
Four-Coordinate Monoboron Complexes with 8-Hydroxyquinolin-5-Sulfonate: Synthesis, Crystal Structures, Theoretical Studies, and Luminescence Properties

 ,
,  , ,
, ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
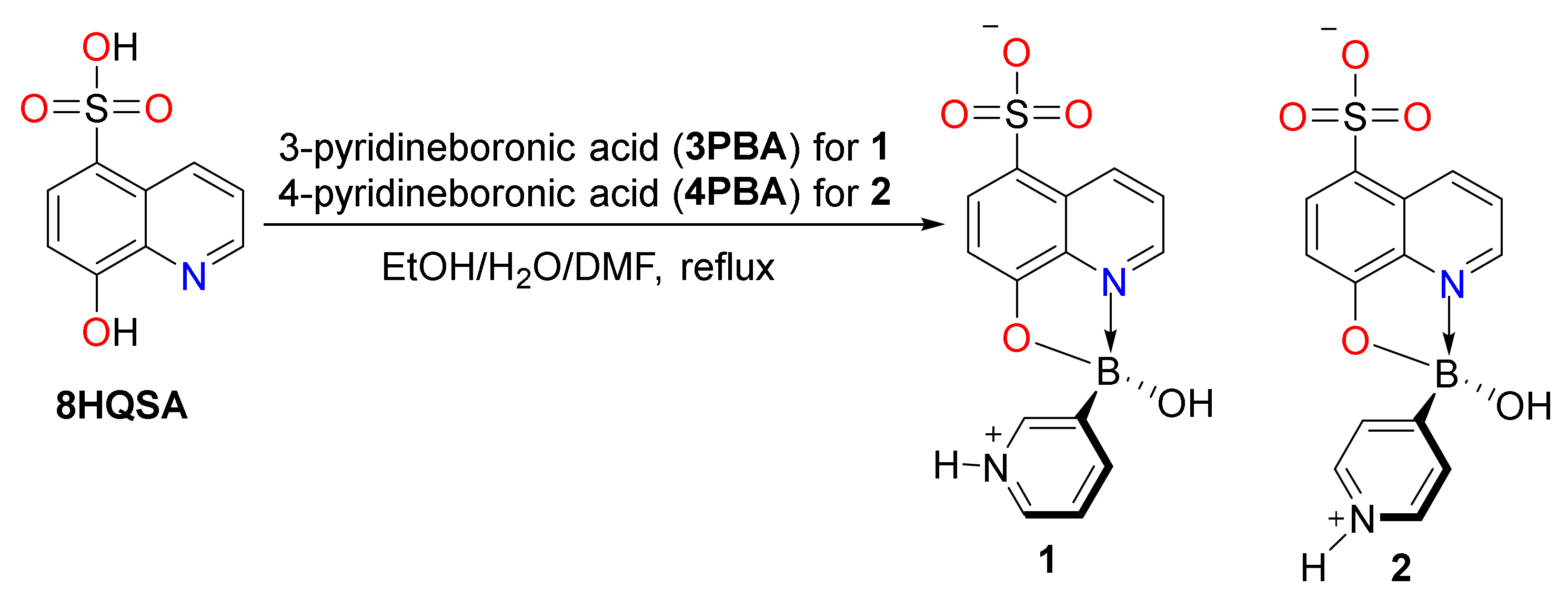
