
Fast and Efficient Synthesis of Racemic Baloxavir Catalyzed by Strong Solid Acid under Microwave Conditions


Abstract
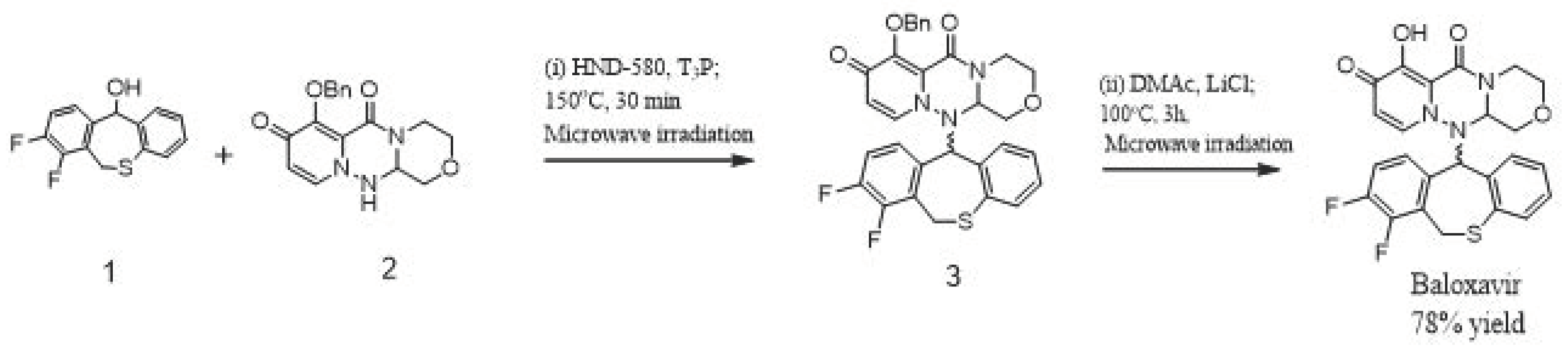
:1. Introduction
2. Materials and Methods
2.1. Instruments and Reagents
2.2. Experimental Method
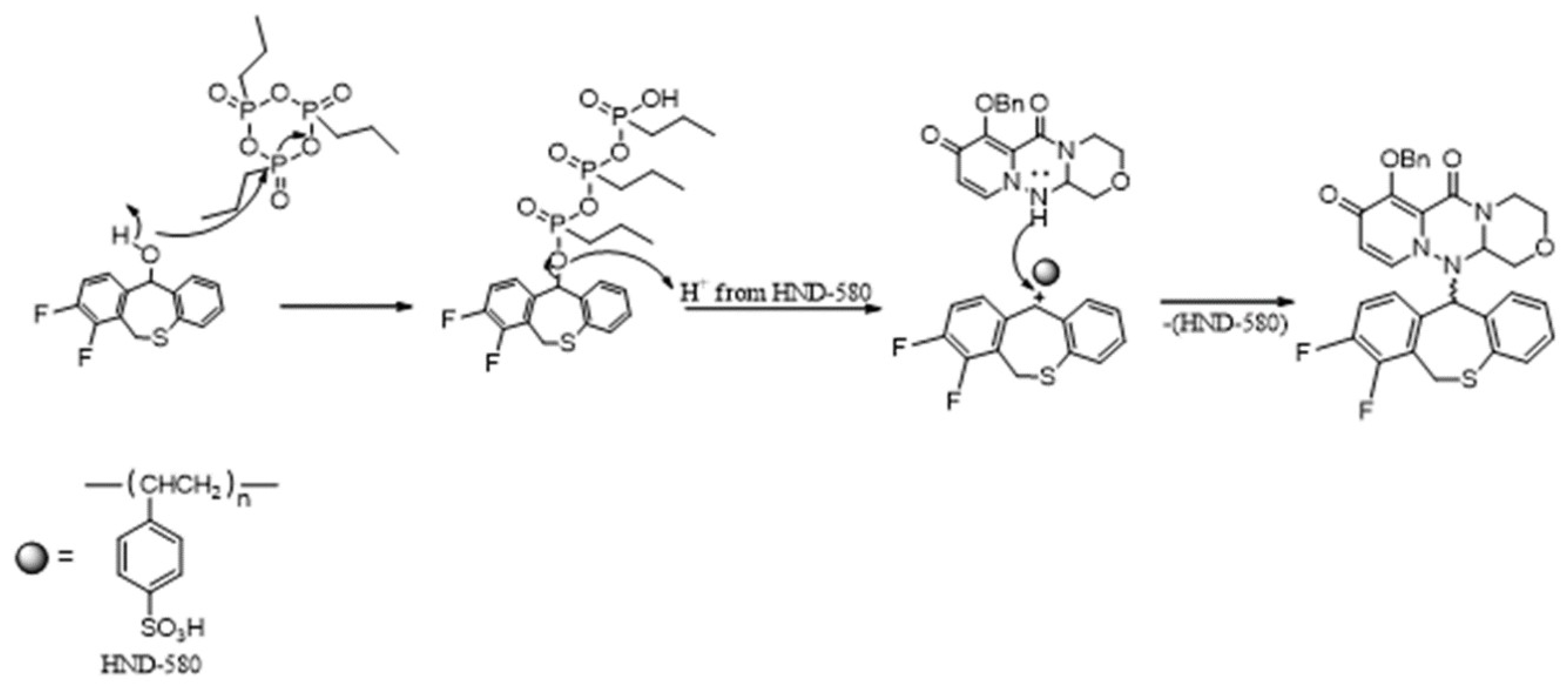
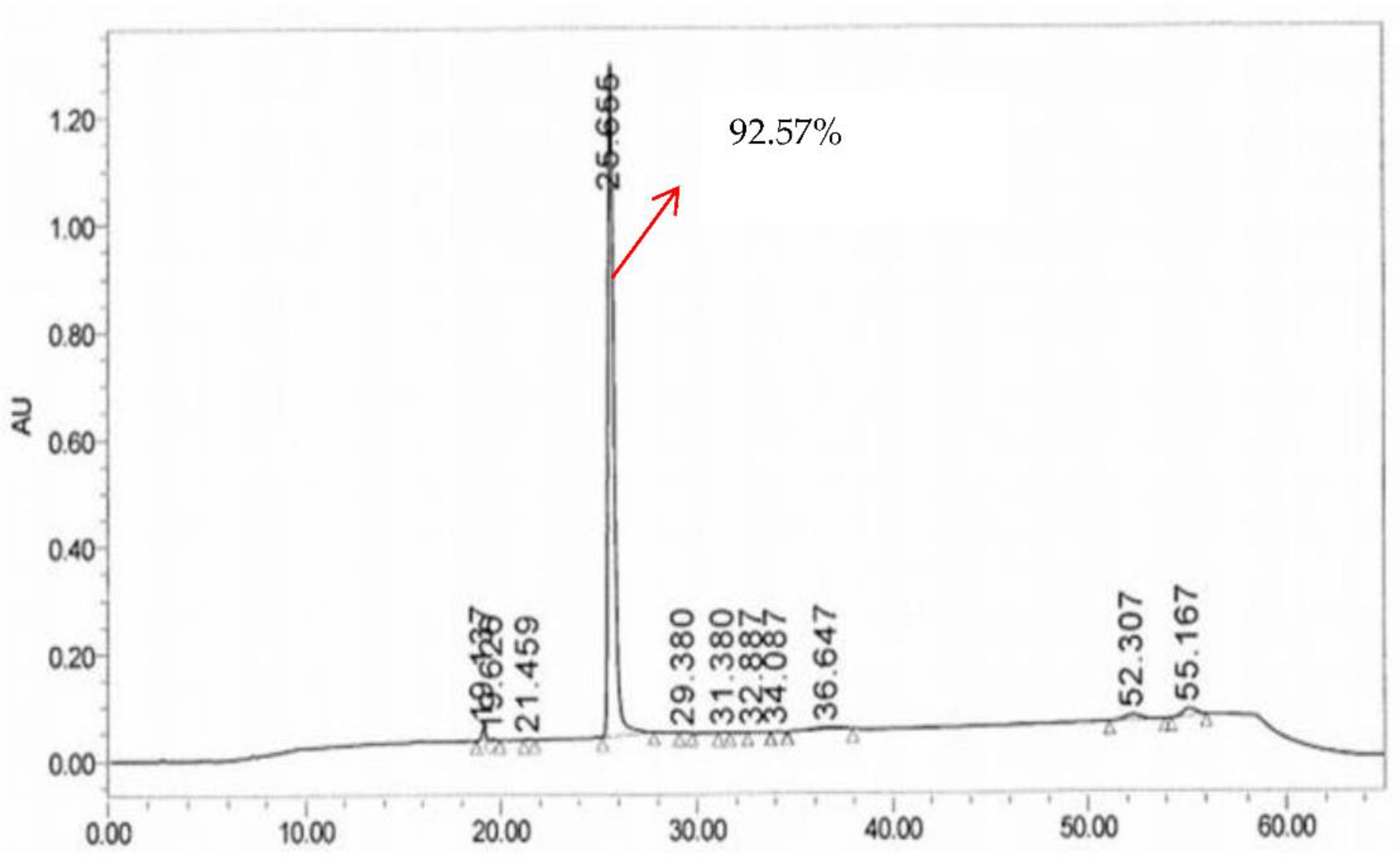
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Ivachtchenko, A.V.; Ivanenkov, Y.A.; Mitkin, O.D.; Yamanushkin, P.M.; Bichko, V.V.; Shevkun, N.A.; Karapetian, R.N.; Leneva, I.A.; Borisova, O.V.; Veselov, M.S. Novel oral anti-influenza drug candidate AV5080. J. Antimicrob. Chemother. 2014, 69, 1892–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Chen, H. Enhancement of influenza virus transmission by gene reassortment. In Influenza Pathogenesis and Control-Volume I.; Compans, R.W., Oldstone, M.B.A., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 185–204. [Google Scholar]
- Imai, M.; Kawaoka, Y. The new anti-influenza drug baloxavir marboxil: Can influenza viruses with reduced susceptibility to baloxavir maintain viral fitness? In Influenza: Advances in Diagnosis and Management; Fujita, J., Ed.; Springer: Singapore, 2021; pp. 211–219. [Google Scholar]
- Tao, J.J.; Lü, Q.Z.; Li, X.Y.; Dai, P.F.; Ye, Y.R.; Shen, Y. Research on new anti-influenza drug baloxavir marboxil. Chin. J. Clin. Med. 2019, 26, 308–311. [Google Scholar]
- Ikematsu, H.; Hayden, F.G.; Kawaguchi, K.; Kinoshita, M.; de Jong, M.D.; Lee, N.; Takashima, S.; Noshi, T.; Tsuchiya, K.; Uehara, T. Baloxavir Marboxil for Prophylaxis against Influenza in Household Contacts. N. Engl. J. Med. 2020, 383, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Noshi, T.; Kitano, M.; Taniguchi, K.; Yamamoto, A.; Omoto, S.; Baba, K.; Hashimoto, T.; Ishida, K.; Kushima, Y.; Hattori, K.; et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase PA subunit. Antivir. Res. 2018, 160, 109–117. [Google Scholar] [CrossRef]
- Heo, Y.-A. Baloxavir: First Global Approval. Drugs 2018, 78, 693–697. [Google Scholar] [CrossRef]
