Abstract
Dengue is an infection transmitted by the Aedes mosquito and is considered a major public health concern in many tropical and Asian countries, including the Philippines. It is caused by the dengue virus (DENV) which belongs to the Flaviviridae family and has four serotypes. The non-structural protein 5 (NS5), which consists of an MTase domain and an RdRp domain, is the largest and most conserved protein among flaviviruses and thus a potential target against DENV. However, there are very limited studies on the functional homodimer structure of NS5. Through molecular dynamics, it was found that residues 458–470, 583–586, 630–637, 743–744, and 890–900 of monomer A and residues 14–24, 311–315, and 462–464 of monomer B undergo essential motions for the conformational changes in the RdRp template tunnel and GTP binding in the MTase domain. Through the analysis of these motions, it was also proposed that in the dimeric structure of NS5 only one pair of domains contribute to the function of the protein. Other essential residues, specifically A-ASP533, A-LYS689, A-ARG620, A-ARG688, A-SER710, B-ARG620, B-LYS689, A-GLU40, A-ARG262, A-GLU267, A-ARG673, and B-ARG673, were also identified to play important roles in the information flow necessary for the function of the protein. In particular, shortest paths analysis led to the identification of ARG673 as an essential residue for the communication between RdRp and MTase catalytic sites. Mutation of this residue led to changes in the conformational flexibility of the RdRp finger subdomain, which may influence the RdRp catalytic function. These findings serve as a basis for future studies on the mechanism and inhibition of the NS5 dimer for dengue drug discovery.
1. Introduction
Dengue is an infection transmitted by the Aedes mosquito and caused by a virus from the Flaviviridae family, which includes the Zika, Yellow Fever, West Nile, and Japanese Encephalitis viruses [1]. It consists of four serotypes (DENV 1–4) and uses a positive-strand RNA genome, with replication occurring on the endoplasmic reticulum membranes of the host cell [2]. Infection with the dengue virus (DENV) leads to a wide range of disease severity. Mild cases of dengue infection are characterized by fevers, acute febrile illness, and joint pain; however, severe dengue infection symptoms, such as dengue hemorrhagic fever and dengue shock syndrome, may also occur [3]. According to the World Health Organization [4], there is an estimated 390 million cases of dengue infections globally per year, with the greatest risk of infection in tropical and Asian countries.
In the Philippines, DENV is considered a major public health problem, as it is endemic throughout the country [5]. As of 2022, there has been an increase in dengue cases in the Philippines, with 193,010 cases reported from January to October 2022, compared to 65,684 cases during the same period in 2021 [4]. Yet despite the health risk that DENV poses, there are neither any specific treatments nor widespread vaccine against the virus, as current measures merely control its symptoms.
The Non-Structural Protein 5 (NS5) is the largest, most conserved protein among flaviviruses, as well as the dengue serotypes, and it is responsible for the replication of viral RNA and evasion from the host immune system [6,7,8]. The major role that NS5 plays in the replication and propagation of the dengue virus, as well as its highly conserved structure, makes it an ideal target for DENV treatment. Thus, information on the key structural regions of its functional structure can be useful in understanding how the protein works and how it can be inhibited in a physiological setting. The functional structure of NS5 exists as a homodimer [9]. Each NS5 monomer that makes up the dimer consists of two domains connected by a linker: a methyltransferase domain (MTase) at the N-terminal and an RNA-dependent RNA polymerase (RdRp) at the C-terminal. Molecular dynamic studies have been done on the two separate domains of NS5; however, there are no studies that analyze its full functional dimeric structure.
The NS5 protein has been the subject of previous drug discovery efforts, resulting in the discovery of N-sulfonyl anthranilic acid derivatives which have been described in allosteric inhibition of NS5 [10]. Direct inhibitors of the protein include beta-d-2′-ethynyl-7-deaza-adenosine triphosphate (2′E-7D-ATP), which can compete with the natural nucleotide binder [11], a bis-chloro-diphenylamine 2-aminoindan-2-carboxyl derivative (NITD-434), and a 1,4-N-bis-substituted-piperazine-3-carboxyl group with a diphenylethane and a biphenyl-tetrazole moiety (NITD-640), which both affect the RdRp domain by changing the organization of the finger and palm subdomains [12].
Despite the availability of known inhibitors against NS5, there are still no known drugs developed for this protein, nor for dengue in general. In this study, the homodimer structure of dengue NS5 was analyzed using molecular dynamics, principal component analysis (PCA), and network analysis, to predict a mechanistic model for its protein function and to identify key residues for dynamics and signal flow. Information garnered from this study can, hopefully, be used in the discovery and design of potent NS5 inhibitors that can potentially be developed into dengue therapeutics in the future.
