
Succinyl and Adipoyl Dihydrazones: A Solid-State, Solution and Antibacterial Study



Abstract
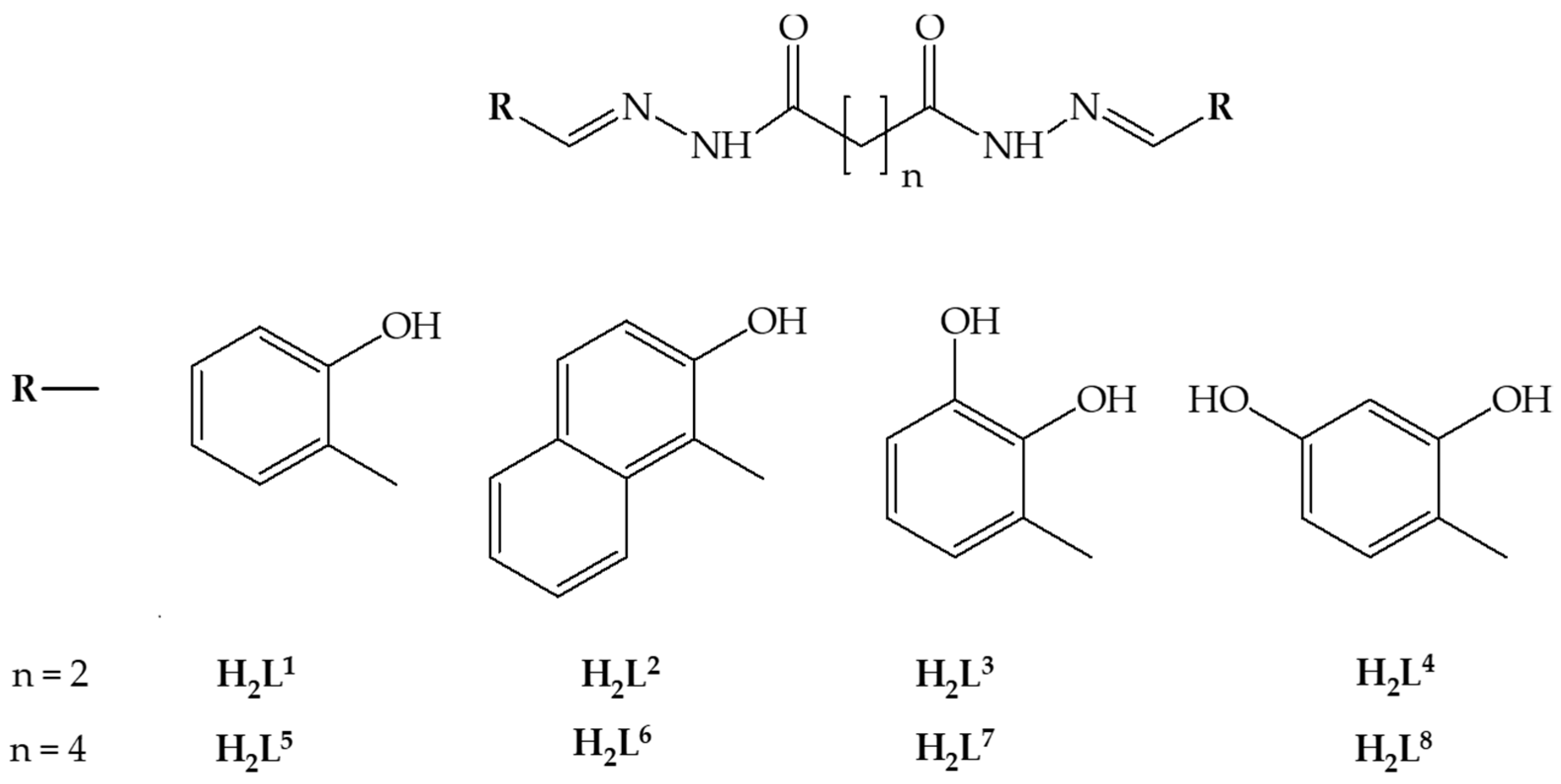
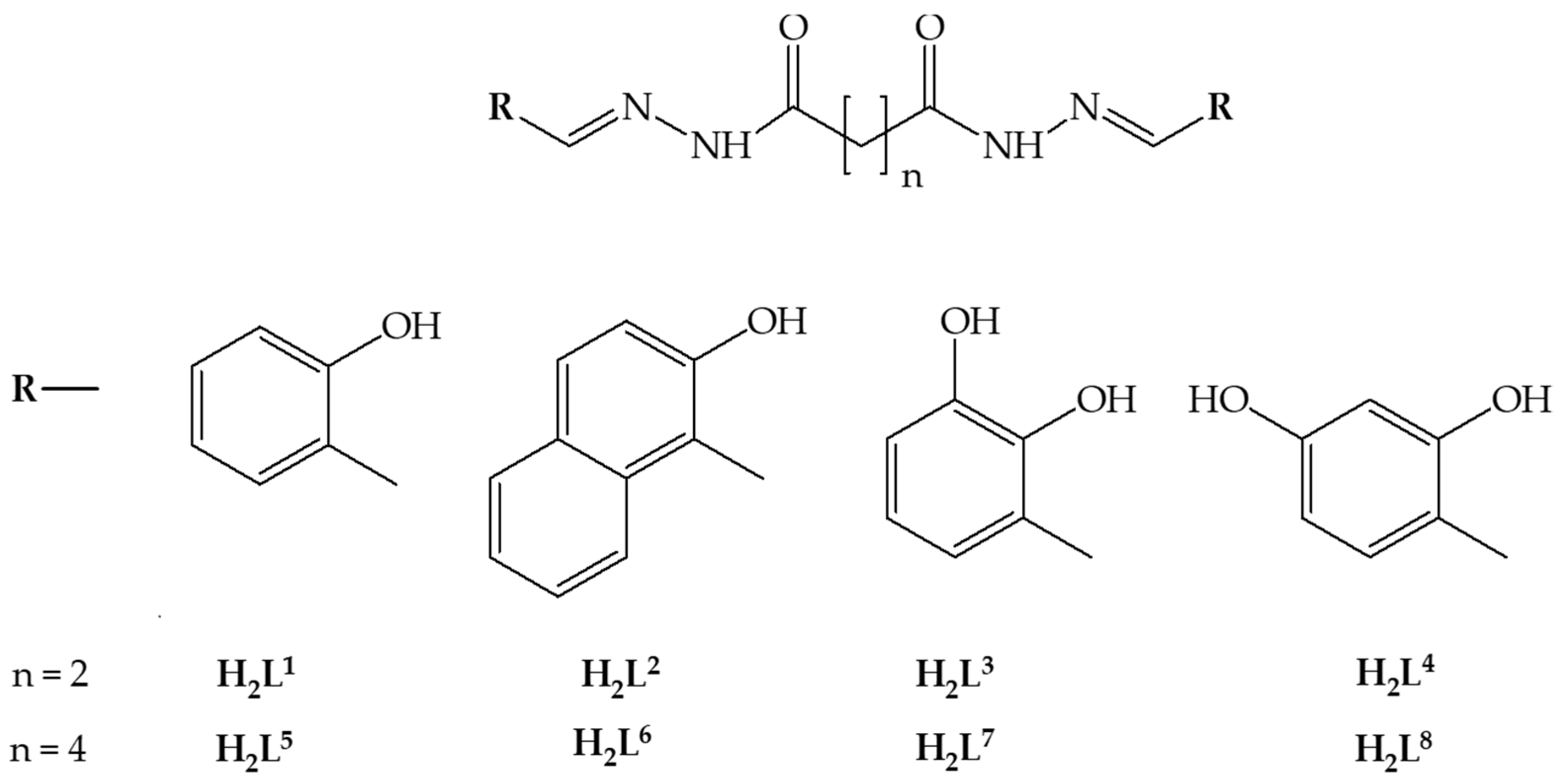
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.1.1. Synthesis of H4L1
2.1.2. Synthesis of H4L2
2.1.3. Synthesis of H4L3
2.1.4. Synthesis of H4L4
2.1.5. Synthesis of H4L5
2.1.6. Synthesis of H4L6
2.1.7. Synthesis of H4L7
2.1.8. Synthesis of H4L8
2.2. Methods
3. Results and Discussion
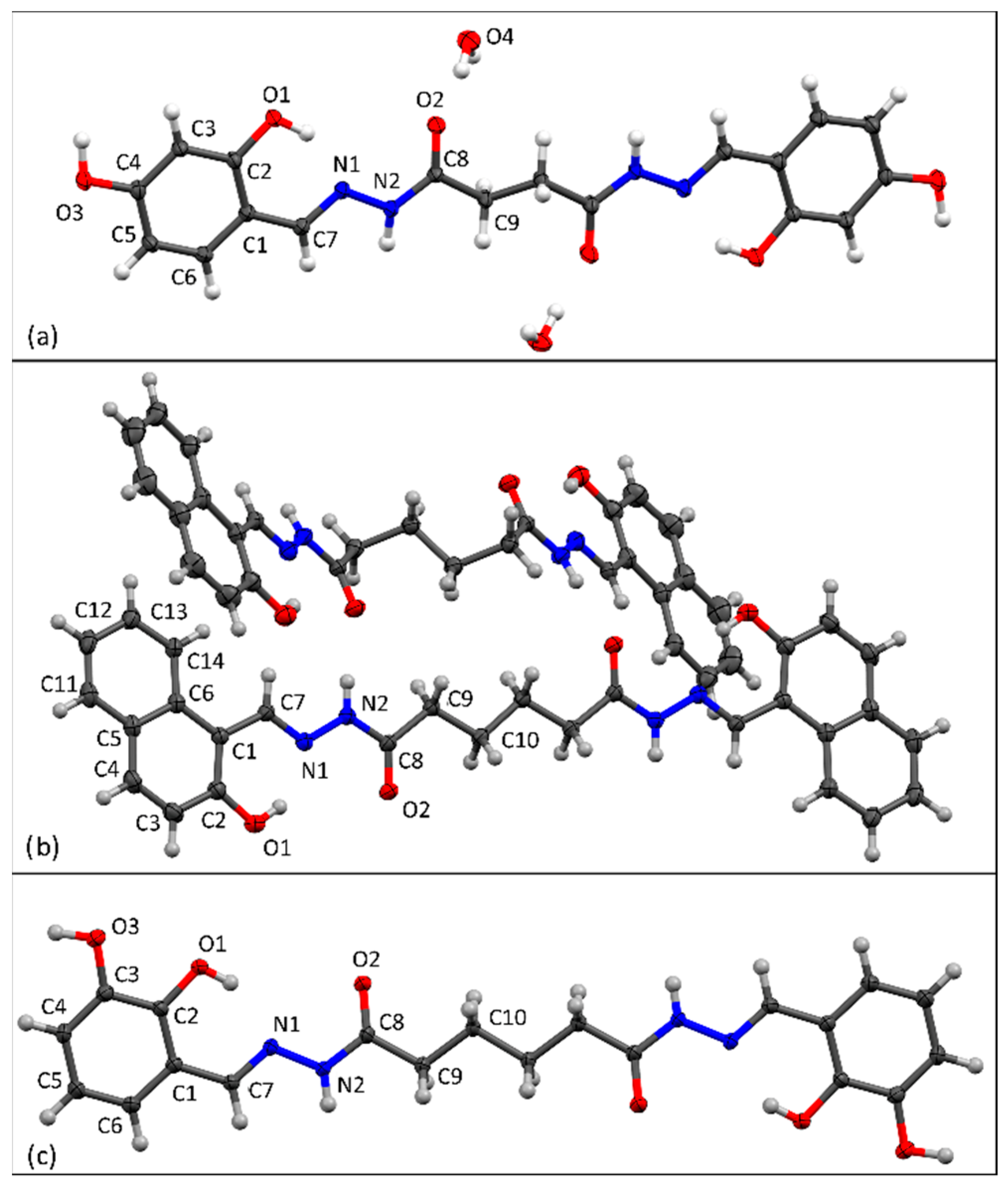
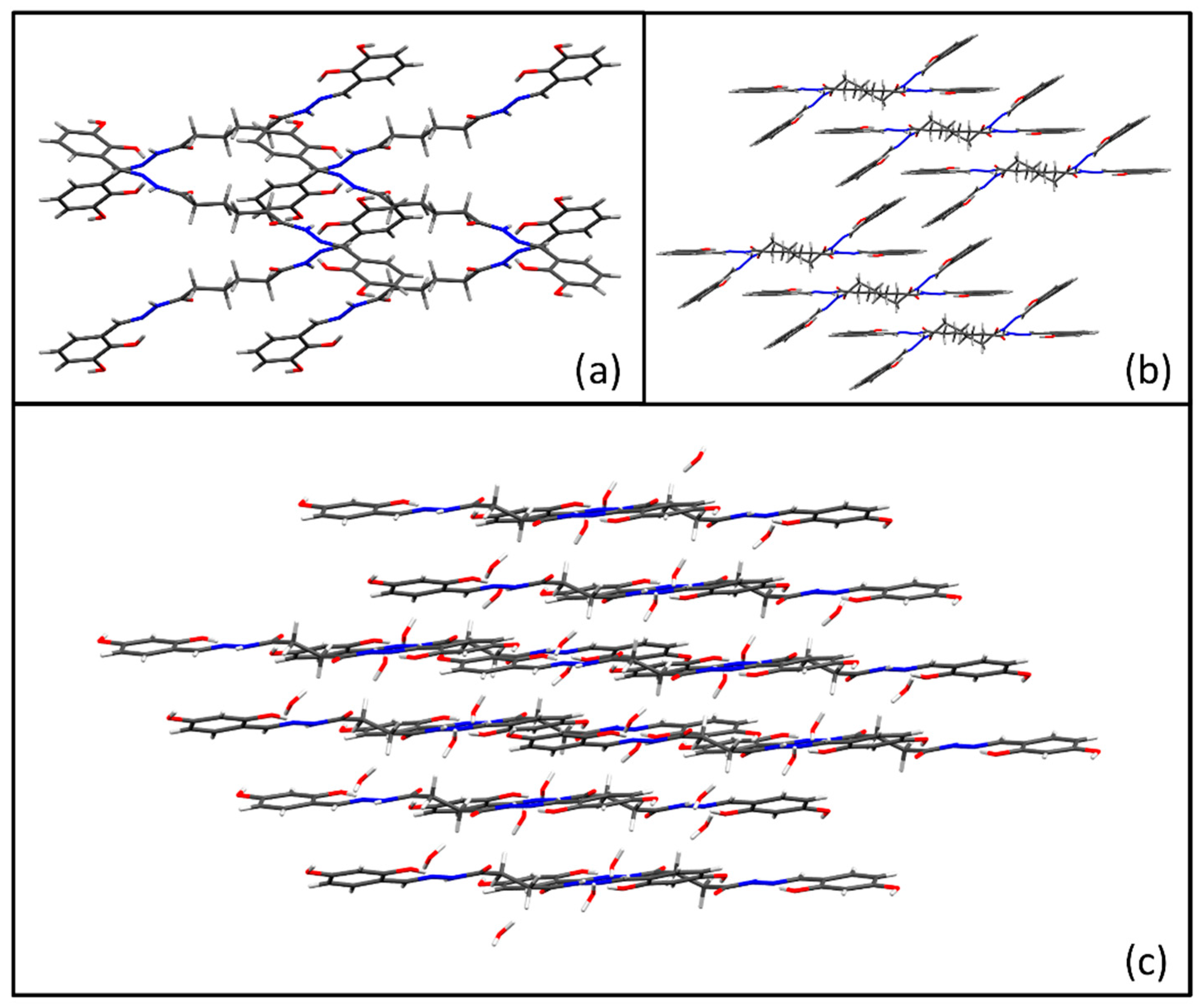
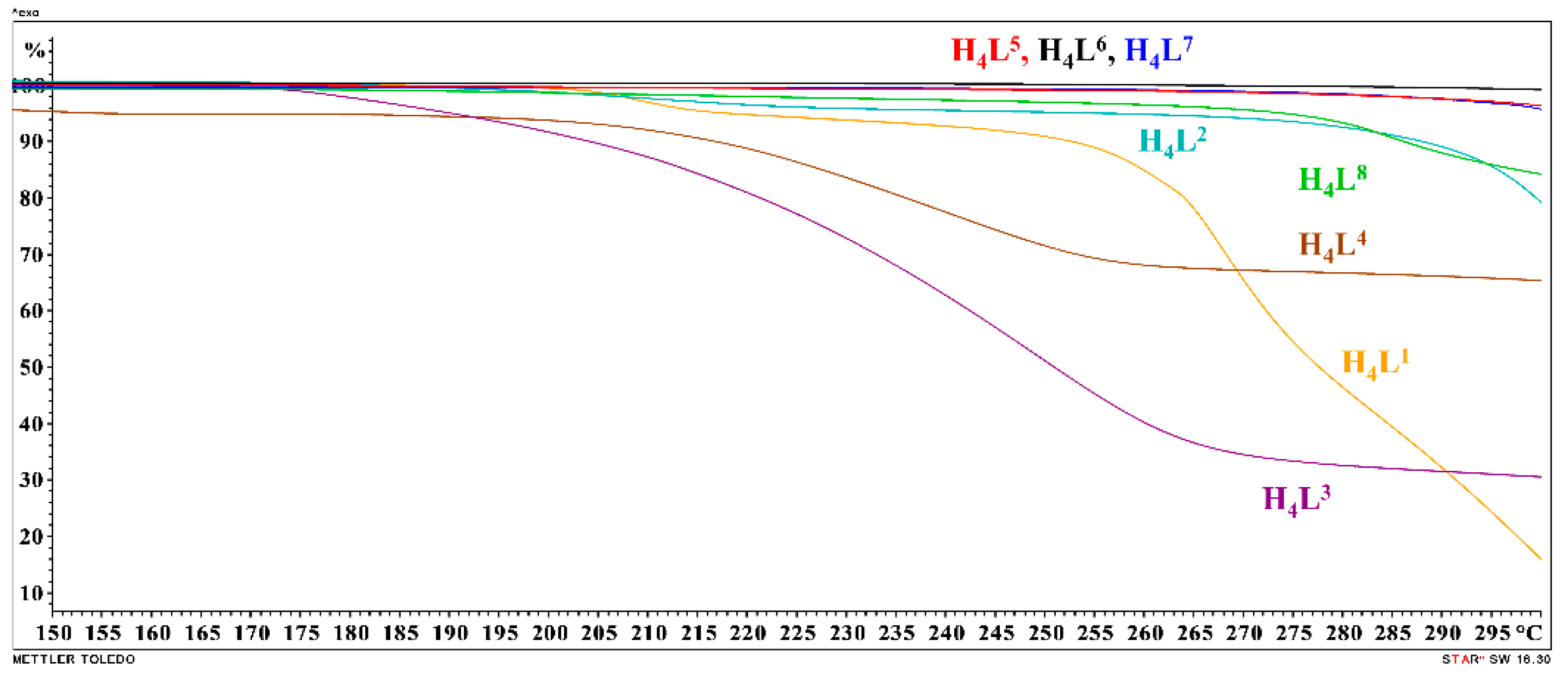
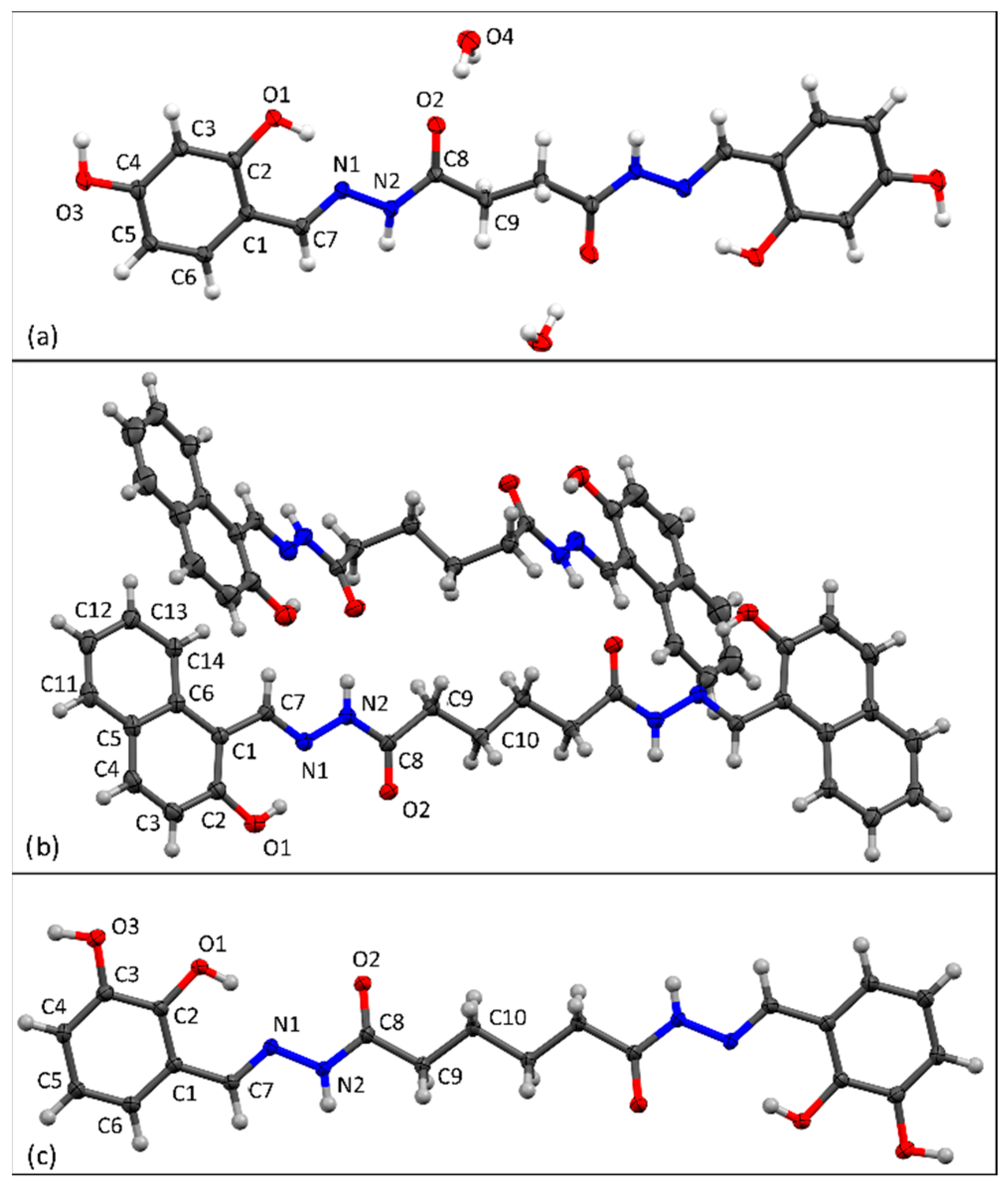
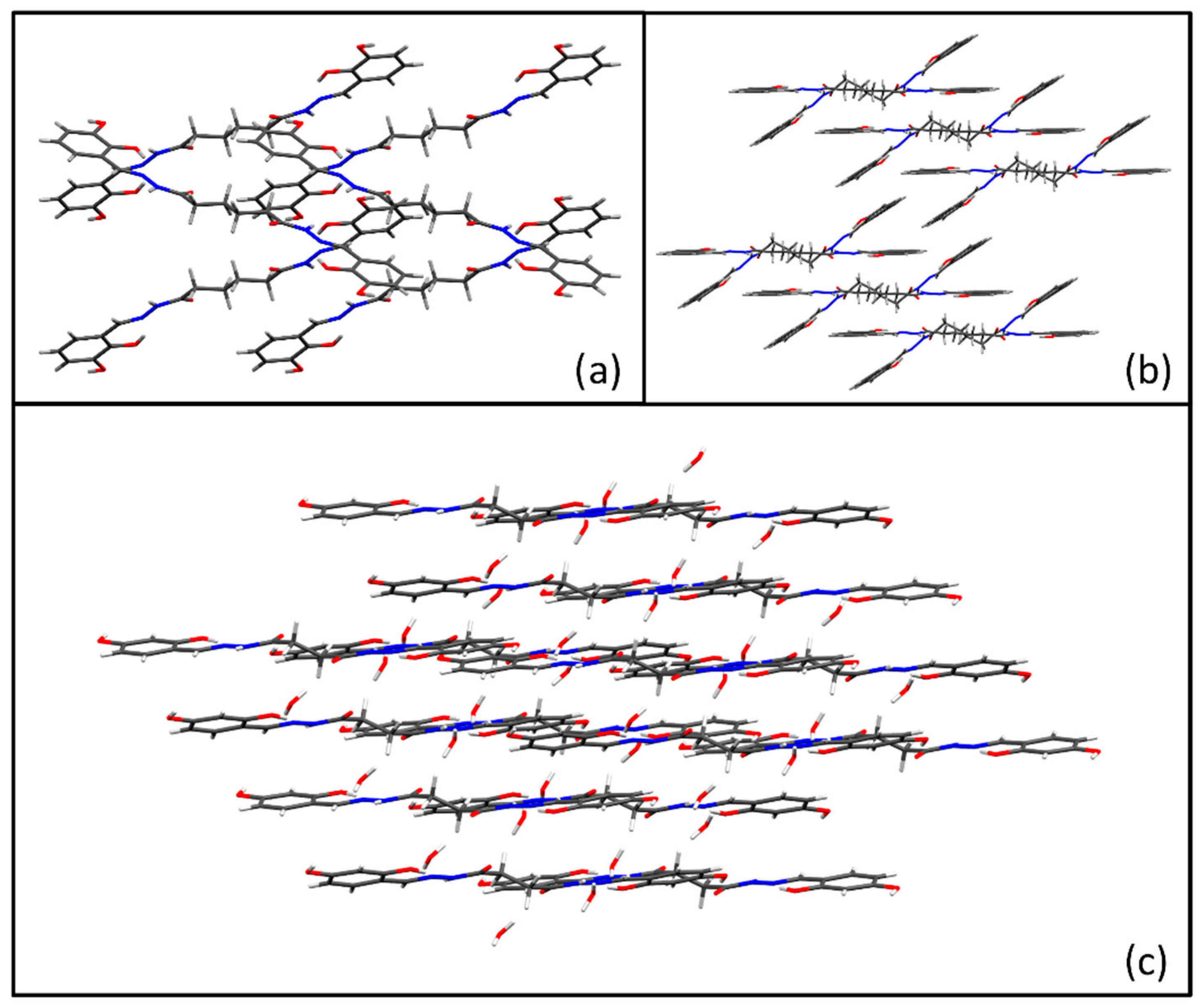
3.1. Synthesis and Solid-State Characterization
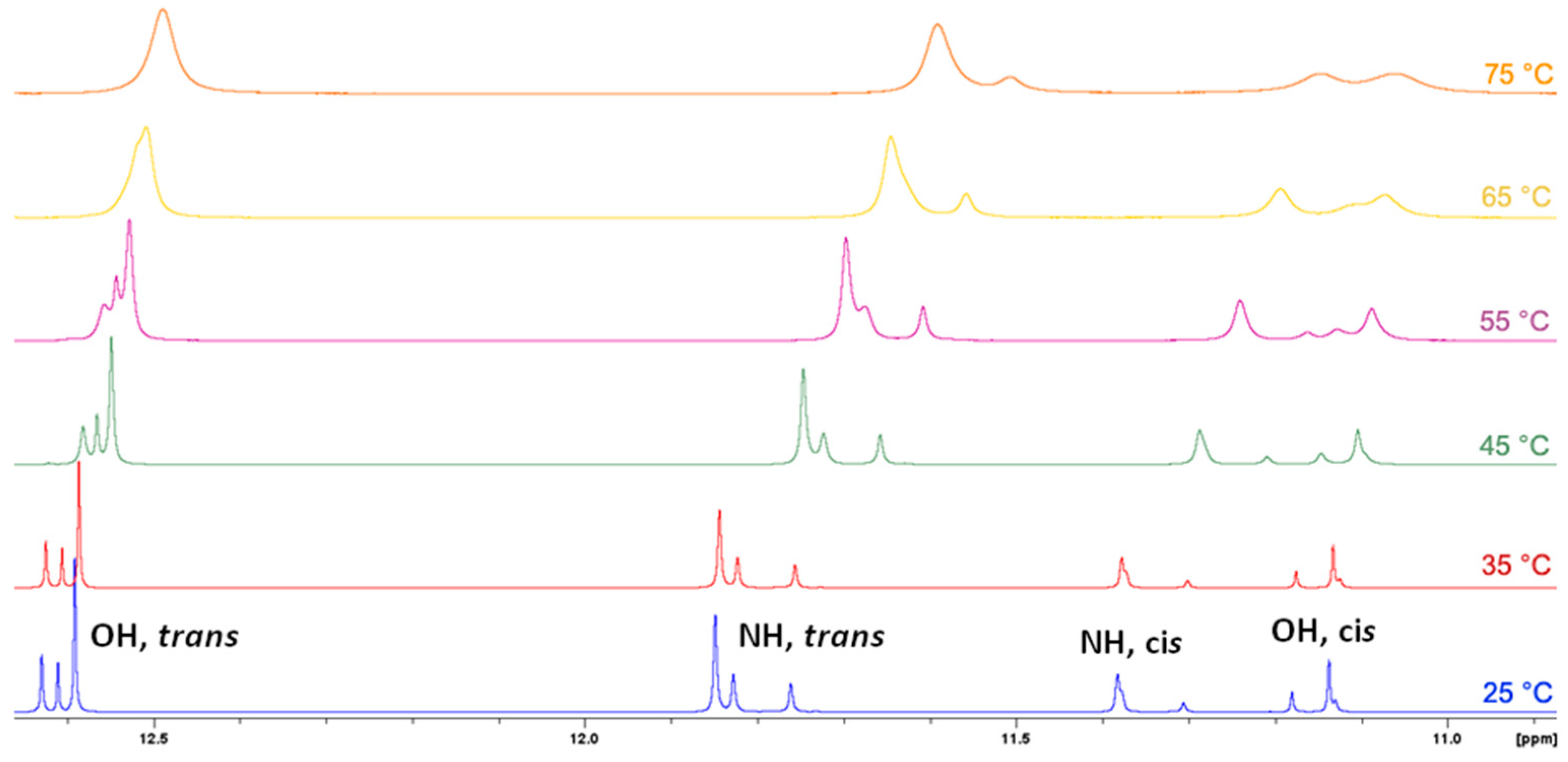
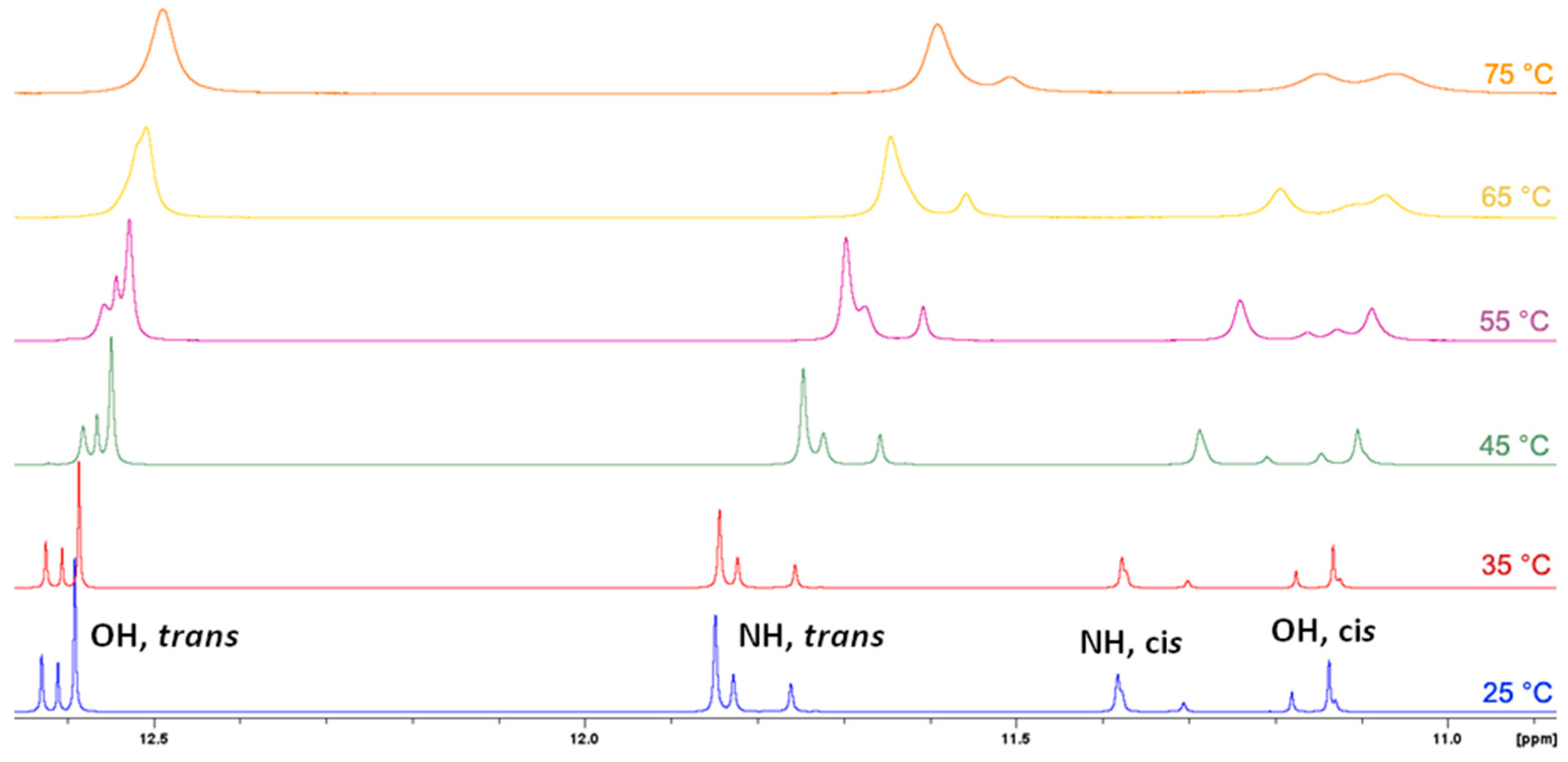
3.2. NMR Spectroscopy
3.3. In Vitro Cytotoxic and Antibacterial Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tatum, L.A.; Su, X.; Aprahamian, I. Simple Hydrazone Building Blocks for Complicated Functional Materials. Acc. Chem. Res. 2014, 47, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Ali, A. Exploring the Pharmacological Activities of Hydrazone Derivatives: A Review. J. Phytochem. Biochem. 