
Computational Study of Benzothiazole Derivatives for Conformational, Thermodynamic and Spectroscopic Features and Their Potential to Act as Antibacterials

 ,
,  , ,
, ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Computational Details
2.2. Molecular Docking Analysis
2.3. Ligand Preparation, Protein Preparation and Grid Generation
3. Results and Discussion
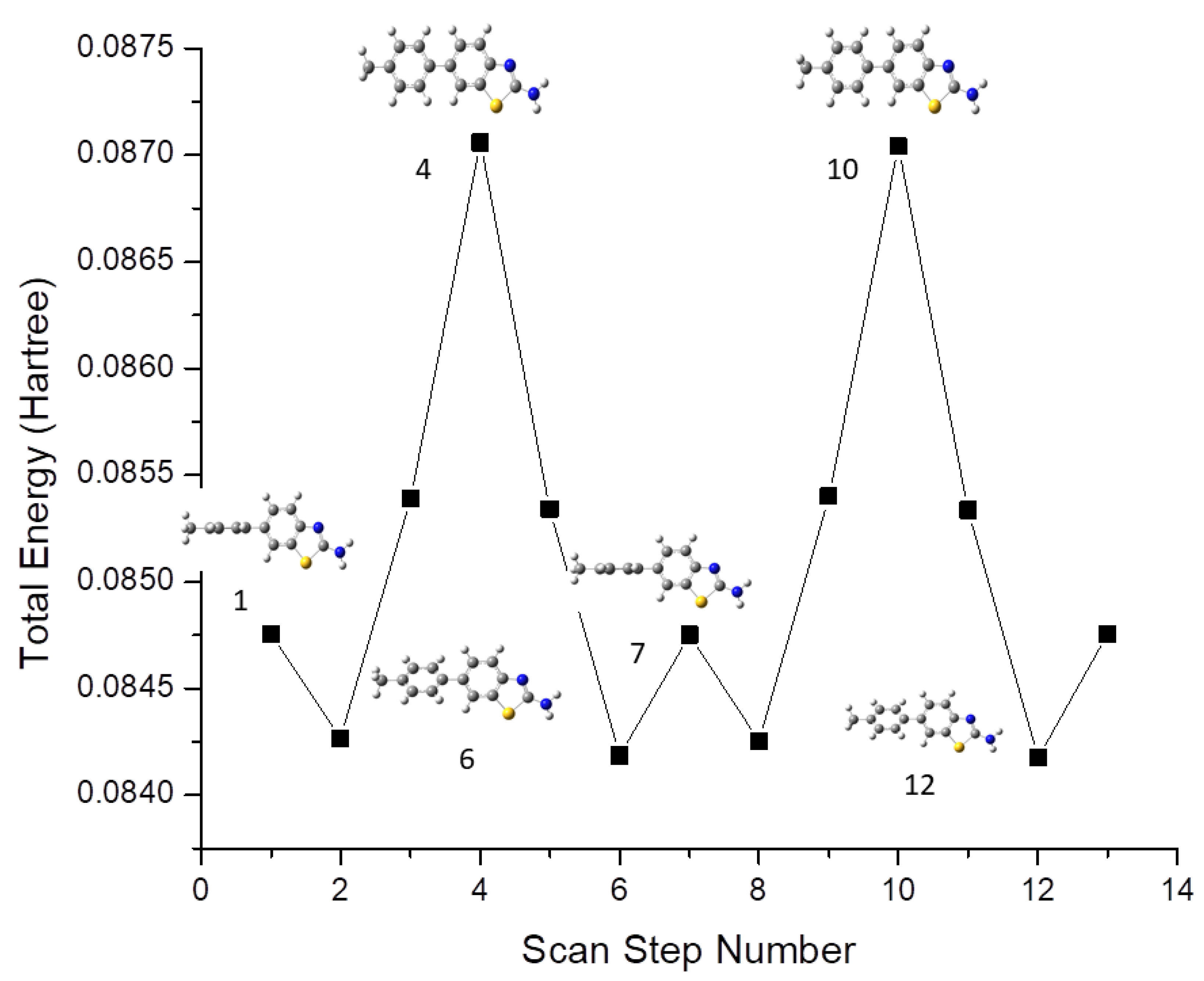
3.1. Conformational Analysis
3.2. Geometrical Parameters
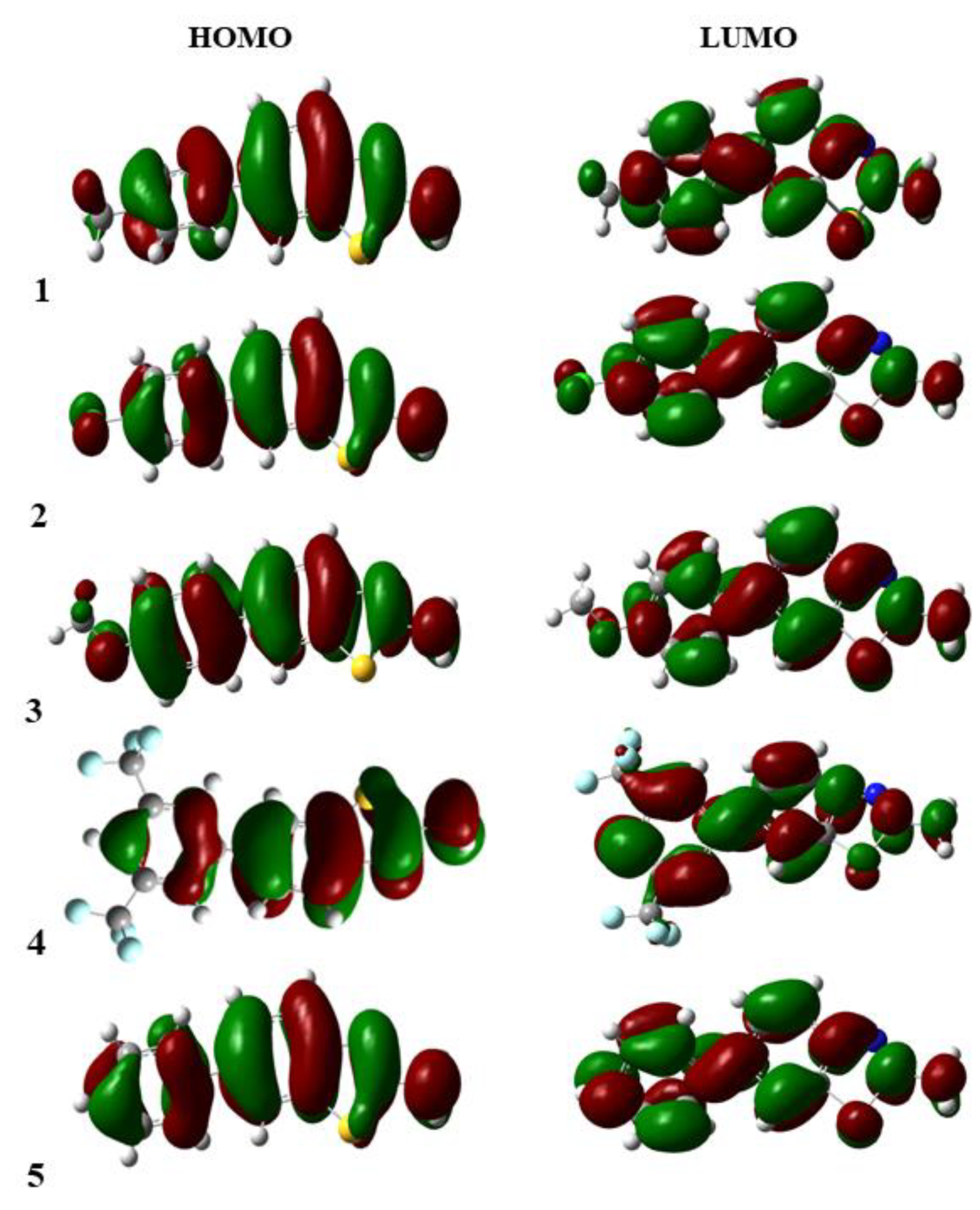
3.3. Frontier Molecular Orbitals
3.4. NLO Properties
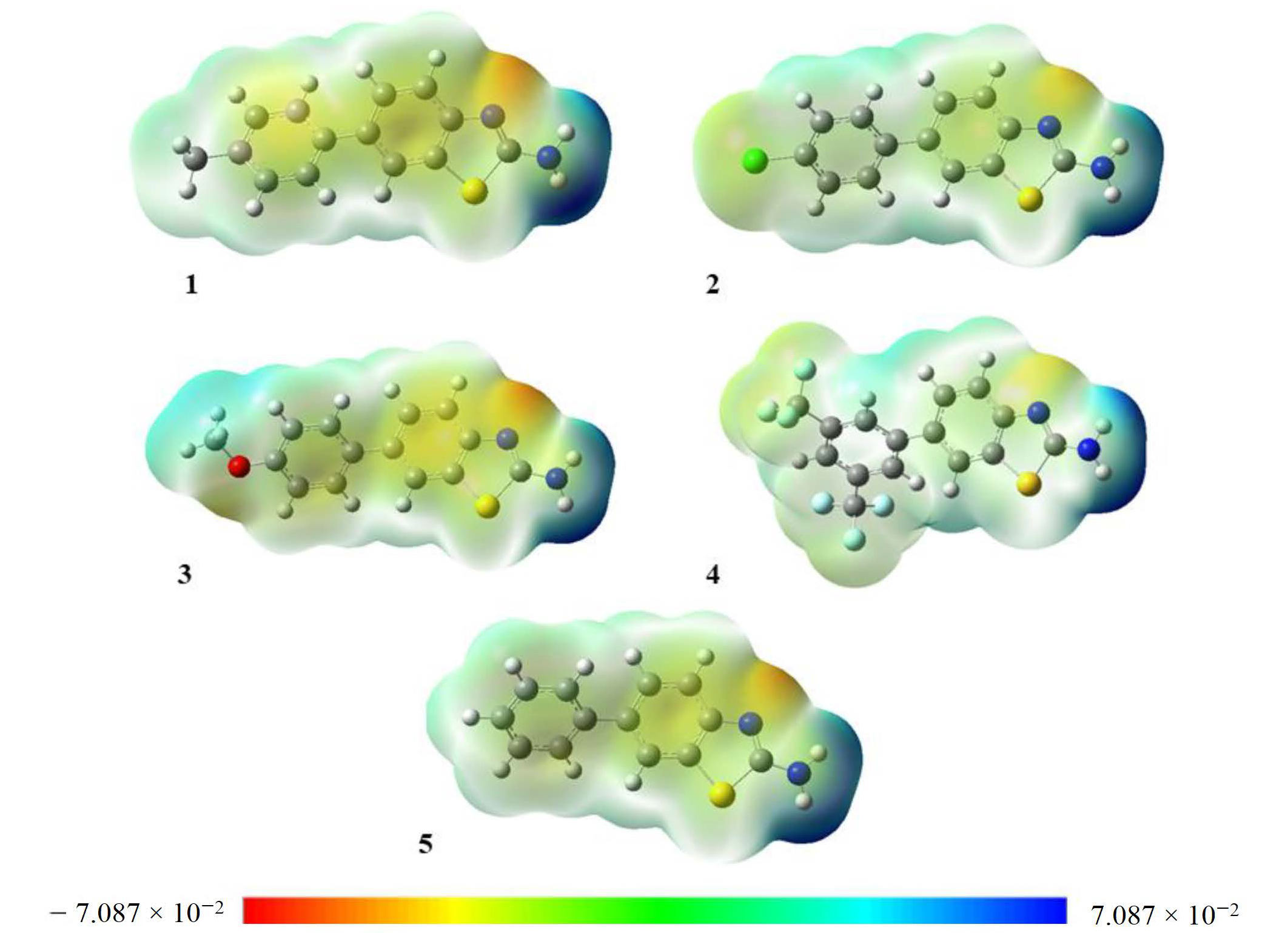
3.5. Molecular Electrostatic Potential (MEP)
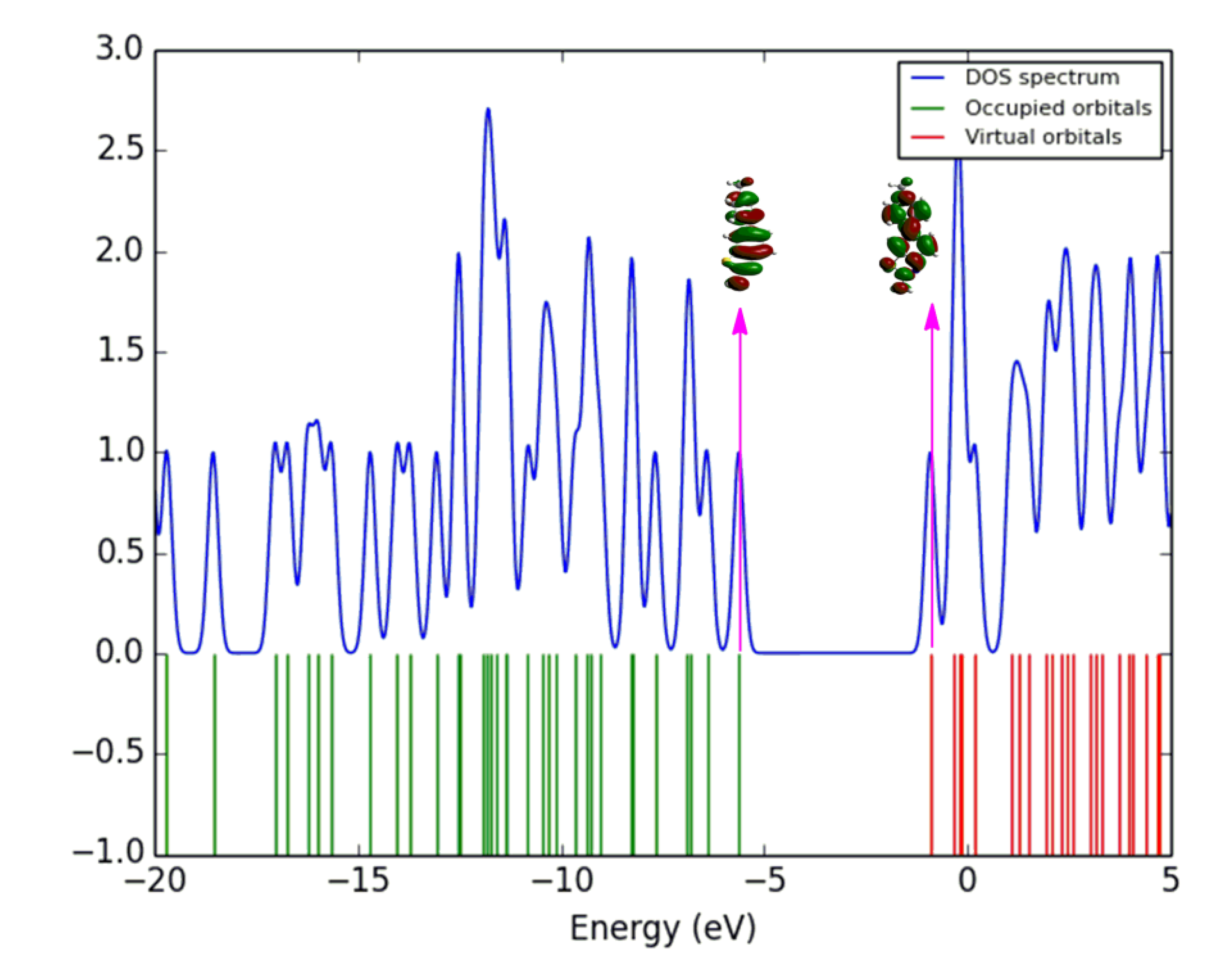
3.6. Density of States
3.7. NMR Spectra
3.8. UV-Visible Spectra
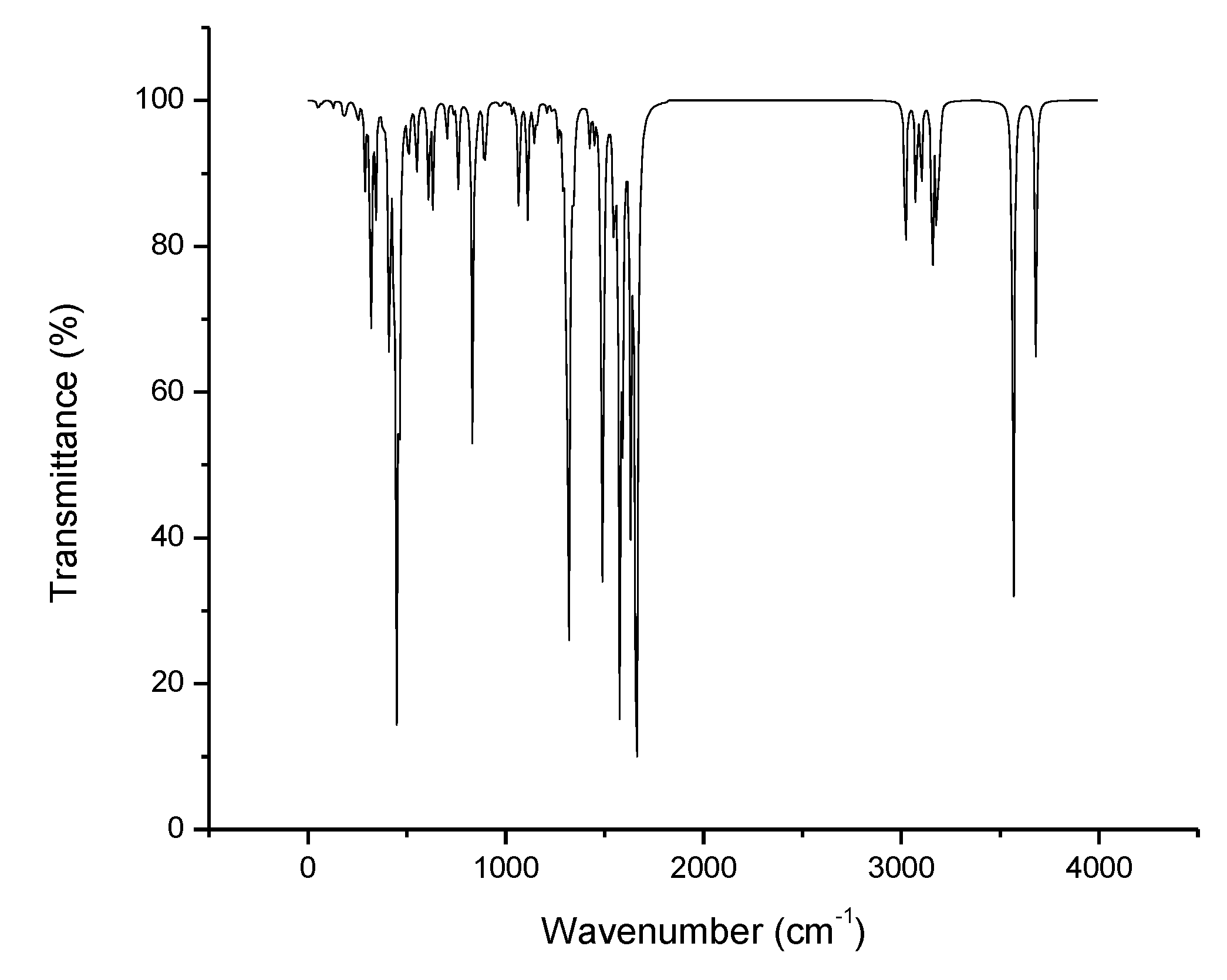
3.9. IR Spectra
3.10. Reactivity Parameters
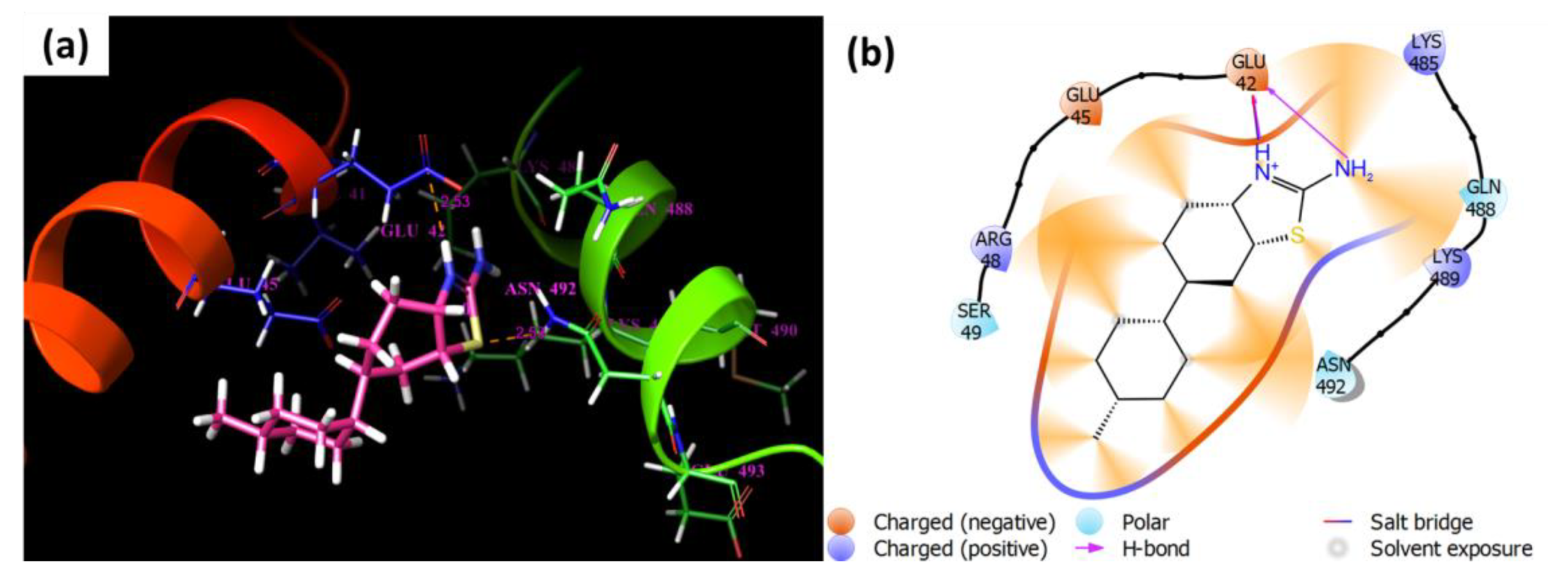
3.11. Molecular Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sathyanarayanmoorthi, V.; Karunathan, R.; Kannappan, V. Molecular modeling and spectroscopic studies of Benzothiazole. J. Chem. 2013, 2013, 258519. [Google Scholar] [CrossRef] [Green Version]
- Giorgioni, G.; Accorroni, B.; Di Stefano, A.; Marucci, G.; Siniscalchi, A.; Claudi, F. Benzimidazole, Benzoxazole and Benzothiazole Derivatives as 5HT 2B Receptor Ligands. Synthesis and Preliminary Pharmacological Evaluation. Med. Chem. Res. 2005, 14, 57–73. [Google Scholar] [CrossRef]
- Gull, Y.; Rasool, N.; Noreen, M.; Altaf, A.A.; Musharraf, S.G.; Zubair, M.; Nasim, F.-U.-H.; Yaqoob, A.; DeFeo, V.; Zia-Ul-Haq, M. Synthesis of N-(6-Arylbenzo[d]thiazole-2-acetamide derivatives and their biological activities: An experimental and computational approach. Molecules 2016, 21, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paramashivappa, R.; Kumar, P.P.; Rao, P.S.; Rao, A.S. Design, synthesis and biological evaluation of benzimidazole/benzothiazole and benzoxazole derivatives as cyclooxygenase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 657–660. [Google Scholar] [CrossRef]
- Mahran, M.A.; William, S.; Ramzy, F.; Sembel, A.M. Synthesis and in vitro evaluation of new benzothiazole derivatives as schistosomicidal agents. Molecules 2007, 12, 622–633. [Google Scholar] [CrossRef]