2.2. Preparation of Boronic Ester Complex 1
2.3. Preparation of Boronic Ester Complex 2
2.4. Crystallography
2.5. Computational Details
3. Results and Discussion
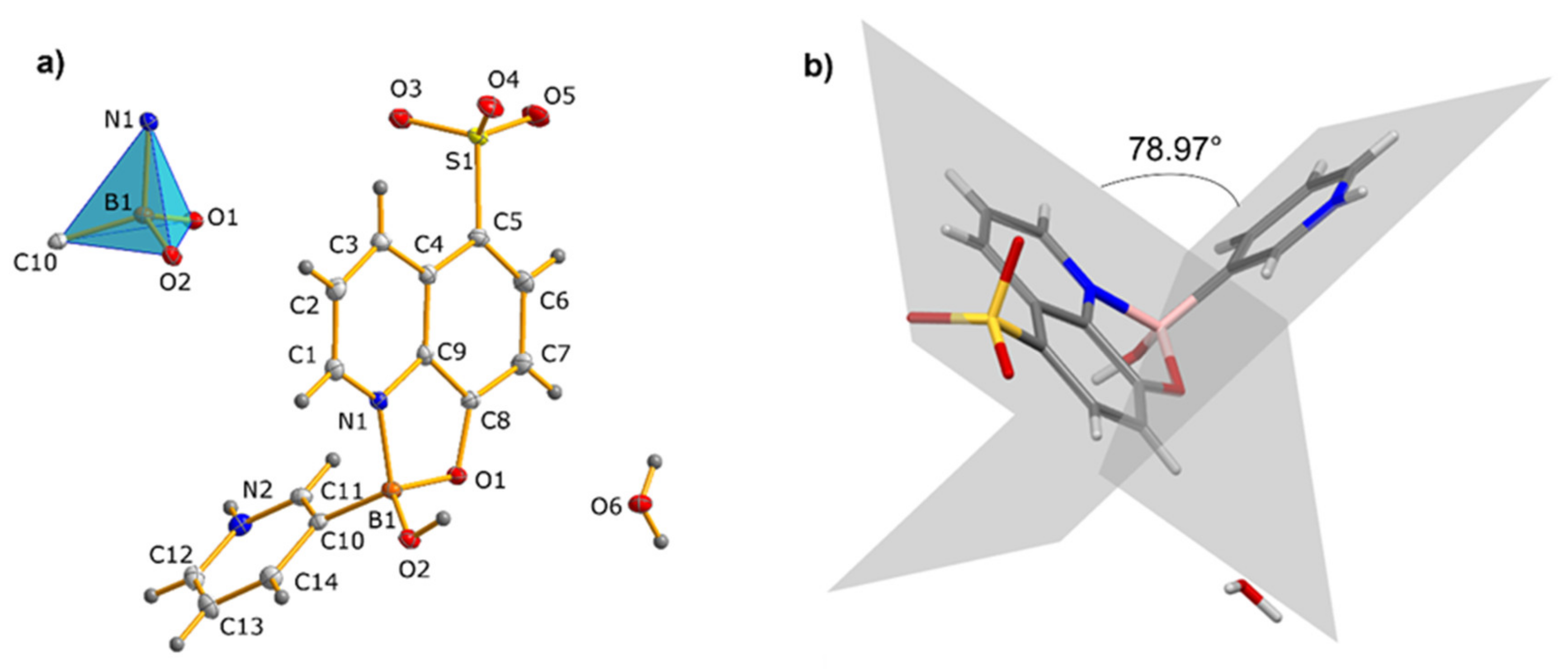
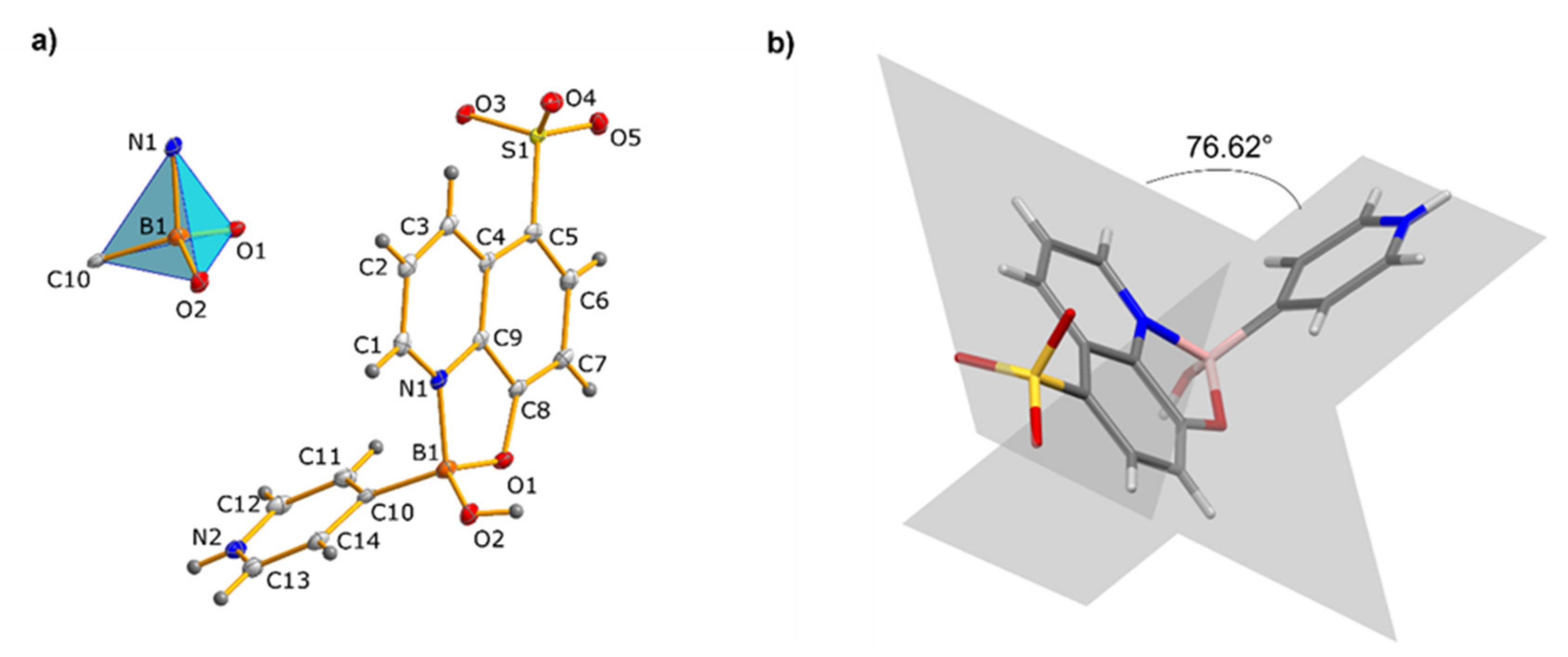
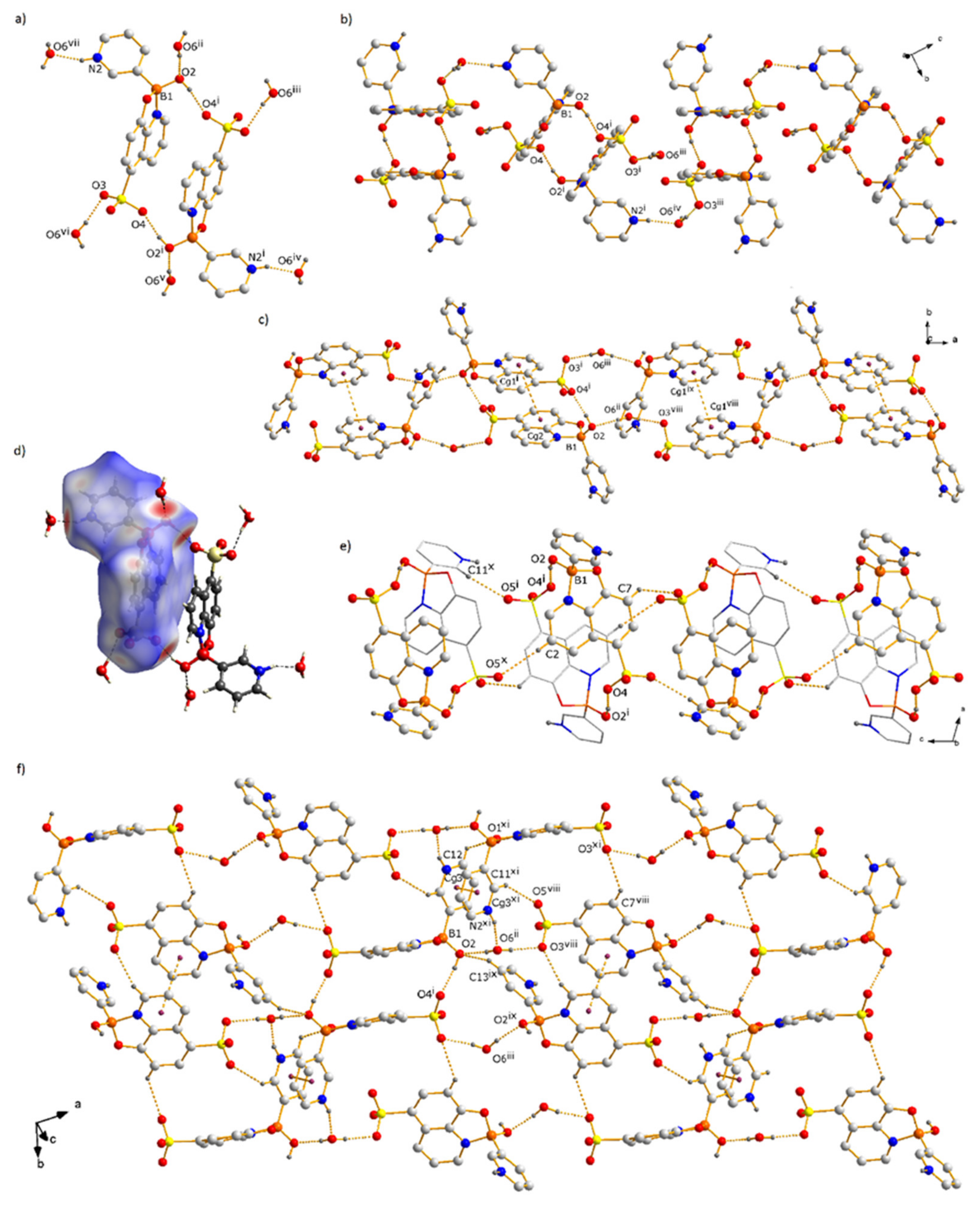