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; de Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. Baloxavir Marboxil for Uncomplicated Influenza in Adults and Adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef]
- Omoto, S.; Speranzini, V.; Hashimoto, T.; Noshi, T.; Yamaguchi, H.; Kawai, M.; Kawaguchi, K.; Uehara, T.; Shishido, T.; Naito, A.; et al. characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci. Rep. 2018, 8, 9633. [Google Scholar] [CrossRef]
- Herlocher, M.L.; Truscon, R.; Elias, S.; Yen, H.-L.; Roberts, N.A.; Ohmit, S.E.; Monto, A.S. Influenza viruses resistant to the antiviral drug oseltamivir: Transmission studies in ferrets. J. Infect. Dis. 2004, 190, 1627–1630. [Google Scholar] [CrossRef] [Green Version]
- Fukao, K.; Ando, Y.; Noshi, T.; Kawai, M.; Yoshida, R.; Shishido, T.; Naito, A. Delayed dosing of S-033188, a novel inhibitor of influenza virus cap-dependent endonuclease, exhibited significant reduction of viral titer and mortality in mice infected with influenza a virus. Open Forum Infect. Dis. 2017, 4, S473. [Google Scholar] [CrossRef] [Green Version]
- Portsmouth, S.; Kawaguchi, K.; Arai, M.; Tsuchiya, K.; Uehara, T. Cap-dependent endonuclease inhibitor S-033188 for the treatment of influenza: Results from a phase 3, randomized, double-blind, placebo- and active-controlled study in otherwise healthy adolescents and adults with seasonal influenza. Open Forum Infect. Dis. 2017, 4, S734. [Google Scholar] [CrossRef] [Green Version]
- Kawai, M. Pharmaceutical Compositions Containing Substituted Polycyclic Pyridone Derivatives and Prodrug Thereof. U.S. Patent 10,759,814, 1 September 2020. [Google Scholar]
- Okamoto, K.; Ueno, T.; Hato, Y.; Hakogi, T.; Majima, S.; Yamao, N.; Ochiai, K. Method for Stereoselectively Producing Substituted Polycyclic Pyridone Derivative. International Patent Application No. WO2019/070059, 10 April 2019. [Google Scholar]