- Rajeeva, B.; Srinivasulu, N.; Shantakumar, S. Synthesis and Antimicrobial Activity of Some New 2-Substituted Benzothiazole Derivatives. E-J. Chem. 2009, 6, 775–779. [Google Scholar] [CrossRef] [Green Version]
- Kini, S.; Swain, S.; Gandhi, A. Synthesis and evaluation of novel benzothiazole derivatives against human cervical cancer cell lines. Indian J. Pharm. Sci. 2007, 69, 46. [Google Scholar] [CrossRef] [Green Version]
- Kaur, H.; Kumar, S.; Singh, I.; Saxena, K.; Kumar, A. Synthesis, characterization and biological activity of various substituted benzothiazole derivatives. Dig. J. Nanomater. Bios. 2010, 5, 67–76. [Google Scholar]
- Vicini, P.; Geronikaki, A.; Incerti, M.; Busonera, B.; Poni, G.; Cabras, C.A.; La Colla, P. Synthesis and biological evaluation of benzo[d]isothiazole, benzothiazole and thiazole Schiff bases. Bioorg. Med. Chem. 2003, 11, 4785–4789. [Google Scholar] [CrossRef]
- Yadav, P.; Devprakash, D.; Senthilkumar, G. Benzothiazole: Different methods of synthesis and diverse biological activities. ChemInform 2011, 42. [Google Scholar] [CrossRef]
- Netalkar, P.P.; Netalkar, S.P.; Budagumpi, S.; Revankar, V.K. Synthesis, crystal structures and characterization of late first row transition metal complexes derived from benzothiazole core: Anti-tuberculosis activity and special emphasis on DNA binding and cleavage property. Eur. J. Med. Chem. 2014, 79, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, S.S.; Thakor, P.; Ray, A.; Doshi, H.; Thakkar, V.R. Benzothiazole analogues: Synthesis, characterization, MO calculations with PM6 and DFT, in silico studies and in vitro antimalarial as DHFR inhibitors and antimicrobial activities. Bioorg. Med. Chem. 2017, 25, 5396–5406. [Google Scholar] [CrossRef] [PubMed]
- Bondock, S.; Fadaly, W.; Metwally, M. Recent trends in the chemistry of 2-aminobenzothiazoles. J. Sulphur Chem. 2009, 30, 74–107. [Google Scholar] [CrossRef]
- Lewars, E. Computational chemistry. In Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Springer: Berlin/Heidelberg, Germany, 2003; p. 318. [Google Scholar]