2017, 1, 1–11. [Google Scholar]
- Sonawane, S.J.; Kalhapure, R.S.; Govender, T. Hydrazone Linkages in pH Responsive Drug Delivery Systems. Eur. J. Pharm. Sci. 2017, 99, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P.A. Applications of Supramolecular Anion Recognition. Chem. Rev. 2015, 115, 8038–8155. [Google Scholar] [CrossRef]
- Su, X.; Aprahamian, I. Hydrazone-based switches, metallo-assemblies and sensors. Chem. Soc. Rev. 2014, 43, 1963–1981. [Google Scholar] [CrossRef] [Green Version]
- Van Dijken, D.J.; Kovaříček, P.; Ihrig, S.P.; Hecht, S. Acylhydrazones as Widely Tunable Photoswitches. J. Am. Chem. Soc. 2015, 137, 14982–14991. [Google Scholar] [CrossRef]
- Li, L.-Y.; Peng, J.-D.; Zhou, W.; Qiao, H.; Deng, X.; Li, Z.-H.; Li, J.-D.; Fu, Y.-D.; Li, S.; Sun, K.; et al. Potent Hydrazone Derivatives Targeting Esophageal Cancer Cells. Eur. J. Med. Chem. 2018, 148, 359–371. [Google Scholar] [CrossRef]