- Hughes, D.L. Review of the patent literature: Synthesis and final forms of antiviral drugs tecovirimat and baloxavir marboxil. Org. Process Res. Dev. 2019, 23, 1298–1307. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, Z.; Kou, J.; Wu, S.; Zhang, J.; Yuan, X.; Wu, X.; Li, C.-h.; Liao, G. Development of a Quality Controllable and Scalable Process for the Preparation of 7,8-Difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-ol: A Key Intermediate for baloxavir marboxil. Org. Process Res. Dev. 2019, 23, 2716–2723. [Google Scholar] [CrossRef]
- Kawaguchi, N.; Koshimichi, H.; Ishibashi, T.; Wajima, T. Evaluation of drug-drug interaction potential between baloxavir marboxil and oseltamivir in healthy subjects. Clin. Drug Investig. 2018, 38, 1053–1060. [Google Scholar] [CrossRef] [Green Version]
- Koshimichi, H.; Ishibashi, T.; Kawaguchi, N.; Sato, C.; Kawasaki, A.; Wajima, T. Safety, tolerability, and pharmacokinetics of the novel anti-influenza agent baloxavir marboxil in healthy adults: Phase I study findings. Clin. Drug Investig. 2018, 38, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Ison, M.G.; Portsmouth, S.; Yoshida, Y.; Shishido, T.; Hayden, F.; Uehara, T. LB16. Phase 3 Trial of Baloxavir Marboxil in High-Risk Influenza Patients (CAPSTONE-2 Study). Open Forum Infect. Dis. 2018, 5, S764–S765. [Google Scholar] [CrossRef]
- Shibahara, S.; Fukui, N.; Maki, T.; Anan, K. Method for Producing Substituted Polycyclic Pyridone Derivative and Crystal. WO2017221869A1, 28 December 2017. [Google Scholar]
- Zheng, X.; Zhang, Y.; Fu, C.; Wu, Y. A synthetic method of novel Tamiflu. CN Patent 109504721A, 22 March 2019. [Google Scholar]
- Tang, C.; Ren, Q.; Luo, H. Influenza Virus Replication Inhibitor and Use Thereof. WO 2019/052565A1,, 21 March 2019. [Google Scholar]
- Tang, L.; Yan, H.; Wu, W.; Chen, D.; Gao, Z.; Hou, J.; Zhang, C.; Jiang, Y. Synthesis and Anti-Influenza Virus Effects of Novel Substituted Polycyclic Pyridone Derivatives Modified from Baloxavir. J. Med. Chem. 2021, 64, 14465–14476. [Google Scholar] [CrossRef]
- Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. The use of microwave ovens for rapid organic synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- Misono, M.; Okuhara., T. Solid superacid catalysts. CHEMTECH 1993, 23. Available online: https://www.osti.gov/biblio/5465998 (accessed on 1 November 1993).
- Hino, M.; Arata, K. Synthesis of solid superacid catalyst with an acid strength of H0⩽–16.04. J. Chem. Soc. Chem. Commun. 1980, 18, 851–852. [Google Scholar] [CrossRef]
- Yamaguchi, T. Recent progress in solid superacid. Appl. Catal. 1990, 61, 1–25. [Google Scholar] [CrossRef]
- Qureshi, Z.S.; Deshmukh, K.M.; Tambade, P.J.; Dhake, K.P.; Bhanage, B.M. Amberlyst-15 in Ionic Liquid: An Efficient and Recyclable Reagent for Nucleophilic Substitution of Alcohols and Hydroamination of Alkenes. Eur. J. Org. Chem. 2010, 2010, 6233–6238. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Catalyst | Catalyst Amount a | Reaction Temperature (°C) b | Temperature Rise Time (min) | Response Time (min) | (±) Baloxavir Yield c,d |
|---|---|---|---|---|---|---|
| 1 | Methanesulfonic acid | 5% | 150 | 50 | 30 | 40% |
| 2 | p-toluenesulfonic acid | 5% | 150 | 50 | 30 | 45% |
| 3 | Amberlyst15 | 5% | 150 | 50 | 30 | 22% |
| 4 | Amberlyst16 | 5% | 150 | 50 | 30 | 28% |
| 5 | Amberlyst31 | 5% | 150 | 50 | 30 | 29% |
| 6 | Amberlyst33 | 5% | 150 | 50 | 30 | 33% |
| 7 | Amberlyst35 | 5% | 150 | 50 | 30 | 72% |
| 8 | Amberlyst36 | 5% | 150 | 50 | 30 | 67% |
| 9 | Amberlyst39 | 5% | 150 | 50 | 30 | 34% |
| 10 | Amberlyst40 | 5% | 150 | 50 | 30 | 53% |
| 11 | Amberlyst70 | 5% | 150 | 50 | 30 | 24% |
| 12 | Amberlyst131 | 5% | 150 | 50 | 30 | 26% |
| 13 | HND-8 | 5% | 150 | 50 | 30 | 20% |
| 14 | HND-580 | 5% | 150 | 50 | 30 | 75% |
| 15 | HND-582 | 5% | 150 | 50 | 30 | 41% |
| 16 | HND-583 | 5% | 150 | 50 | 30 | 46% |
| 17 | HND-586 | 5% | 150 | 50 | 30 | 65% |
| 18 | HND-587 | 5% | 150 | 50 | 30 | 55% |
| No. | Catalyst | Catalyst Amount a | Reaction Temperature (°C) b | Temperature Rise Time (min) | Response Time (min) | (±) Baloxavir Yield c |
|---|---|---|---|---|---|---|
| 1 | HND-580 | 1% | 150 | 50 | 30 | 18% |