- Ramachandran, K.; Deepa, G.; Namboori, K. Computational Chemistry and Molecular Modeling: Principles and Applications; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Gull, Y.; Rasool, N.; Noreen, M.; Nasim, F.-u.-H.; Yaqoob, A.; Kousar, S.; Rashid, U.; Bukhari, I.H.; Zubair, M.; Islam, M. Efficient synthesis of 2-amino-6-arylbenzothiazoles via Pd (0) Suzuki cross coupling reactions: Potent urease enzyme inhibition and nitric oxide scavenging activities of the products. Molecules 2013, 18, 8845–8857. [Google Scholar] [CrossRef] [Green Version]
- Hashmi, M.A.; Khan, A.; Ayub, K.; Farooq, U. Spectroscopic and density functional theory studies of 5, 7, 3′, 5′-tetrahydroxyflavanone from the leaves of Olea ferruginea. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 128, 225–230. [Google Scholar] [CrossRef]
- Mubarik, A.; Rasool, N.; Hashmi, M.A.; Mansha, A.; Zubair, M.; Shaik, M.R.; Sharaf, M.A.; Awwad, E.M.; Abdelgawad, A. Computational Study of Structural, Molecular Orbitals, Optical and Thermodynamic Parameters of Thiophene Sulfonamide Derivatives. Crystals 2021, 11, 211. [Google Scholar] [CrossRef]
- Ahmad, G.; Rasool, N.; Mubarik, A.; Zahoor, A.F.; Hashmi, M.A.; Zubair, M.; Bilal, M.; Hussien, M.; Akhtar, M.S.; Haider, S. Facile Synthesis of 5-Aryl-N-(pyrazin-2-yl) thiophene-2-carboxamides via Suzuki Cross-Coupling Reactions, Their Electronic and Nonlinear Optical Properties through DFT Calculations. Molecules 2021, 26, 7309. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, Y.; Lu, Z.; Huang, Z. Structural basis of Staphylococcus aureus Cas9 inhibition by AcrIIA14. Nucleic Acids Res. 2021, 49, 6587–6595. [Google Scholar] [CrossRef]
- Jabri, E.; Carr, M.B.; Hausinger, R.P.; Karplus, P.A. The crystal structure of urease from Klebsiella aerogenes. Science 1995, 268, 998–1004. [Google Scholar] [CrossRef]
- Bhachoo, J.; Beuming, T. Investigating protein–peptide interactions using the Schrödinger computational suite. In Modeling Peptide-Protein Interactions; Springer: Berlin/Heidelberg, Germany, 2017; pp. 235–254. [Google Scholar]
- Lu, J.; Sigalovsky, D.; Hango, C.R.; Devaney, K.J. Computational Chemistry in the High School Classroom. Ph.D. Thesis, Worcester Polytechnic Institute, Worcester, MA, USA, 2014. [Google Scholar]