- Verma, G.; Marella, A.; Shaquiquzzaman, M.; Akhtar, M.; Rahmat Ali, M.; Mumtaz Alam, M. A review exploring biological activities of hydrazones. J. Pharm. Bioallied Sci. 2014, 6, 69–80. [Google Scholar]
- Rollas, S.; Küçükgüzel, S.G. Biological activities of hydrazone derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira Carneiro Brum, J.; Costa França, T.C.; LaPlante, S.R.; Figueroa Villar, J.D. Synthesis and Biological Activity of Hydrazones and Derivatives: A Review. Mini-Rev. Med. Chem. 2020, 20, 342–368. [Google Scholar] [CrossRef]
- Ullah, H.; Previtali, V.; Mihigo, H.B.; Twamley, B.; Rauf, M.K.; Javed, F.; Waseem, A.; Baker, R.J.; Rozas, I. Structure-activity relationships of new Organotin(IV) anticancer agents and their cytotoxicity profile on HL-60, MCF-7 and HeLa human cancer cell lines. Eur. J. Med. Chem. 2019, 181, 111544. [Google Scholar] [CrossRef] [PubMed]
- Omidi, S.; Kakanejadifard, A. A review on biological activities of Schiff base, hydrazone, and oxime derivatives of curcumin. RSC Adv. 2020, 10, 30186–30202. [Google Scholar] [CrossRef] [PubMed]