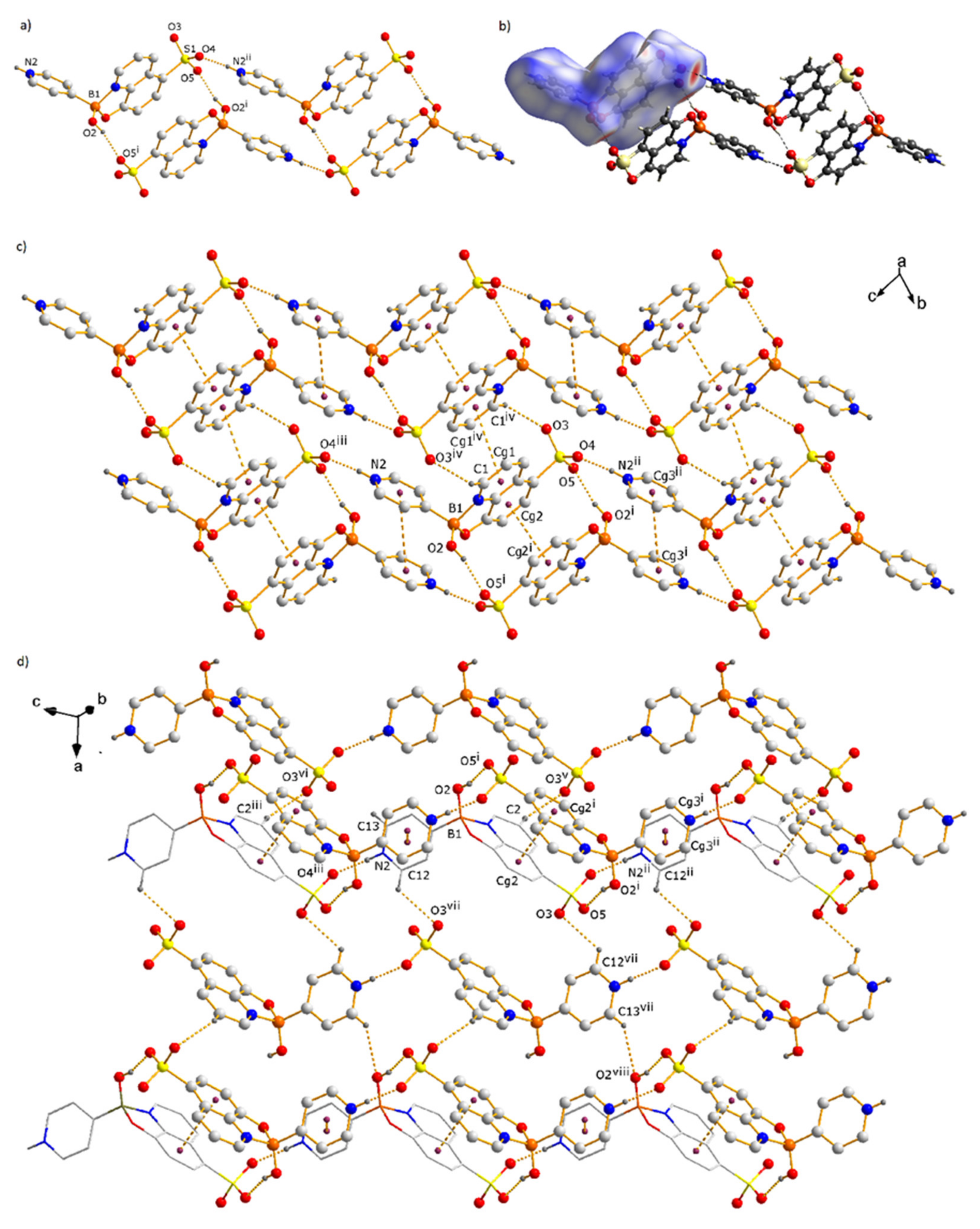
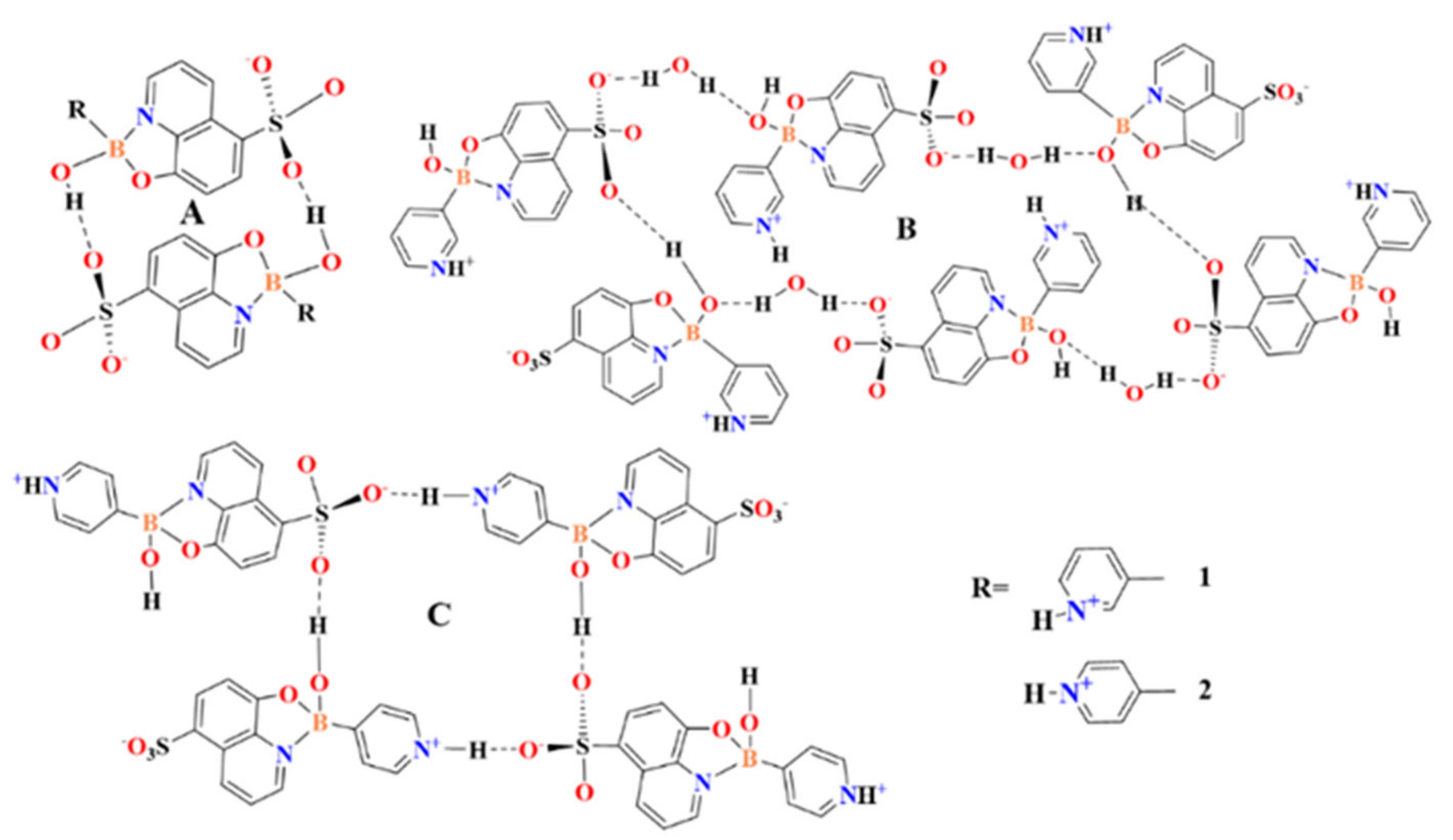
3.1. Molecular and Supramolecular Structures of 1 and 2