- Mahmood, N.; Rasool, N.; Ikram, H.M.; Hashmi, M.A.; Mahmood, T.; Zubair, M.; Ahmad, G.; Rizwan, K.; Rashid, T.; Rashid, U. Synthesis of 3, 4-Biaryl-2, 5-Dichlorothiophene through Suzuki Cross-Coupling and Theoretical Exploration of Their Potential Applications as Nonlinear Optical Materials. Symmetry 2018, 10, 766. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.-R. The Principles of Nonlinear Optics; OSTI: Oak Ridge, TN, USA, 1984. [Google Scholar]
- Politzer, P.; Truhlar, D.G. Chemical Applications of Atomic and Molecular Electrostatic Potentials: Reactivity, Structure, Scattering, and Energetics of Organic, Inorganic, and Biological Systems; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Pandey, U.; Srivastava, M.; Singh, R.; Yadav, R. DFT study of conformational and vibrational characteristics of 2-(2-hydroxyphenyl) benzothiazole molecule. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 129, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Wang, Q.; Long, X.; Hu, S.; Gao, T. Density Function Theory Study on the Reaction Mechanism of Cerium with Oxygen for Ce-bearing Aerosol Particle Formation. J. Wuhan Univ. Technol. Sci. Ed. 2020, 35, 501–505. [Google Scholar] [CrossRef]
- Magyar, R.; Tretiak, S.; Gao, Y.; Wang, H.-L.; Shreve, A. A joint theoretical and experimental study of phenylene–acetylene molecular wires. Chem. Phys. Lett. 2005, 401, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Siehl, H.U.; Müller, T.; Gauss, J. NMR Spectroscopic and quantum chemical characterization of the (E)− and (Z)− isomers of the penta-1, 3-dienyl-2-cation. J. Phys. Org. Chem. 2003, 16, 577–581. [Google Scholar] [CrossRef]
- Xavier, R.J.; Dinesh, P. Spectroscopic (FTIR, FT-Raman, 13C and 1H NMR) investigation, molecular electrostatic potential, polarizability and first-order hyperpolarizability, FMO and NBO analysis of 1-methyl-2-imidazolethiol. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 118, 999–1011. [Google Scholar] [CrossRef]