- Wahbeh, J.; Milkowski, S. The Use of Hydrazones for Biomedical Applications. SLAS Technol. 2019, 24, 161–168. [Google Scholar] [CrossRef]
- Ray, D.; Foy, J.T.; Hughes, R.P.; Aprahamian, I. A switching cascade of hydrazone-based rotary switches through coordination-coupled proton relays. Nat. Chem. 2012, 4, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Ruben, M.; Lehn, J.-M.; Müller, P. Addressing metal centres in supramolecular assemblies. Chem. Soc. Rev. 2006, 35, 1056–1067. [Google Scholar] [CrossRef]
- Hardy, J.G. Metallosupramolecular grid complexes: Towards nanostructured materials with high-tech applications. Chem. Soc. Rev. 2013, 42, 7881–7899. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Niel, V.; Thompson, L.K.; Xu, Z.; Milway, V.A.; Harvey, R.G.; Miller, D.O.; Wilson, C.; Leech, M.; Howard, J.A.K.; et al. Self-assembled polynuclear clusters derived from some flexible polydentate dihydrazide ligands. Dalton Trans. 2004, 9, 1446–1455. [Google Scholar] [CrossRef]
- Stadler, A.-M.; Harrowfield, J. Bis-acyl-/aroyl-hydrazones as multidentate ligands. Inorg. Chim. Acta 2009, 362, 4298–4314. [Google Scholar] [CrossRef]