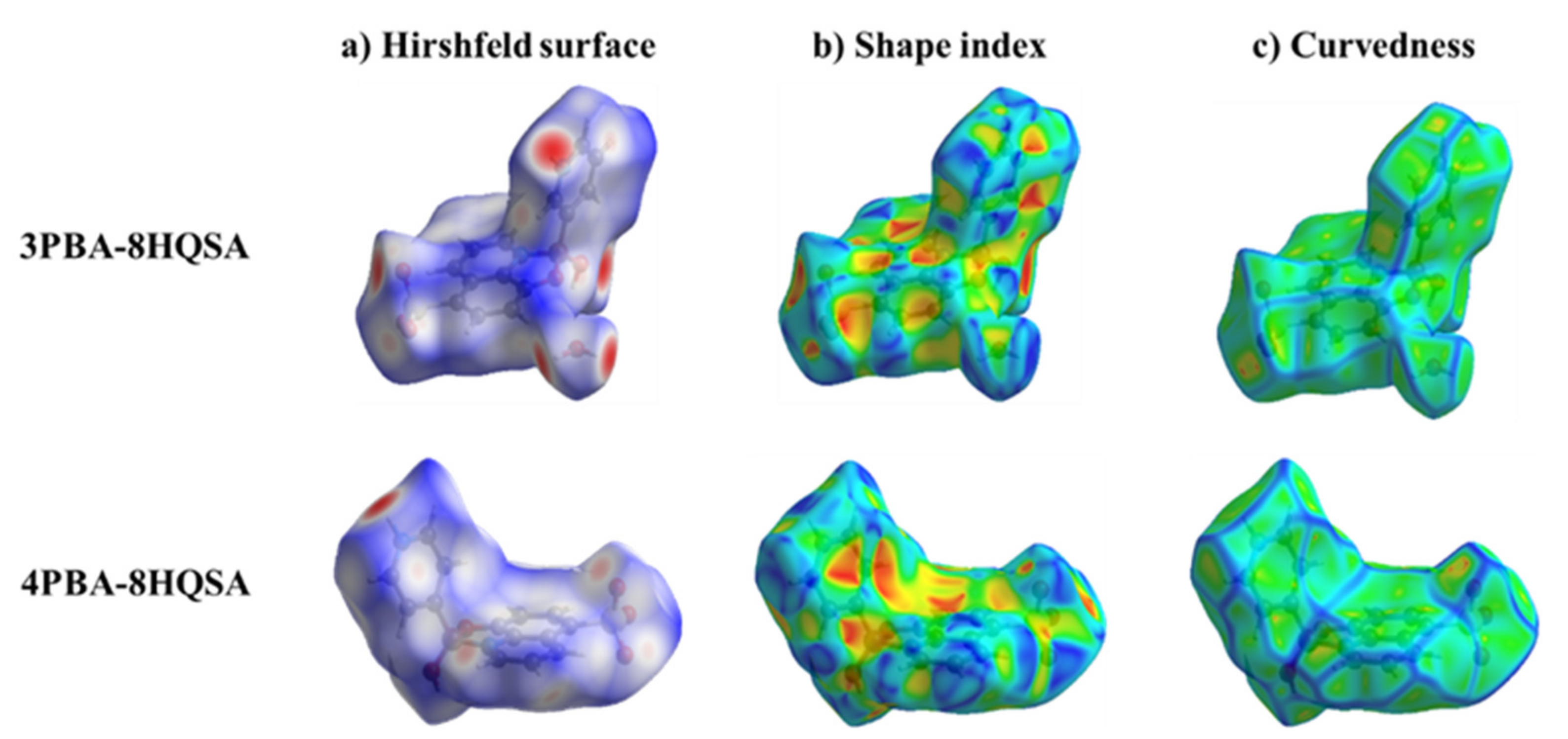
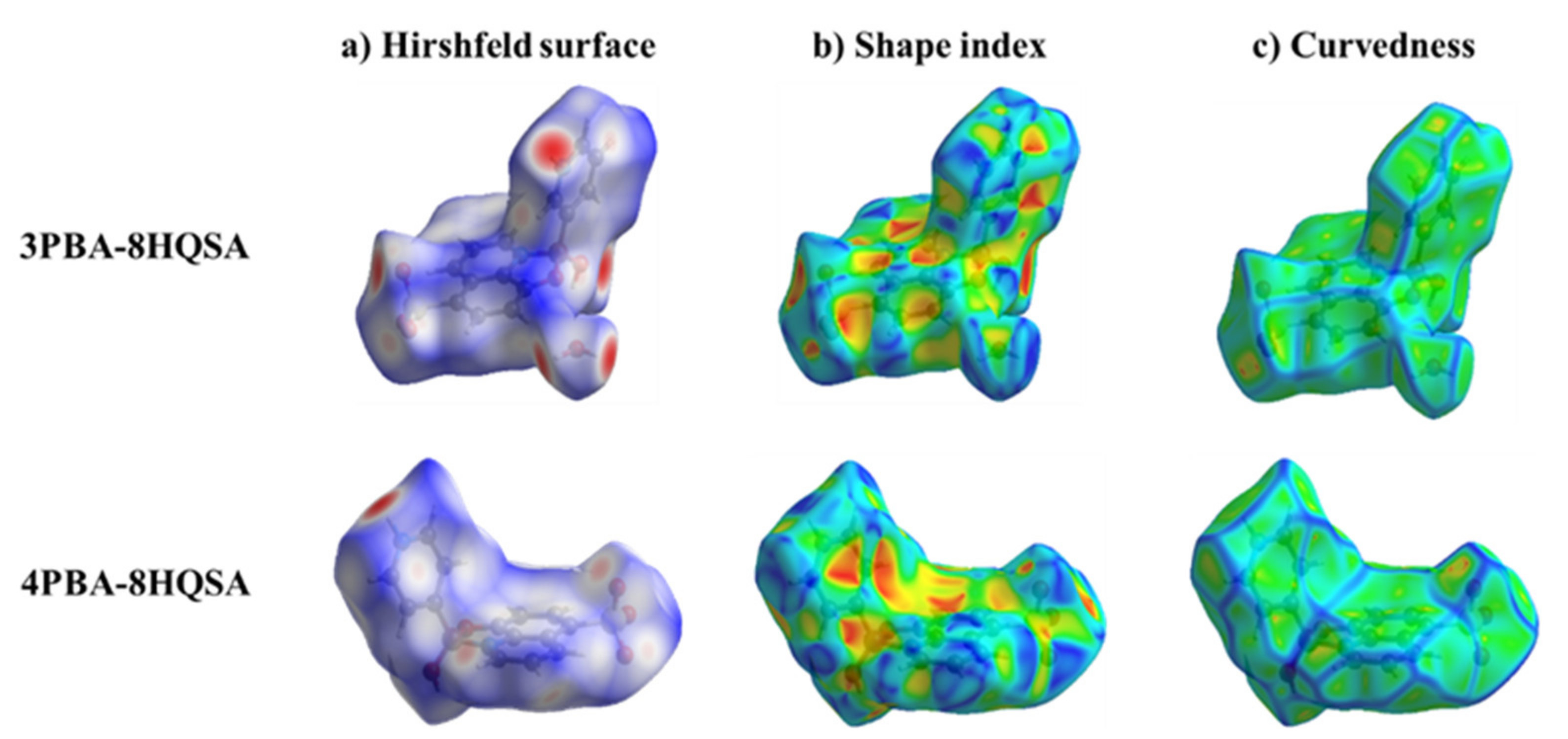
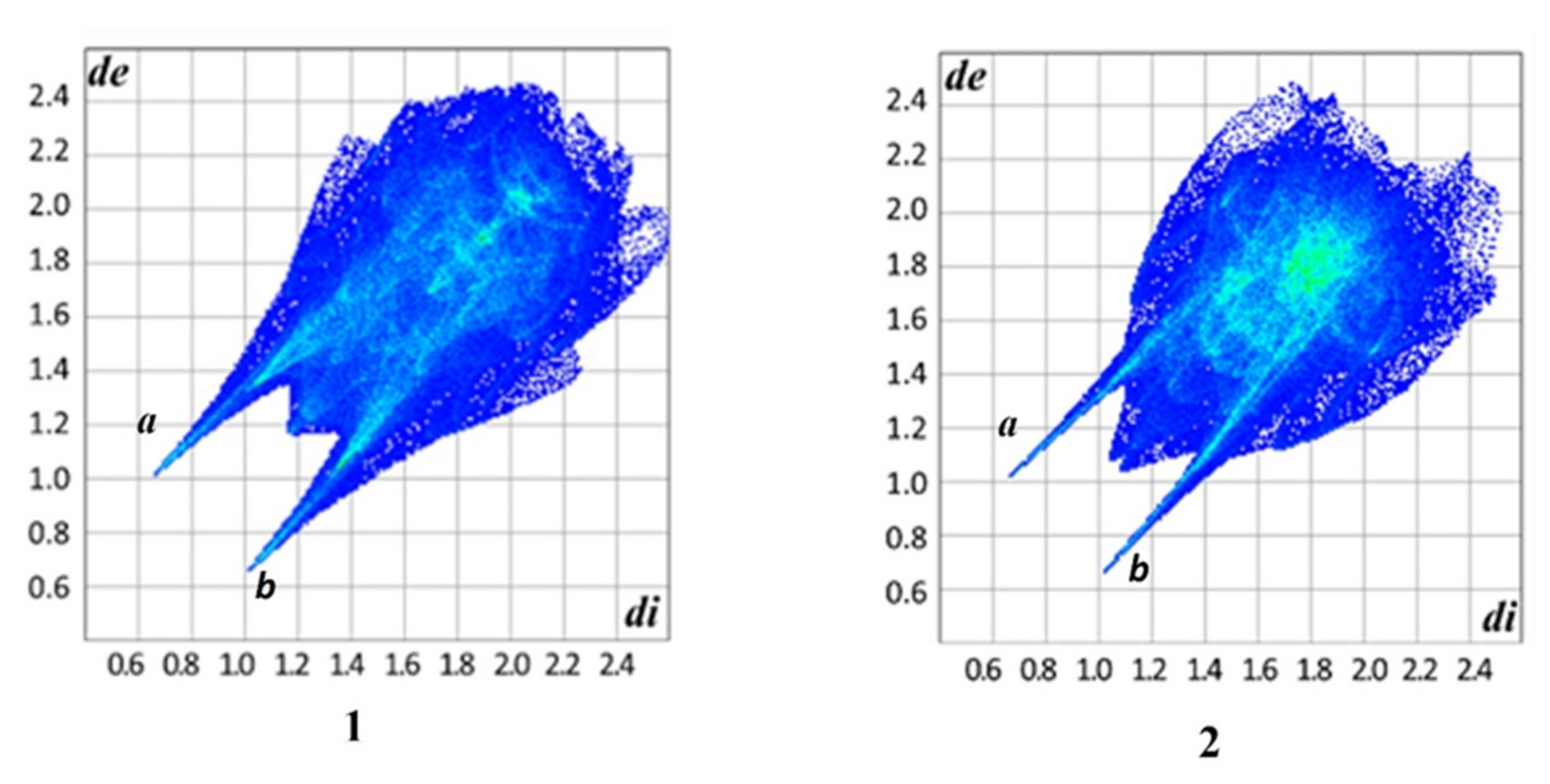
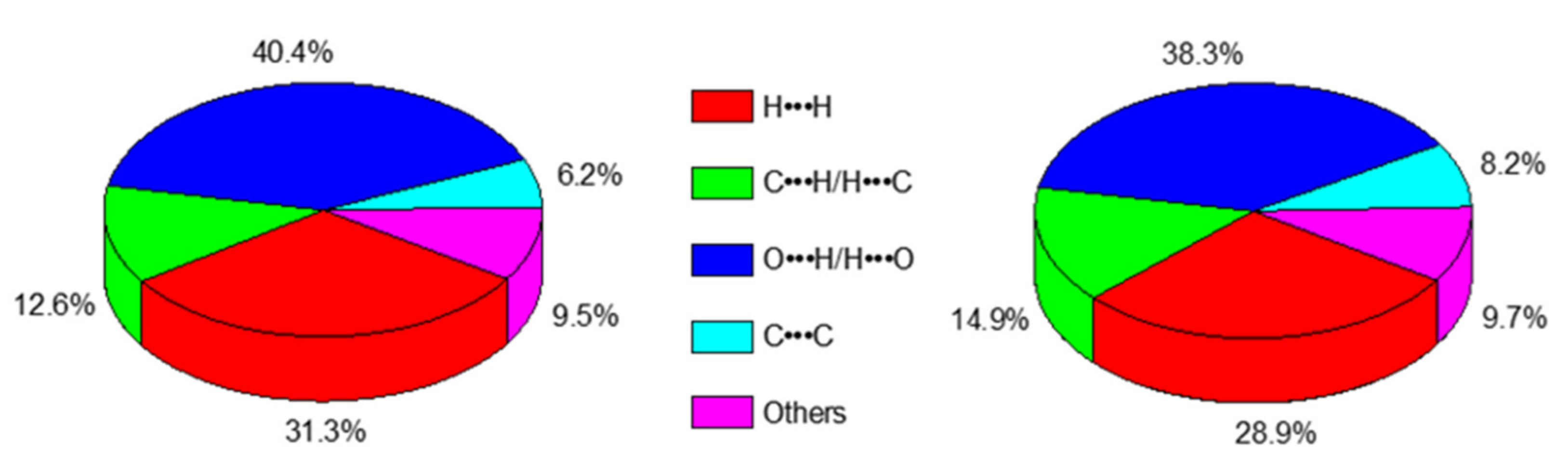