- Schmid, F.X. Biological macromolecules: UV-visible spectrophotometry. In eLS; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Obi-Egbedi, N.; Obot, I.; El-Khaiary, M.; Umoren, S.; Ebenso, E. Computational simulation and statistical analysis on the relationship between corrosion inhibition efficiency and molecular structure of some phenanthroline derivatives on mild steel surface. Int. J. Electrochem. Sci. 2011, 6, e5675. [Google Scholar]
- Tsuneda, T.; Song, J.-W.; Suzuki, S.; Hirao, K. On Koopmans’ theorem in density functional theory. J. Chem. Phys. 2010, 133, 174101. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | |||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Bond lengths | |||||
| S1–C2 | 1.79 | 1.79 | 1.79 | 1.79 | 1.79 |
| C2–N3 | 1.29 | 1.29 | 1.29 | 1.29 | 1.29 |
| C2–NH2 | 1.36 | 1.35 | 1.36 | 1.35 | 1.36 |
| N3–C3a | 1.38 | 1.38 | 1.38 | 1.38 | 1.38 |
| C3a–C4 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 |
| C7–C7a | 1.38 | 1.38 | 1.38 | 1.38 | 1.38 |
| C7a–C3a | 1.41 | 1.41 | 1.41 | 1.41 | 1.41 |
| C7a–S1 | 1.76 | 1.76 | 1.76 | 1.76 | 1.76 |
| C6–C1’ | 1.48 | 1.48 | 1.48 | 1.48 | 1.48 |
| C1’–C2’ | 1.40 | 1.40 | 1.39 | 1.40 | 1.40 |
| Bond angles | |||||
| S1–C2–N3 | 115.83 | 115.82 | 115.80 | 115.83 | 115.82 |
| S1–C2–NH2 | 119.56 | 119.64 | 119.51 | 119.58 | 119.53 |
| H2N–C2–N3 | 124.52 | 124.45 | 124.58 | 124.50 | 124.56 |
| C2–N3–C3a | 111.21 | 111.22 | 111.22 | 111.19 | 111.22 |
| N3–C3a–C7a | 115.91 | 115.92 | 115.90 | 115.92 | 115.90 |
| C3a–C7a–S1 | 109.10 | 109.10 | 109.10 | 109.17 | 109.12 |
| C7a–S1–C2 | 87.91 | 87.91 | 87.93 | 87.86 | 87.91 |
| C5–C6–C1’ | 120.89 | 120.81 | 120.97 | 120.81 | 120.85 |
| C7–C6–C1’ | 120.43 | 120.38 | 120.46 | 120.20 | 120.41 |
| Dihedral angles | |||||
| S1–C2–N3–C3a | −0.01 | −0.10 | −0.11 | 0.08 | −0.19 |
| S1–C2–N–H | 26.08 | 24.56 | 27.10 | 23.95 | 25.96 |
| C2–N3–C3a–C7a | −0.08 | −0.60 | −0.77 | −0.58 | −0.67 |
| N3–C3a–C7a–S1 | 0.14 | 1.01 | 1.27 | 0.79 | 1.21 |
| C3a–C7a–S1–C2 | −0.11 | −0.83 | −1.04 | −0.58 | −1.03 |
| C7a–S1–C2–NH2 | 177.03 | 177.60 | 177.48 | 177.34 | 177.65 |
| Compounds | EHOMO (eV) | ELUMO (eV) | ∆E (eV) | Hyperpolarizability (βo) (Hartree) |
|---|---|---|---|---|
| 1 | −5.63 | −0.92 | 4.71 | 1200.67 |
| 2 | −5.78 | −1.16 | 4.62 | 1989.38 |
| 3 | −5.46 | −0.82 | 4.64 | 153.51 |
| 4 | −5.99 | −1.53 | 4.46 | 3825.91 |