2. Materials and Methods
2.1. System Preparation
The crystal structure of DENV3 Non-Structural Protein 5 (NS5) (PDB ID: 5CCV) was prepared using the Solution Builder of the Input Generator in CHARMM-GUI [13,14]. The PDB file was uploaded, followed by the selection of chains A and B included in a single homodimer of NS5 with a Type I dimer interface [9]. The ligands included in the dimer were two zinc ions in each of the RdRp domains and one S-adenosyl-l-homocysteine (SAH) molecule in each of the MTase domains. The GalaxyFill program [15] in CHARMM-GUI was then utilized to resolve any missing residues in the middle of the structure; missing terminal residues for both chains were no longer modeled. The dimer was solvated in a rectangular water box using the TIP3P water model, neutralized with 0.15 M NaCl, and afterward, the CHARMM36 force field was applied. The final system contained 254 sodium ions, 286 chloride ions, and 9996 water molecules.
The R673 mutant was prepared separately using the same PDB structure, chains, and ligands. ARG673 was mutated to alanine and the rest of the set-up was consistent with the wild-type (WT) system. The final system contained 231 sodium ions, 255 chloride ions, and 9999 water molecules.
2.2. Molecular Dynamic Simulation
The WT and R673A mutant protein system was subjected to energy minimization in 5000 steps, with the steepest descent algorithm using GROMACS 5.1.4 [16]. Three MD trials were performed for the WT system with the NVT and NPT equilibration, both at 500,000 steps, done using a temperature of 310 K and a pressure of 1 bar. Each production run consists of 300 ns with 2 fs time step. Structural plots were generated using GROMACS, while hydrogen bond analysis was performed using VMD.
2.3. Principal Component Analysis
The trajectories obtained from the WT MD simulation were subjected to PCA using GROMACS 5.1.4. A covariance matrix was produced from the atomic coordinates of the trajectory; from this matrix, the eigenvectors or essential modes were taken. The principal components (PC) of the protein backbone were then identified from the projection and analyzed.
2.4. Network Analysis
The WT MD simulation trajectory was also subjected to network analysis using gRINN [17]. The topology, run input, and trajectory files obtained from GROMACS were entered into the gRINN New Calculation interface. A trajectory stride of 10 and a filtering distance cutoff of 15 Å was utilized.
3. Results
3.1. Molecular Dynamics Simulations of the Wild-Type NS5 Homodimer
Prior to the 300 ns production run, the system was minimized and equilibrated to 310 K and 1 bar. To evaluate the stability and conformational fluctuations of NS5 during the MD production, the root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) were obtained from the resulting trajectories of the three MD runs. In Figure 1a, it can be observed that the NS5 production systems have equilibrated in solution at around 100 ns. All RMSD profiles converged and reached a plateau at around 100 ns, indicating that the systems reached stability during the production run. The RMSF plots (Figure 1b) of the NS5 homodimer show that, while the two monomers have a mostly similar fluctuation pattern, which may be attributed to their shared structure and function, there are regions where MTase is more flexible in one chain and RdRp is more flexible in the other. As seen in the plot, when MTase is more flexible in chain A, RdRp is more flexible in chain B, and vice versa. The greatest fluctuations are found at residues 456–472, 584–585, 630–637, 743–745, 801–803, and 890–900 of monomer A and residues 19–23, 312–315, and 462–464 of monomer B.
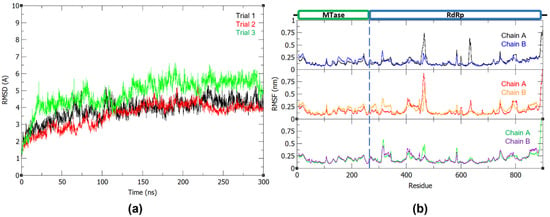
Figure 1.
(a) Backbone RMSD plot of the three 300 ns dengue NS5 production runs and (b) RMSF plot of the dengue NS5 protein chain A and B with trial 1, trial 2, and trial 3 in the first, middle, and last panel, respectively.
The number of hydrogen bonds did not greatly fluctuate throughout the MD simulation (Figure 2). The hydrogen bond occupancy for the whole structure was also determined and key H-bonds are summarized in Table 1. Approximately 3000 H-bonds with more than 5% occupancy were found, along with multiple bonds showing occupancy values ≥ 100% (Supplementary Table S1), wherein the donors and acceptors form more than one H-bond with each other. It was found that the H-bonds identified by Klema et al. [9] in the crystal structure of the NS5 homodimer were present in the simulated protein in high occupancy throughout the 300 ns runs, specifically, the H-bond network involving ARG262 and GLU267, which connects the linker found between the two domains to the MTase core residues LEU94 and TYR119. Furthermore, there are H-bonds between the linker residue GLU269 and the finger residues ARG361 and LYS595. The H-bond interactions between the MTase residue SER128 and the RdRp residue GLY526, which are also involved in the linkage of the overall dimeric structure, were additionally found to have high occupancy. Thus, it can be posited that these interactions play important roles in the linkage, structural stability, and function of the NS5 dimer.