3.2. Analysis of the Hirshfeld Surface
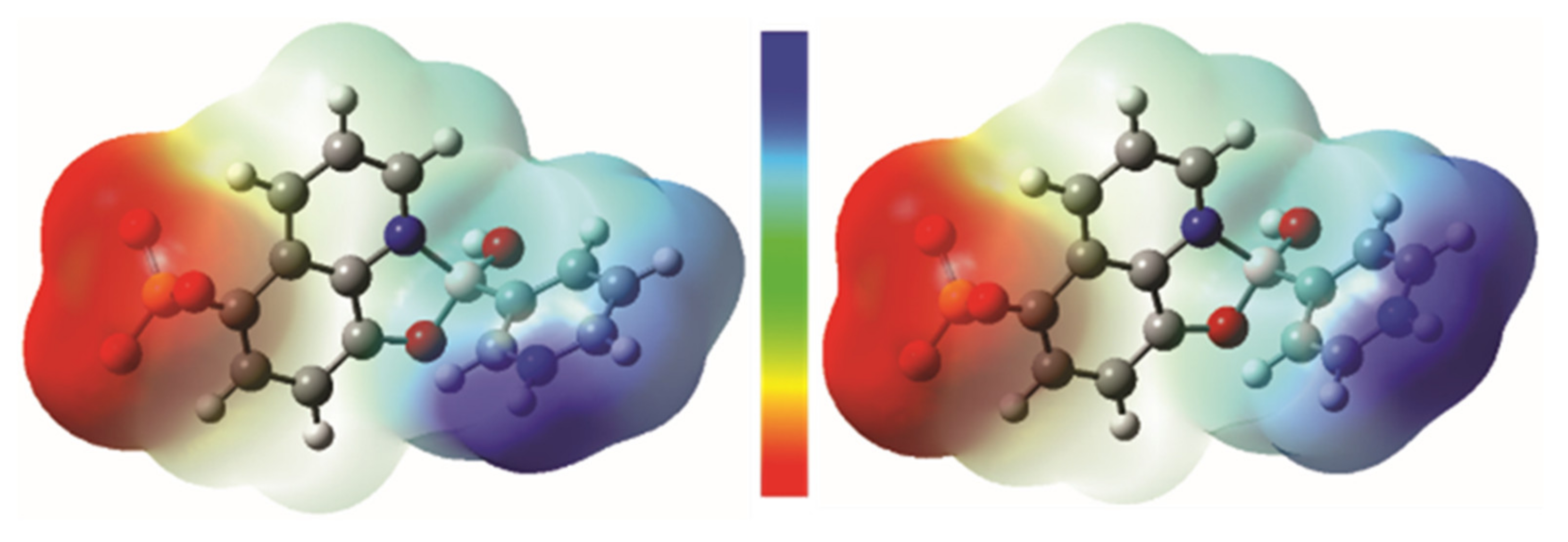
3.3. Analysis of the Molecular Electrostatic Potential Maps