| 5 | −5.71 | −0.98 | 4.73 | 2031.01 |
| Compounds | |||||
|---|---|---|---|---|---|
| Position of H-Atom | 1 | 2 | 3 | 4 | 5 |
| 4 | 7.68 | 7.67 | 7.66 | 7.74 | 7.90 |
| 5 | 7.58 | 7.55 | 7.54 | 7.69 | 7.59 |
| 7 | 7.87 | 7.87 | 7.85 | 7.98 | 7.69 |
| 2’ | 7.64 | 7.65 | 7.67 | 8.22 | 7.68 |
| 3’ | 7.51 | 7.52 | 6.92 | – | 7.66 |
| 4’ | – | – | – | 8.05 | 7.53 |
| 5’ | 7.50 | 7.52 | 7.17 | – | 7.66 |
| 6’ | 7.58 | 7.60 | 7.60 | 8.16 | 7.73 |
| Position of C-Atom | |||||
| C2 | 170.5 | 170.9 | 170.6 | 171.5 | 170.8 |
| C3a | 155.7 | 156.4 | 155.2 | 157.3 | 155.9 |
| C4 | 122.6 | 122.7 | 122.6 | 123.1 | 122.7 |
| C5 | 129.4 | 129.4 | 128.9 | 129.6 | 129.6 |
| C6 | 141.8 | 139.9 | 141.6 | 137.9 | 141.8 |
| C7 | 123.2 | 123.3 | 122.8 | 123.8 | 123.5 |
| C7a | 141.9 | 141.9 | 141.9 | 142.3 | 141.8 |
| C1’ | 145.1 | 146.9 | 139.2 | 149.9 | 148.3 |
| C2’ | 131.6 | 133.1 | 132.8 | 133.8 | 131.8 |
| C3’ | 133.1 | 133.0 | 111.7 | 136.3 | 132.6 |
| C4’ | 143.0 | 144.9 | 149.2 | 124.3 | 130.6 |
| C5’ | 133.1 | 133.0 | 120.9 | 136.1 | 132.6 |
| C6’ | 131.4 | 133.8 | 132.7 | 133.1 | 131.5 |
| Compounds | Major Contribution | λmax (nm) | Oscillator Strengths (f) | Excitation Energies (eV) |
|---|---|---|---|---|
| (ES1) H → L 70% | 382.25 | 0.2735 | 3.24 | |
| 1 | (ES2) H → L 60% | 265.23 | 0.0033 | 4.67 |
| (ES3) H → L 66% | 247.58 | 0.0431 | 5.007 | |
| (ES1) H → L 69% | 383.06 | 0.2773 | 3.23 | |
| 2 | (ES2) H → L 67% | 330.41 | 0.0423 | 3.75 |
| (ES3) H → L 67% | 301.83 | 0.0195 | 4.10 | |
| (ES1) H → L 69% | 382.55 | 0.2750 | 3.24 | |
| 3 | (ES2) H → L 67% | 330.92 | 0.0424 | 3.74 |
| (ES3) H → L 67% | 301.29 | 0.0189 | 4.11 | |
| (ES1) H → L 69% | 384.48 | 0.2729 | 3.22 | |
| 4 | (ES2) H → L 66% | 330.32 | 0.0411 | 3.75 |
| (ES3) H → L 69% | 311.23 | 0.0051 | 3.98 | |
| (ES1) H → L 69% | 382.35 | 0.2691 | 3.24 | |
| 5 | (ES2) H → L 67% | 331.06 | 0.0424 | 3.74 |
| (ES3) H → L 67% | 301.11 | 0.0192 | 4.11 |
| Compounds | Bond/Groups | Vibrational Modes | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| – | 3680 | 3686 | 3690 | 3682 | NH2 (amine) | Scissoring |
| – | 3568 | 3570 | 3568 | 3572 | NH2 (amine) | Rocking |
| 3681 | – | – | – | – | NH2 (amine) | Antisymmetric Stretching |