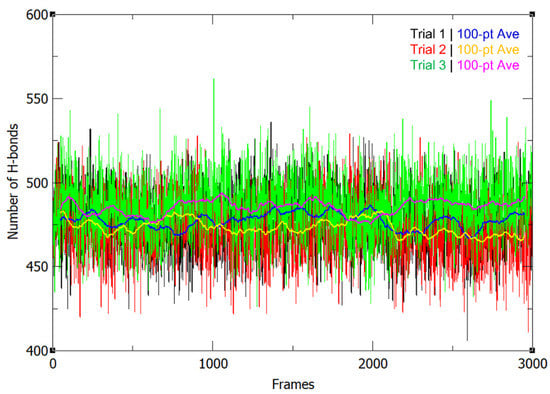
Figure 2.
Number of H-bonds in the NS5 homodimer structure throughout the 300 ns production run for 3 trials, along with their 100-pt running averages.

Table 1.
Hydrogen bond donor and acceptor occupancy within the NS5 protein for three production trials. “Main” signifies that the main chain or the backbone participates in hydrogen bonding, while “Side” signifies that the residue side chain participates in hydrogen bonding.
3.2. Principal Component Analysis
From the covariance matrix, a set of eigenvectors arranged by descending eigenvalues was obtained. From there, it was identified that the first five eigenvectors are responsible for 47.62%, 16.68%, 13.88%, 8.46%, and 7.27% of the total motion of the protein and thus define the essential motions of the system. This agrees with the 2D projections (Figure 3), where it can be observed that the clusters are more spread along the eigenvector 1 projection, suggesting that the fluctuations in protein conformation are indeed largely influenced by PC1. Moreover, the compact clusters shown in the other 2D projections show the high correlation of the different principal components, as well as the localized conformational stability involving these eigenvectors.
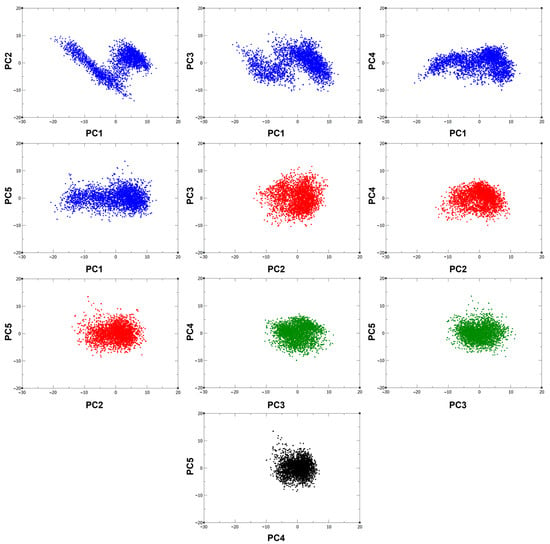
Figure 3.
2D projections of the 300 ns trajectory on the first five eigenvectors.
To further analyze these projections, the RMSF values of these eigenvectors were obtained (Figure 4a) to identify the specific regions related to the observed atomic displacements and motions. Using the RMSF plot, the PC1 can be related to the motions in the α17 helix (residues 630–637) and the loop between the α21 and α22 helices (residues 743–744) found in the palm and thumb subdomains of the RdRp in one monomer, as well as motions in the RdRp L1 fingertip (residues 311–315) of the other monomer. Both monomers also exhibit motions at the RdRp motif F (residues 458–470), which plays a role in the stabilization of nascent base pairs during replication. These movements are thus essential to the conformational changes in the RNA template tunnel for the entry and exit to the GDD (Gly-Asp-Asp) catalytic site. Aside from these, PC1 was also found to be related to motions in the MTase α1 and α2 helices (residues 14–24) in the partner chain, which indicates their role in the opening of the GTP binding pocket for the formation of an enzyme-GMP complex, in turn needed for the formation of the cap on the replicated RNA. Similarly, PC2 shows motions at the RdRp α17 helix and the RdRp motif F in the first monomer. On the other hand, PC3 and PC4 represent motions in RdRp α17 helix of the first monomer, while PC5 shows similar motions as PC2. The similarities between the motions of the PCs result in the more clustered regions seen in the 2D projections.
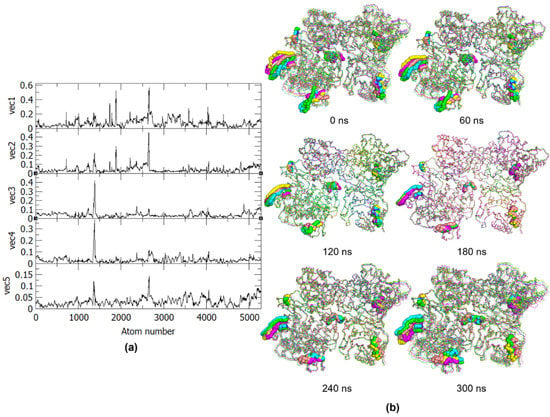
Figure 4.
