294770-2294773) and can be found at https://www.ccdc.cam.ac.uk/structures.
Acknowledgments
Computational support and infrastructure were provided by the “Centre for Information and Media Technology” (ZIM) at the University of Duesseldorf (Germany).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Haj Hassani Sohi, T.; Maass, F.; Czekelius, C.; Suta, M.; Vasylyeva, V. Co-crystallization of organic chromophore roseolumiflavin and effect on its optical characteristics. CrystEngComm 2022, 24, 7315–7325. [Google Scholar] [CrossRef]
- Bera, M.K.; Pal, P.; Malik, S. Solid-state emissive organic chromophores: Design, strategy and building blocks. J. Mater. Chem. C 2020, 8, 788–802. [Google Scholar] [CrossRef]
- Singh, M.; Liu, K.; Qu, S.; Ma, H.; Shi, H.; An, Z.; Huang, W. Recent advances of cocrystals with room temperature phosphorescence. Adv. Opt. Mater. 2021, 9, 2002197. [Google Scholar] [CrossRef]
- Griesser, U.J. The importance of solvates. In Polymorphism in the Pharmaceutical Industry; Hilfiker, R., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 211–233. ISBN 9783527311460. [Google Scholar]
- Boothroyd, S.; Kerridge, A.; Broo, A.; Buttar, D.; Anwar, J. Why do some molecules form hydrates or solvates? Cryst. Growth Des. 2018, 18, 1903–1908. [Google Scholar] [CrossRef]
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv. Drug Deliv. Rev. 2017, 117, 25–46. [Google Scholar] [CrossRef]
- Seddon, K.R. Pseudo polymorph: A polemic. Cryst. Growth Des. 2004, 4, 1087. [Google Scholar] [CrossRef]
- Desiraju, G.R. Counterpoint: What’s in a name? Cryst. Growth Des. 2004, 4, 1089–1090. [Google Scholar] [CrossRef]
- Bernstein, J. And another comment on pseudo-polymorphism. Cryst. Growth Des. 2005, 5, 1661–1662. [Google Scholar] [CrossRef]
- Nangia, A. Pseudopolymorph: Retain this widely accepted term. Cryst. Growth Des. 2006, 6, 2–4. [Google Scholar] [CrossRef]
- Grothe, E.; Meekes, H.; Vlieg, E.; Ter Horst, J.H.; de Gelder, R.D. Solvates, salts, and cocrystals: A proposal for a feasible classification system. Cryst. Growth Des. 2016, 16, 3237–3243. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Vittal, J.J.; Ramanan, A. Crystal Engineering: A Textbook; World Scientific: Singapore, 2011; ISBN 978-981-4338-75-2. [Google Scholar]
- Aakeröy, C.B.; Seddon, K.R. The hydrogen bond and crystal engineering. Chem. Soc. Rev. 1993, 22, 397–407. [Google Scholar] [CrossRef]
- Videnova-Adrabińska, V. The hydrogen bond as a design element in crystal engineering. Two- and three-dimensional building blocks of crystal architecture. J. Mol. Struct. 1996, 374, 199–222. [Google Scholar] [CrossRef]
- Komisarek, D.; Pallaske, M.; Vasylyeva, V. Crystal structure and thermal properties of phenibut, phenibut H2O and phenibut HCl: A case for phase stability based on structural considerations. Z. Anorg. Allg. Chemie 2021, 647, 984–991. [Google Scholar] [CrossRef]
- Heinen, T.; Hoelscher, S.; Vasylyeva, V. Structural study of anhydrous and hydrated 5-fluorouracil co-crystals with nicotinamide and isonicotinamide. Z. Kristallogr.—Cryst. Mater. 2022, 237, 109–116. [Google Scholar] [CrossRef]
- Karasulu, B.; Thiel, W. Photoinduced intramolecular charge transfer in an electronically modified flavin derivative: Roseoflavin. J. Phys. Chem. B 2015, 119, 928–943. [Google Scholar] [CrossRef] [PubMed]
- Scarbrough, F.E.; Shieh, H.-S.; Voet, D. The X-ray crystal structure of the molecular complex bis(lumiflavin–2,6-diamino-9-ethylpurine)–ethanol–water. Acta Crystallogr. B Struct. Sci. 1977, 33, 2512–2523. [Google Scholar] [CrossRef]
- Wells, J.L.; Trus, B.L.; Johnston, R.M.; Marsh, R.E.; Fritchie, C.J. Crystal structure of the yellow molecular complex lumiflavin–bis(naphthalene-2,3-diol). Acta Crystallogr. B Struct. Sci. 1974, 30, 1127–1134. [Google Scholar] [CrossRef]
- Kuo, M.C.; Dunn, J.B.R.; Fritchie, C.J. The crystal structure of a flavin molecular complex: 10-propylisoalloxazine–bis(naphthalene-2,3-diol). Acta Crystallogr. B Struct. Sci. 1974, 30, 1766–1771. [Google Scholar] [CrossRef]
- Voet, D.; Rich, A. The crystal and molecular structure of an intermolecular complex between riboflavin and an adenosine derivative. Proc. Natl. Acad. Sci. USA 1971, 68, 1151–1156. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPRO, v171.42; Oxford Diffraction/Agilent Technologies UK Ltd.: Yarnton, UK, 2022.
- APEX2, v2012; Bruker AX Inc.: Madison, WI, USA, 2012.
- SAINT, v2018; Bruker Analytical X-ray Systems: Madison, WI, USA, 2018.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Komisarek, D.; Haj Hassani Sohi, T.; Vasylyeva, V. Co-crystals of zwitterionic GABA API’s pregabalin and phenibut: Properties and application. CrystEngComm 2022, 24, 8390–8398. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring nature and predicting strength of hydrogen bonds: A correlation analysis between Atoms-in-Molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. B 1990, 46, 256–262. [Google Scholar] [CrossRef]
- Bader, R.; Nguyen-Dang, T.T. Quantum Theory of Atoms in Molecules–Dalton Revisited; Elsevier: Amsterdam, The Netherlands, 1981; pp. 63–124. ISBN 9780120348145. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).