| 3566 | – | – | – | – | NH2 (amine) | Antisymmetric Stretching |
| 3230 | 3240 | 3226 | 3165 | 3330 | C–H (phenyl) | Deformation |
| 3155 | 3165 | 3170 | 3195 | 3150 | C–H (phenyl) | Stretching |
| 3132 | – | – | – | – | C–H (methyl) | Deformation |
| 3074 | – | – | – | – | H–C–H (methyl) | Antisymmetric Stretching |
| 3020 | – | – | – | – | H–C–H (methyl) | Symmetric Stretching |
| 1590 | – | – | – | – | NH2 (amine) | Scissoring |
| 1590 | 1645 | 1679 | 1700 | 1620 | NH2 (amine) | Antisymmetric Stretching |
| 1510 | 1560 | 1635 | 1660 | 1500 | C–C (phenyl) | Stretching |
| 1565 | 1590 | 1625 | 1650 | 1550 | C–C (benzo) | Stretching |
| 1490 | – | – | – | – | H–C–H (methyl) | Deformation |
| 1320 | 1460 | 1430 | 1530 | 1380 | C–H (phenyl) | Deformation |
| – | – | – | 1382 | – | C-CF3 | Stretching |
| 1345 | 1390 | 1425 | 1490 | 1310 | C–H (benzo) | Stretching |
| – | 1310 | 1315 | 1325 | 1310 | NH2 (amine) | Symmetric Stretching |
| – | – | 1300 | 1320 | – | C–N (benzo) | Stretching |
| – | 1275 | 1265 | 1290 | 1260 | C–H (benzo) | Stretching |
| – | – | 1279 | – | – | C–OCH3 | Stretching |
| 1260 | – | – | – | – | C–H (benzo) | Deformation |
| – | 1100 | 1115 | 1120 | 1105 | NH2 (amine) | Symmetric Stretching |
| 1110 | – | – | – | – | NH2 (amine) | Deformation |
| 1150 | 1155 | 1155 | 1160 | 1150 | C–H (benzo) | Stretching |
| – | – | – | 1119 | – | C-F | Stretching |
| – | 1099 | – | – | – | C–Cl | Stretching |
| – | – | 1059 | – | – | O-CH3 | Stretching |
| 1020 | 1025 | 1035 | 1040 | 1015 | C–C (phenyl) | Stretching |
| 1055 | 1075 | 1075 | 1080 | 1060 | C–C (benzo) | Stretching |
| 8010 | 885 | 915 | 985 | 8025 | C–H (phenyl) | Deformation |
| Compounds | I | A | ƞ | σ | μ | ω |
|---|---|---|---|---|---|---|
| 1 | 5.62 | 0.91 | 2.35 | 0.42 | −3.27 | 2.27 |
| 2 | 5.77 | 1.16 | 2.30 | 0.43 | −3.47 | 2.61 |
| 3 | 5.46 | 0.81 | 2.32 | 0.43 | −3.14 | 2.12 |
| 4 | 5.98 | 1.53 | 2.22 | 0.45 | −3.76 | 3.17 |
| 5 | 5.70 | 0.97 | 2.36 | 0.42 | −3.34 | 2.35 |
| Compounds | Docking Score | ∆G Edvw | ∆G Coloumb | ∆G Energy | ∆G Internal | ∆G Model |
|---|---|---|---|---|---|---|
| 1 | −2.71 | −26.155 | −0.429 | −26.584 | 0.09 | −34.161 |