(a) RMSF of the first five eigenvectors from PCA and (b) Frames from the 300 ns trajectory for every 60 ns. The residues with essential motions are represented in spheres and the five principal components projections are colored as follows: PC1, green; PC2, cyan; PC3, magenta; PC4, yellow; PC5, pink.
The essential motions exhibited by the eigenvector RMSF, as well as the PCs, correspond to the residues identified during MD analyses, except for residues 794–803 of monomer A, which make up the priming loop. The essential dynamics of the above-mentioned regions can also be better observed in Figure 4b, which presents the motions of the PCs for every 60 ns of the trajectory. From these frames, it can also be observed that the dimer exhibits an inward motion where the partner domains from each monomer are moving closer to one another. Based on the identified essential motions and the frame-by-frame analysis, only the RdRp of one chain and the MTase of the other chain exhibit significant movements as a pair, while the two other domains in the dimer do not have many relevant movements that may contribute to the protein function. The minimal movement of the other RdRp-MTase partner in the homodimer may be due to conformational and steric hindrances resulting from the movement of the first pair, i.e., MTase of chain A has limited dynamics to allow freedom in the RdRp domain and vice versa for chain B. This suggests that for the dengue NS5 protein to function, only one pair of RdRp-MTase is needed.
3.3. Network Analysis
To determine the importance of the individual amino acids in a protein to its dynamics and function, network analysis via gRINN is performed to calculate the interaction energies from MD results and create a network that describes the roles of these residues [17]. From the network, the centrality measures, like betweenness centrality (BC) and closeness centrality (CC), were calculated (Figure 5). Residues with high BC represent the most essential residues in the NS5 information flow, while those with high CC indicate their high efficiency in transferring information (Table 2) [18].
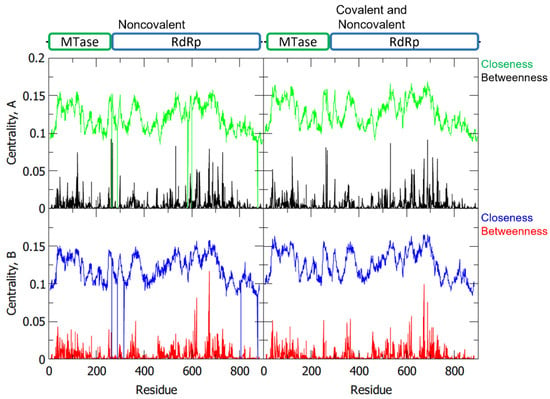
Figure 5.
Betweenness and closeness centrality of the NS5 homodimer residues with separate plots for chain A and chain B, and for noncovalent only and covalent with noncovalent interactions.
Table 2.
Residues with ≥0.05 BC and ≥0.13 CC for covalent and noncovalent interactions.
Among the listed essential residues, it was identified that A-ASP533, A-ARG620, A-ARG688, A-LYS689, A-SER710, B-ARG620, and B-LYS689 are part of the RdRp palm domain. Moreover, ASP533 was proposed to be an interacting residue for the 2′E-7D-ATP NS5 RdRp inhibitor [11]. Another residue with high BC is the GLU40 of both monomers which is part of the MTase domain. The residues B-ARG673, A-ARG262, A-GLU267, and A-ARG673 are also of great interest because, aside from their high BC values, they are involved as linkers in the NS5 protein. In particular, A-ARG262 and A-GLU267 are residues that play a role in linking together the two domains of monomer A, while A-ARG673 and B-ARG673 aid in linking together the individual monomers to form the dimeric structure of NS5. Other residues that have not been noted compared to the previous sections may have other functions within the protein network that affects structural stability and function.
Through network analysis, the shortest paths from the residues in the catalytic sites of the RdRp (GLY662-ASP663-ASP664) and MTase (LYS61-ASP146-LYS180-GLU216) were also analyzed to determine which residues potentially play roles in the information flow involved with the overall mechanism of the protein. Shown in Table 3 are the shortest paths from the monomer A RdRp to the monomer B MTase, as well as the shortest path from the monomer B RdRp to the monomer A MTase. Both paths make use of residue ARG673 which, as previously mentioned, is a residue involved in the linkage of the two monomers with high BC.
Table 3.
Shortest path between RdRp and MTase catalytic sites.
3.4. Molecular Dynamics Simulations of the R673A Mutant
Due to the noted significance of ARG673 in both chains from the raw centrality measures and shortest paths analysis, its mutation to alanine was also studied to obtain more information on how it controls the NS5 protein network.