- Lu, C.; Htan, B.; Ma, C.; Liao, R.-Z.; Gan, Q. Acylhydrazone Switches: E/Z Stability Reversed by Introduction of Hydrogen Bonds. Eur. J. Org. Chem. 2018, 48, 7046–7050. [Google Scholar] [CrossRef]
- Landge, S.M.; Tkatchouk, E.; Benítez, D.; Lanfranchi, D.A.; Elhabiri, M.; Goddard, W.A.; Aprahamian, I. Isomerization Mechanism in Hydrazone-Based Rotary Switches: Lateral Shift, Rotation, or Tautomerization? J. Am. Chem. Soc. 2011, 133, 9812–9823. [Google Scholar] [CrossRef] [Green Version]
- Cvrtila, I.; Fanlo-Virgós, H.; Schaeffer, G.; Monreal Santiago, G.; Otto, S. Redox Control over Acyl Hydrazone Photoswitches. J. Am. Chem. Soc. 2017, 139, 12459–12465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borthakur, R.; Kumar, A.; Lemtur, A.; Lal, R.A. Synthesis, characterization and electrochemical properties of bis(μ2-perchlorato)tricopper(II) complexes derived from succinoyldihydrazones. RSC Adv. 2013, 3, 15139–15147. [Google Scholar] [CrossRef]