| 2 | −3.064 | −25.524 | −3.234 | −28.758 | 0.189 | −34.231 |
| 3 | −3.45 | −25.911 | −4.781 | −30.692 | 4.231 | −36.37 |
| 4 | −2.053 | −30.72 | −1.026 | −31.747 | 0.952 | −43.277 |
| 5 | −2.416 | −24.444 | −1.118 | −25.562 | 0.041 | −32.142 |
| Thiourea | −2.46 | −6.481 | −8.095 | −14.576 | 0 | −15.732 |
| Binding Energies (kcal/mol) of Staphylococcus aureus bacterium (PDB: 7EL1) | ||||||
| 1 | −3.345 | −17.9 | −4.783 | −22.683 | 0.03 | −27.757 |
| 2 | −3.26 | −19.708 | −4.279 | −23.986 | 0.028 | −29.282 |
| 3 | −3.309 | −20.484 | −4.307 | −24.791 | 0.082 | −30.202 |
| 4 | −2.58 | −19.34 | −3.73 | −23.069 | 0.227 | −26.847 |
| 5 | −2.844 | −17.946 | −2.751 | −20.697 | 0.123 | −24.357 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mubarik, A.; Mahmood, S.; Rasool, N.; Hashmi, M.A.; Ammar, M.; Mutahir, S.; Ali, K.G.; Bilal, M.; Akhtar, M.N.; Ashraf, G.A. Computational Study of Benzothiazole Derivatives for Conformational, Thermodynamic and Spectroscopic Features and Their Potential to Act as Antibacterials. Crystals 2022, 12, 912. https://doi.org/10.3390/cryst12070912
Mubarik A, Mahmood S, Rasool N, Hashmi MA, Ammar M, Mutahir S, Ali KG, Bilal M, Akhtar MN, Ashraf GA. Computational Study of Benzothiazole Derivatives for Conformational, Thermodynamic and Spectroscopic Features and Their Potential to Act as Antibacterials. Crystals. 2022; 12(7):912. https://doi.org/10.3390/cryst12070912
Chicago/Turabian StyleMubarik, Adeel, Sajid Mahmood, Nasir Rasool, Muhammad Ali Hashmi, Muhammad Ammar, Sadaf Mutahir, Kulsoom Ghulam Ali, Muhammad Bilal, Muhammad Nadeem Akhtar, and Ghulam Abbas Ashraf. 2022. "Computational Study of Benzothiazole Derivatives for Conformational, Thermodynamic and Spectroscopic Features and Their Potential to Act as Antibacterials" Crystals 12, no. 7: 912. https://doi.org/10.3390/cryst12070912
APA StyleMubarik, A., Mahmood, S., Rasool, N., Hashmi, M. A., Ammar, M., Mutahir, S., Ali, K. G., Bilal, M., Akhtar, M. N., & Ashraf, G. A. (2022). Computational Study of Benzothiazole Derivatives for Conformational, Thermodynamic and Spectroscopic Features and Their Potential to Act as Antibacterials. Crystals, 12(7), 912. https://doi.org/10.3390/cryst12070912