Similar to WT structure, the mutant system was minimized and equilibrated at 310 K and 1 bar, and stabilized at around 100 ns of the production run (Figure 6a). Based on the comparison of the WT and mutant NS5 RMSF, monomer B remains more flexible in the MTase domain and monomer A has a more flexible RdRp domain. However, RdRp finger subdomain residues 360–420 of the mutant protein are observed to be more flexible in the mutant chain A, while losing the flexibility in residues 456–472 that was previously observed in the wild-type structure (Figure 6b).
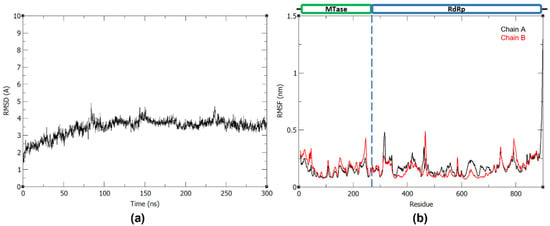
Figure 6.
(a) Backbone RMSD plot of the R673A NS5 mutant and (b) RMSF plot of the R673A NS5 mutant chain A and B.
Visual observation of the NS5 protein structure shows that ARG673 is outside of the RdRp catalytic site, despite its observed influence on the finger subdomain (Figure 7a). Interestingly, surface analysis shows that ARG673 is positioned in a small pocket behind the MTase catalytic site (Figure 7b), suggesting that it can be a new potential allosteric site, distinct from the proposed allosteric site for N-sulfonylanthranilic acid derivative inhibitor [10].
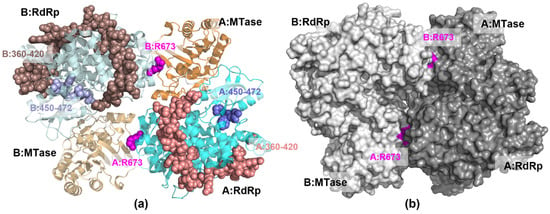
Figure 7.
The 3D structure of NS5 homodimer structure showing (a) the position of ARG673 (magenta spheres) relative to the RdRp finger subdomain residues 360–420 (light brown spheres) and 450–472 (light blue spheres) (b) the protein surface with the pockets containing ARG673.
4. Discussion
Dengue continues to be a major public health concern, especially in tropical countries like the Philippines. Despite this, no drugs have been developed and approved for dengue, and treatments continue to target the symptoms rather than the root of the condition.
The DENV proteome consists of 10 proteins, specifically three structural proteins and seven nonstructural proteins. The nonstructural proteins include NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. Out of these, NS5 is the largest and most conserved protein responsible for viral replication and capping, making it a possible target against which to design drugs inhibiting multiple DENV serotypes.
NS5 consists of two domains, specifically the MTase domain and RdRp domain. The MTase domain is found in the N-terminal of NS5 and is responsible for the capping of the viral DNA by utilizing an S-adenosyl-L-methionine (SAM) as a methyl donor, as well as its proposed guanylyltransferase (GTase) activity. In the C-terminal of NS5 is the RdRp domain that replicates the viral genome. While dynamic studies have been done on these NS5 domains, they were conducted separately and, as far as the researchers know, computational analyses are yet to be performed for the full functional dimeric structure. Therefore, to fully understand the protein structure-function relationship of the DENV NS5, the dynamics of the full protein structure and how one domain can affect the dynamics of the other has to be considered.
In a previous study [9], NS5 was reported to preferentially arrange itself into homodimers with the RdRp of one monomer placed beside the MTase of the other monomer (Figure 7). It was determined that this arrangement would allow better coordination between the RNA synthesis of the RdRp and the capping of the MTase because it would require smaller conformational changes to complete the process due to the position of the domains. In this study, the difference in RMSF between the MTase, as well as that of the RdRp, in monomer A compared to monomer B indicates that only one MTase-RdRp partner is moving or working at a time.