- Ranford, J.D.; Vittal, J.J.; Wang, Y.M. Dicopper(II) Complexes of the Antitumor Analogues Acylbis(salicylaldehyde hydrazones) and Crystal Structures of Monomeric [Cu2(1,3-propanedioyl bis(salicylaldehydehydrazone))(H2O)2]⋅(ClO4)2·3H2O and Polymeric [{Cu2(1,6-hexanedioyl bis(salicylaldehydehydrazone))(C2H5OH)2}m]⋅(ClO4)2m·m(C2H5OH). Inorg. Chem. 1998, 37, 1226–1231. [Google Scholar] [PubMed]
- Popiołek, Ł. Hydrazide–hydrazones as potential antimicrobial agents: Overview of the literature since 2010. Med. Chem. Res. 2017, 26, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Narasimhan, B. Hydrazides/hydrazones as antimicrobial and anticancer agents in the new millennium. Mini Rev. Med. Chem. 2013, 13, 971–987. [Google Scholar] [CrossRef] [PubMed]
- Shujah, S.; Khalid, N.; Ali, S. Homobimetallic Organotin(IV) Complexes with Succinohydrazide Schiff Base: Synthesis, Spectroscopic Characterization, and Biological Screening. Russ. J. Gen. Chem. 2017, 87, 515–522. [Google Scholar] [CrossRef]
- Sedaghat, T.; Aminian, M.; Bruno, G.; Amiri Rudbari, H. Binuclear organotin(IV) complexes with adipic dihydrazones: Synthesis, spectral characterization, crystal structures and antibacterial activity. J. Organomet. Chem. 2013, 737, 26–31. [Google Scholar] [CrossRef]
- Sharma, M.P.; Varshney, V.K.; Sharma, R.C. Synthesis and antimicrobial study of some hydrazone metal complexes. Proc. Natl. Acad. Sci. USA 1991, 61, 447–452. [Google Scholar]
- González-García, C.; Mata, A.; Zani, F.; Antonia Mendiola, M.; López-Torres, E. Synthesis and antimicrobial activity of tetradentate ligands bearing hydrazone and/or thiosemicarbazone motifs and their diorganotin(IV) complexes. J. Inorg. Biochem. 2016, 163, 118–130. [Google Scholar] [CrossRef]
- CrysAlisPro Software System, version 1.171.41.92a; Rigaku Oxford Diffraction: Oxford, UK, 2020.
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Rubčić, M.; Pisk, J.; Pičuljan, K.; Damjanović, V.; Lovrić, J.; Vrdoljak, V. Symmetrical disubstituted carbohydrazides: From solid-state structures to cytotoxic and antibacterial activity. J. Mol. Struct. 2019, 1178, 222–228. [Google Scholar] [CrossRef]
- CLSI Document M07-A8, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2009.