| 2 | HND-580 | 2% | 150 | 50 | 30 | 31% |
| 3 | HND-580 | 3% | 150 | 50 | 30 | 57% |
| 4 | HND-580 | 5% | 150 | 50 | 30 | 78% |
| 5 | HND-580 | 5% | 140 | 50 | 30 | 61% |
| 6 | HND-580 | 5% | 160 | 50 | 30 | 66% |
| 7 | HND-580 | 5% | 150 | 10 | 40 | 65% |
| 8 | HND-580 | 5% | 150 | 20 | 30 | 72% |
| 9 | HND-580 | 5% | 150 | 30 | 20 | 71% |
| 10 | HND-580 | 5% | 150 | 40 | 10 | 56% |
| 11 | HND-580 | 5% | 150 | 50 | 0 | 58% |
| 12 | HND-580 | 5% | 150 | 60 | 0 | 59% |
| 13 | HND-580 | 5% | 150 | 50 | 20 | 70% |
| NO. | Compound 1 | Compound 2 | Dosage of 1-Propylphosphoric Anhydride | (±) Baloxavir Yield a |
|---|---|---|---|---|
| 1 | 1 mmol | 1 mmol | 2 mmol | 73% |
| 2 | 1 mmol | 1 mmol | 2 mmol + 2 mL ethyl acetate | 78% |
| 3 | 1 mmol | 1 mmol | 55 mmol | 75% |
| NO. | Reaction Temperature (°C) b | Reaction Time (h) b | (±) Baloxavir Yield c |
|---|---|---|---|
| 1 | 80 | 3 | 10% |
| 2 | 80 | 4 | 25% |
| 3 | 100 | 1.5 | 40% |
| 4 | 100 | 3 | 78% |
| 5 | 100 | 4 | 79% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Lv, X.; Meng, Z.; Xu, Z.; Zheng, Z.; Li, J.; Xue, M. Fast and Efficient Synthesis of Racemic Baloxavir Catalyzed by Strong Solid Acid under Microwave Conditions. Crystals 2022, 12, 891. https://doi.org/10.3390/cryst12070891
Wang Y, Lv X, Meng Z, Xu Z, Zheng Z, Li J, Xue M. Fast and Efficient Synthesis of Racemic Baloxavir Catalyzed by Strong Solid Acid under Microwave Conditions. Crystals. 2022; 12(7):891. https://doi.org/10.3390/cryst12070891
Chicago/Turabian StyleWang, Yiyun, Xiaofang Lv, Zihui Meng, Zhibin Xu, Zhonghui Zheng, Jiarong Li, and Min Xue. 2022. "Fast and Efficient Synthesis of Racemic Baloxavir Catalyzed by Strong Solid Acid under Microwave Conditions" Crystals 12, no. 7: 891. https://doi.org/10.3390/cryst12070891
APA StyleWang, Y., Lv, X., Meng, Z., Xu, Z., Zheng, Z., Li, J., & Xue, M. (2022). Fast and Efficient Synthesis of Racemic Baloxavir Catalyzed by Strong Solid Acid under Microwave Conditions. Crystals, 12(7), 891. https://doi.org/10.3390/cryst12070891