The residues that make up the RdRp finger subdomain are expected to be highly flexible to allow conformational changes relevant to RdRp activity. In particular, residues 311–315 of monomer B are part of the Loop 1 (L1) fingertip, which surrounds the RdRp active site and connects the finger and thumb domains [2]. The observed flexibility of this loop agrees with its noted function as a modulator during RdRp replication by transitioning the template tunnel between open and closed conformations [19]. The highly flexible finger subdomain residues 457–472 and 462–464 noted in monomer A and B, respectively, are also of significant interest, as these residues are also known to be part of the conserved motif F which functions to stabilize the nascent base pairs. Aside from the finger subdomain, highly flexible residues 630–637 of monomer A were identified in the palm subdomain of the RdRp and are part of the α17 helix. This secondary structure serves as a link between motif B, which aids in the passage of RNA through the RdRp tunnel, and motif C, which contains the catalytic residues GLY662, ASP663, and ASP664 [2]. The RMSF also shows the flexibility of residues 743–744, 794–803, and 890–900 in monomer A, which are all part of the thumb subdomain of the RdRp that houses the catalytic site. Residues 743–744 are part of the loop that connects the α21 and α22 helix of the RdRp and acts similarly to Loop 1 in the palm subdomain such that it can also adopt confirmations that influence the shape of the RNA template tunnel [20]. For residues 794–803, these amino acids are known to be part of the priming loop which is an important structure that stabilizes de novo initiation by regulating access to and exit from the tunnel and thus the active site [9]. The MTase domain was also found to have flexibility at residues 18–24 of monomer B. These residues are associated with the α1 and α2 helices which help form the GTP binding pocket of the MTase N-terminal [21]. Overall, the RMSF analysis was able to identify key flexible residues needed for the function and dynamics of the protein, which may possibly serve as target residues in inhibiting the activity of the NS5 homodimer.
In conjunction with the RMSF results, the essential motions observed from the eigenvector analysis show that only one RdRp domain moves in concert with its partner MTase domain from the other monomer in an inward motion, similar to a protein binding site changing conformation to allow for tighter substrate binding. It is possible that, due to the orientations of the two domains in the homodimer, only one MTase-RdRp partner is needed to allow the full function of NS5. This is because the inward conformation of one partner leads to the other being unable to do so without affecting the conformation of the former. These observations agree with the dimer model presented by Klema et al. [9], which proposes that the RdRp of one monomer interacts with the MTase of the adjacent monomer to complete the function of the protein. Moreover, the motions from the PCA indicate that the NS5 dimer is formed not to increase replication but for the more efficient orientation of the domains, given that only one pair of domains undergoes the replication and capping mechanism.
The RMSF and the PCA results are mostly consistent with each other, except for the dynamics of residues 794–803 of monomer A, which are found in the priming loop. Although the priming loop has been identified to aid in the access to and exit from the template tunnel during de novo initiation, it is possible that its motion is not considered essential because the motions by the other secondary structures in the RdRp domain allow for the necessary conformational changes required for the RdRp activity.
Aside from the protein motions, it is important to consider how information is sent throughout the protein structure, especially for targets like NS5 where separate domains within the same protein exhibit different but concerted functions. Protein energy networks can represent the interactions and connectivity between the residues in a protein which can have biological importance, such as involvement in structural stability, protein dynamics, and signaling [22]. From the protein network analysis, the betweenness and closeness centralities were measured to predict the importance of each residue within the protein signaling framework. BC describes the importance of a residue as a path toward other residues and their influence on the flow of information in the protein, while CC identifies residues with the highest efficiency of transferring information [18]. From the listed residues with high BC and CC, while a number of residues display high significance within the network due to their positions as linkers, several other residues are identified as crucial for other functions. A-ASP533 is a residue found in the RdRp motif A, which acts as a cation binding site and coordinates the Mg2+ ion towards the GDD catalytic site to allow its use in the formation of new phosphodiester bonds during replication. Meanwhile, ARG620 makes up the α17 helix, which contributes to the functional architecture of the catalytic site [2,20]. Thus, these residues may serve to signal when Mg2+ is present in the pocket and ready for the reaction with the GDD residues. Targeting these during drug design efforts can significantly affect the catalytic mechanism of the RdRp domain. A-ARG688, A-LYS689, and B-LYS689 make up the motif D, which is responsible for the release of the PPi by-product from ATP after replication, while the A-SER710 residue helps make up motif E, which binds a zinc ion relevant to maintaining the structural architecture of the protein [2]. These residues may serve to communicate the need for a change in conformation to allow the exit of the new RNA upon completion of replication. Hence, targeting the signal flow in these residues can hinder the formation or release of new RNA by DENV. Another residue with high BC is the GLU40 of both monomers, which is part of the MTase domain. This residue is of great interest, as it is also known to be part of the α3 helix of the MTase region, which helps form the GTP binding pocket needed for the cap formation in the replicated RNA. This residue may be involved in communicating to the protein that GTP is available for the capping process to allow the RdRp to release the RNA and transfer it to the MTase domain. Compounds targeting these residues may function as an obstacle in the capping mechanism of NS5. Further to this, one of the most interesting residues is ARG673, a residue involved in the linkage of the two monomers in NS5. ARG673 can be used by both monomers when the MTase and RdRp domains are communicating with each other and targeting the signal flow to and from this residue may prove to be an excellent way to inhibit NS5 activity.
Interestingly, mutating ARG673 in the NS5 structure to alanine led to notable changes in the flexibility of the RdRp domain. The increase in flexibility of residues 360–420 and decrease in flexibility of residues 456–472 in the R673A mutant suggest that ARG673 can influence the movement of the finger subdomain and thus affect RdRp function.