- Rubčić, M.; Galić, N.; Halasz, I.; Jednačak, T.; Judaš, N.; Plavec, J.; Šket, P.; Novak, P. Multiple solid forms of 1,5-bis(salicylidene)carbohydrazide: Polymorph-modulated thermal reactivity. Cryst. Growth Des. 2014, 14, 2900–2912. [Google Scholar] [CrossRef]
- Borthakur, R.; Kumar, A.; Lal, R.A. Synthesis and characterization of heterotrinuclear bis(μ2-chlorido)dicopper (II) mono zinc(II) complexes derived from succinoyldihydrazones. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 118, 94–101. [Google Scholar] [CrossRef]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Cryst. B 1990, 46, 256–262. [Google Scholar] [CrossRef]
- Hansen, P.E.; Rozwadowski, Z.; Dziembowska, T. NMR Studies of Hydroxy Schiff Bases. Curr. Org. Chem. 2009, 13, 194–215. [Google Scholar] [CrossRef]
- Claramunt, R.M.; López, C.; Santa María, M.D.; Sanz, D.; Elguero, J. The use of NMR spectroscopy to study tautomerism. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 49, 169–206. [Google Scholar] [CrossRef]
- Novak, P.; Jednačak, T.; Parlov Vuković, J.; Zangger, K.; Rubčić, M.; Galić, N.; Hrenar, T. Synthesis, Structural Characterization and Hydrogen Bonding of Mono(salicylidene)carbohydrazide. Croat. Chem. Acta 2012, 85, 451–456. [Google Scholar] [CrossRef]
- Schilf, W.; Kamieński, B.; Užarević, K. Nitrogen and carbon CPMAS NMR investigations of keto–enol tautomerism in asymmetric o-hydroxy Schiff bases. J. Mol. Struct. 2013, 1031, 211–215. [Google Scholar] [CrossRef]
- Užarević, K.; Rubčić, M.; Stilinović, V.; Kaitner, B.; Cindrić, M. Keto–enol tautomerism in asymmetric Schiff bases derived from p-phenylenediamine. J. Mol. Struct. 2010, 984, 232–239. [Google Scholar] [CrossRef]
- Božić, A.R.; Filipović, N.R.; Verbić, T.Ž.; Milčić, M.K.; Todorović, T.R.; Cvijetić, I.N.; Klisurić, O.R.; Radišić, M.M.; Marinković, A.D. A detailed experimental and computational study of monocarbohydrazones. Arab. J. Chem. 2020, 13, 932–953. [Google Scholar] [CrossRef]
- Caprice, K.; Aster, A.; Cougnon, F.B.L.; Kumpulainen, T. Untying the Photophysics of Quinolinium-Based Molecular Knots and Links. Chem. Eur. J. 2020, 26, 1576–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, G. Chemical aspects of peptide bond isomerisation. Chem. Soc. Rev. 2000, 29, 119–127. [Google Scholar] [CrossRef]
- Hamzi, I.; Barhoumi-Slimi, T.M.; Abidi, R. Synthesis, Characterization, and Conformational Study of Acylhydrazones of α,β-Unsaturated Aldehydes. Heteroat. Chem. 2016, 27, 139–148. [Google Scholar] [CrossRef]
- Kumar, P.; Kadyan, K.; Duhan, M.; Sindhu, J.; Singh, V.; Singh Saharan, B. Design, synthesis, conformational and molecular docking study of some novel acyl hydrazone based molecular hybrids as antimalarial and antimicrobial agents. Chem. Cent. J. 2017, 11, 115. [Google Scholar] [CrossRef] [Green Version]
- In principle, E/Z isomerisation can also occur in hydrazones. However, this process usually proceeds under the influence of external stimuli, e.g., UV irradiation, and it is crucial that structure contains functionality that can stabilize generally less stable Z isomer through e.g., intramolecular hydrogen bond. For example see references 1, 5, 14 and 20. For the structures investigated here, the observed chemical shifts do not suggest the occurrence of Z isomer in solution. Also considering the structure of molecules, occurrence of Z isomer here should be disfavored due to the sterical hindrance it should sustain.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50/µmol L−1 | MIC (μg mL−1) | ||||
|---|---|---|---|---|---|---|
| THP-1 | HepG2 | S. aureus | E. faecalis | E. coli | M. catarrhalis | |
| H4L1 | >100 | >100 | >256 | 64 | >256 | 16 |
| H4L2 | >100 | >100 | >256 | 16 | 128 | 8 |
| H4L3 | >100 | >100 | >256 | >256 | >256 | 128 |
| H4L4 | >100 | >100 | >256 | >256 | >256 | >256 |
| H4L5 | >100 | >100 | >256 | 16 | >256 | 256 |
| H4L6 | >100 | >100 | >256 | 16 | 16 | 16 |
| H4L7 | >100 | >100 | >256 | >256 | >256 | >256 |
| H4L8 | >100 | >100 | >256 | >256 | >256 | 128 |
| staurosporine | 0.32 | 30.75 | - | - | - | - |
| azithromycin | - | - | 2 | 8 | 0.25 | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Topić, E.; Damjanović, V.; Pičuljan, K.; Vrdoljak, V.; Rubčić, M. Succinyl and Adipoyl Dihydrazones: A Solid-State, Solution and Antibacterial Study. Crystals 2022, 12, 1175. https://doi.org/10.3390/cryst12081175
Topić E, Damjanović V, Pičuljan K, Vrdoljak V, Rubčić M. Succinyl and Adipoyl Dihydrazones: A Solid-State, Solution and Antibacterial Study. Crystals. 2022; 12(8):1175. https://doi.org/10.3390/cryst12081175
Chicago/Turabian StyleTopić, Edi, Vladimir Damjanović, Katarina Pičuljan, Višnja Vrdoljak, and Mirta Rubčić. 2022. "Succinyl and Adipoyl Dihydrazones: A Solid-State, Solution and Antibacterial Study" Crystals 12, no. 8: 1175. https://doi.org/10.3390/cryst12081175
APA StyleTopić, E., Damjanović, V., Pičuljan, K., Vrdoljak, V., & Rubčić, M. (2022). Succinyl and Adipoyl Dihydrazones: A Solid-State, Solution and Antibacterial Study. Crystals, 12(8), 1175. https://doi.org/10.3390/cryst12081175