5. Conclusions
The resulting trajectory from the MD production, RMSD, and RMSF, as well as the number of H-bonds and occupancy, were identified in order to determine the structural stability and flexibility of the protein. It was found that the most flexible residues as identified by MD analyses are the RdRp residues 458–470, 583–586, 630–637, 743–744, 794–803, and 890–900 of Monomer A and residues 311–315 and 462–464 of Monomer B, as well as the MTase residues 14–24 of Monomer B. To further confirm that the aforementioned residues undergo essential motions, PCA was conducted on the trajectory and it was found that only residues 794–803 did not exhibit essential motion. Thus, the other residues mentioned display necessary and structurally important dynamics for the NS5 homodimer, particularly for the informational changes of the template tunnel in RdRp, as well as for GTP binding in the MTase.
Through MD analyses and PCA, it was also proposed that in the mechanism of the NS5 dimer, only one pair of RdRp and MTase of opposite monomers function to replicate and cap the viral RNA. This was theorized after observing the motions of the protein wherein monomer A had a more flexible RdRp region, while monomer B was more flexible at the MTase region.
Network analysis was also done to determine the residues essential for the flow of information in the protein. These were found to be residues A-ASP533, A-LYS689, A-ARG620, A-ARG688, A-SER710, B-ARG620, and B-LYS689 of the RdRp domain, as well as A-GLU40 of the MTase domain, all of which play important roles in the catalytic mechanisms of each domain. Moreover, residues A-ARG262, A-GLU267, A-ARG673, and B-ARG673, which are involved in the linkage of the protein, are also found to be essential in the protein signal flow. Shortest path analysis between the catalytic sites of MTase and RdRp further showed that the ARG673 residue plays an important role in the transfer of information between the MTase and RdRp binding sites, aiding in the overall mechanism of the protein.
This study was able to identify essential residues related to the function of the DENV NS5 homodimer through in silico methods, as well as propose a mechanism for the dimer RdRp and MTase interaction. The proposed mechanism can also be further studied and validated to better understand how the DENV NS5 protein functions. The identification of essential motions and residues that are crucial in the information flow of NS5 may aid future studies on dengue treatments and vaccines by providing viable targets for inhibition.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst13010063/s1, Table S1: Hydrogen bonds of NS5 dimer with ≥100% occupancy.
Author Contributions
Conceptualization, D.A.F., S.J.Y.M. and M.C.O.C.; Formal analysis, D.A.F., S.J.Y.M. and G.P.II; Methodology, D.A.F. and S.J.Y.M.; Resources, S.J.Y.M.; Supervision, S.J.Y.M. and M.C.O.C.; Visualization, D.A.F., S.J.Y.M. and G.P.II; Writing—original draft, D.A.F. and S.J.Y.M.; Writing—review & editing, S.J.Y.M., G.P.II and M.C.O.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part via the Research Fellowship Program of De La Salle University (DLSU) RFC-010-T121-22_T321-23 to S.J.Y.M.
Data Availability Statement
The data presented in this study are available in the main text and in the Supplementary Information.
Acknowledgments
This study was made possible by the Department of Science and Technology Advanced Science and Technology Institute (DOST-ASTI) who provided the data storage and high performance (HPC) facilities through the Computing and Archiving Research Environment (COARE).
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- dos Santos Nascimento, I.J.; da Silva Santos-Júnior, P.F.; de Aquino, T.M.; de Araújo-Júnior, J.X.; da Silva-Júnior, E.F. Insights on Dengue and Zika NS5 RNA-dependent RNA polymerase (RdRp) inhibitors. Eur. J. Med. Chem. 2021, 224, 113698. [Google Scholar] [CrossRef] [PubMed]
- El Sahili, A.; Lescar, J. Dengue Virus Non-Structural Protein 5. Viruses 2017, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Bäck, A.T.; Lundkvist, A. Dengue viruses—An overview. Infect. Ecol. Epidemiol. 2013, 3, 19839. [Google Scholar] [CrossRef]
- World Health Organization. Dengue Situation Updates 2022. Available online: http://apps.who.int/iris/handle/10665/352792 (accessed on 28 October 2021).
- Undurraga, E.A.; Edillo, F.E.; Erasmo, J.N.V.; Alera, M.T.P.; Yoon, I.-K.; Largo, F.M.; Shepard, N.S. Disease Burden of Dengue in the Philippines: Adjusting for Underreporting by Comparing Active and Passive Dengue Surveillance in Punta Princesa, Cebu City. Am. J. Trop. Med. Hyg. 2017, 96, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Soh, T.S.; Chan, K.W.K.; Fung, S.S.Y.; Swaminathan, K.; Lim, S.P.; Shi, P.-Y.; Huber, T.; Lescar, J.; Luo, D.; et al. Flexibility of NS5 Methyltransferase-Polymerase Linker Region Is Essential for Dengue Virus Replication. J. Virol. 2015, 89, 10717–10721. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Soh, T.S.; Lim, S.P.; Chung, K.Y.; Swaminathan, K.; Vasudevan, S.G.; Shi, P.-Y.; Lescar, J.; Luo, D. Molecular basis for specific viral RNA recognition and 2′-O-ribose methylation by the dengue virus nonstructural protein 5 (NS5). Proc. Natl. Acad. Sci. USA 2015, 112, 14834–14839. [Google Scholar] [CrossRef]
- Zhao, Y.; Soh, T.S.; Zheng, J.; Chan, K.W.K.; Phoo, W.W.; Lee, C.C.; Tay, M.; Swaminathan, K.; Cornvik, T.C.; Lim, S.P.; et al. A Crystal Structure of the Dengue Virus NS5 Protein Reveals a Novel Inter-domain Interface Essential for Protein Flexibility and Virus Replication. PLoS Pathog. 2015, 11, e1004682. [Google Scholar] [CrossRef]
- Klema, V.J.; Ye, M.; Hindupur, A.; Teramoto, T.; Gottipati, K.; Padmanabhan, R.; Choi, K.H. Dengue Virus Nonstructural Protein 5 (NS5) Assembles into a Dimer with a Unique Methyltransferase and Polymerase Interface. PLoS Pathog. 2016, 12, e1005451. [Google Scholar] [CrossRef]
- Yin, Z.; Chen, Y.-L.; Kondreddi, R.R.; Chan, W.L.; Wang, G.; Ng, R.H.; Lim, J.Y.H.; Lee, W.Y.; Jeyaraj, D.A.; Niyomrattanakit, P.; et al. N-Sulfonylanthranilic Acid Derivatives as Allosteric Inhibitors of Dengue Viral RNA-Dependent RNA Polymerase. J. Med. Chem. 2009, 52, 7934–7937. [Google Scholar] [CrossRef]
- Latour, D.R.; Jekle, A.; Javanbakht, H.; Henningsen, R.; Gee, P.; Lee, I.; Tran, P.; Ren, S.; Kutach, A.K.; Harris, S.F.; et al. Biochemical characterization of the inhibition of the dengue virus RNA polymerase by beta-d-2′-ethynyl-7-deaza-adenosine triphosphate. Antivir. Res. 2010, 87, 213–222. [Google Scholar] [CrossRef]
- Arora, R.; Liew, C.W.; Soh, T.S.; Otoo, D.A.; Seh, C.C.; Yue, K.; Nilar, S.; Wang, G.; Yokokawa, F.; Noble, C.G.; et al. Two RNA Tunnel Inhibitors Bind in Highly Conserved Sites in Dengue Virus NS5 Polymerase: Structural and Functional Studies. J. Virol. 2020, 94, e01130-20. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2015, 12, 405–413. [Google Scholar] [CrossRef]
- Coutsias, E.A.; Seok, C.; Jacobson, M.P.; Dill, K.A. A kinematic view of loop closure. J. Comput. Chem. 2004, 25, 510–528. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Serçinoğlu, O.; Ozbek, P. gRINN: A tool for calculation of residue interaction energies and protein energy network analysis of molecular dynamics simulations. Nucleic Acids Res. 2018, 46, W554–W562. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.; Li, Z.; Na Wu, X.; Rohr, L.; Gombos, S.; Harter, K.; Schulze, W.X. Comparison of path-based centrality measures in protein-protein interaction networks revealed proteins with phenotypic relevance during adaptation to changing nitrogen environments. J. Proteom. 2021, 235, 104114. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.; Kuhn, R.J. Structural proteomics of dengue virus. Curr. Opin. Microbiol. 2008, 11, 369–377. [Google Scholar] [CrossRef]
- Yap, T.L.; Xu, T.; Chen, Y.-L.; Malet, H.; Egloff, M.-P.; Canard, B.; Vasudevan, S.G.; Lescar, J. Crystal Structure of the Dengue Virus RNA-Dependent RNA Polymerase Catalytic Domain at 1.85-Angstrom Resolution. J. Virol. 2007, 81, 4753–4765. [Google Scholar] [CrossRef]
- Kroschewski, H.; Lim, S.P.; Butcher, R.E.; Yap, T.L.; Lescar, J.; Wright, P.J.; Vasudevan, S.; Davidson, A.D. Mutagenesis of the Dengue Virus Type 2 NS5 Methyltransferase Domain. J. Biol. Chem. 2008, 283, 19410–19421. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Parekh, N. NAPS: Network Analysis of Protein Structures. Nucleic Acids Res. 2016, 44, W375–W382. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).