3.4. IR and NMR Spectroscopic Analysis
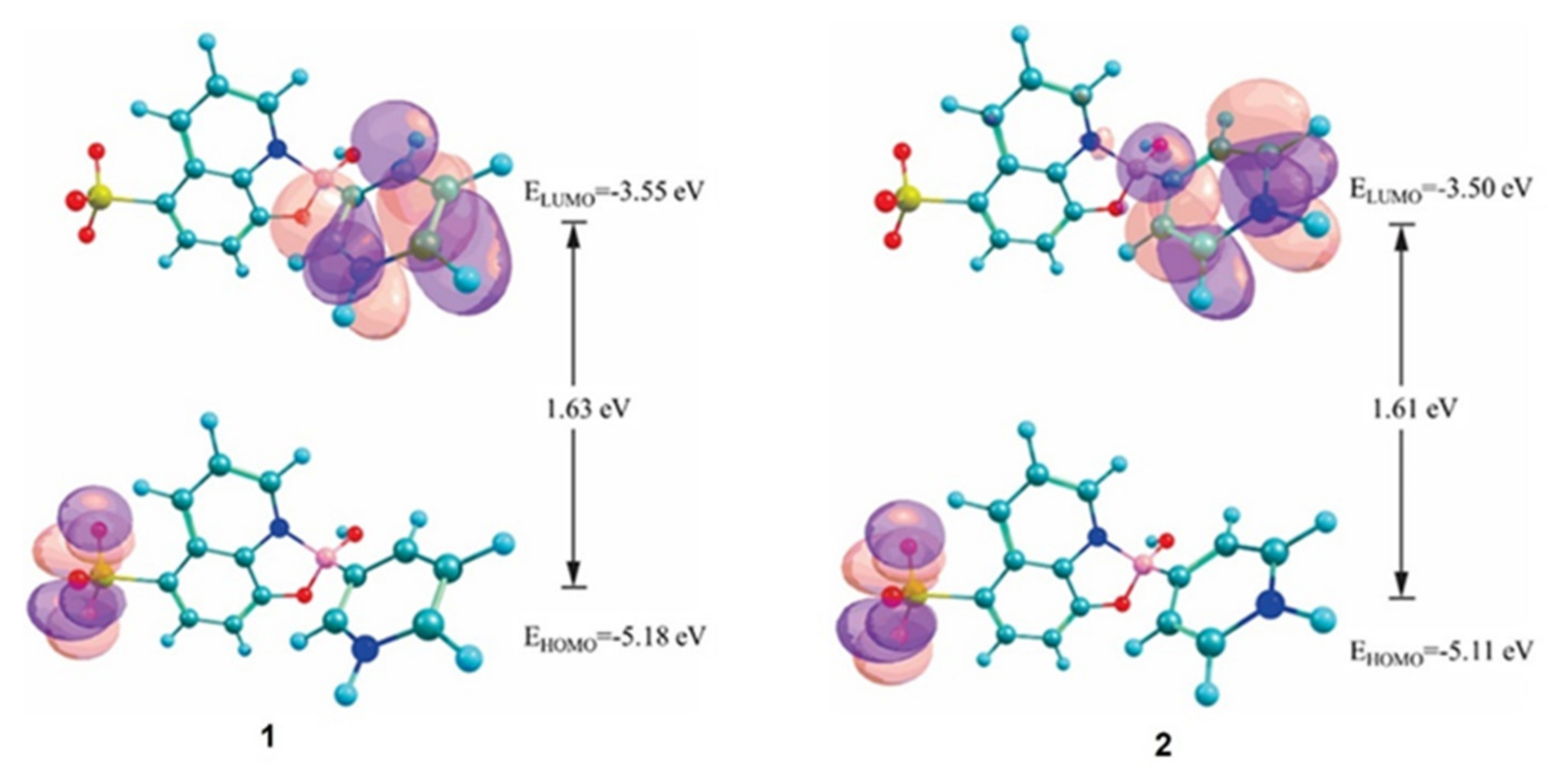
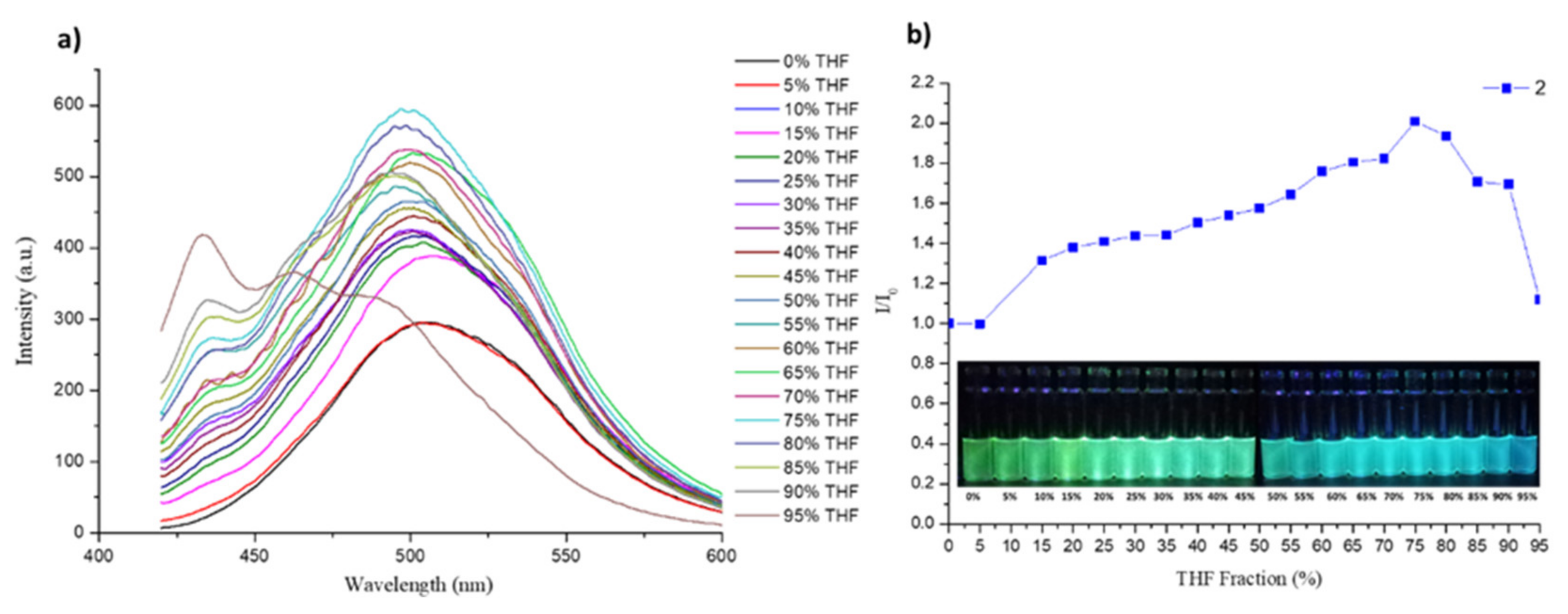
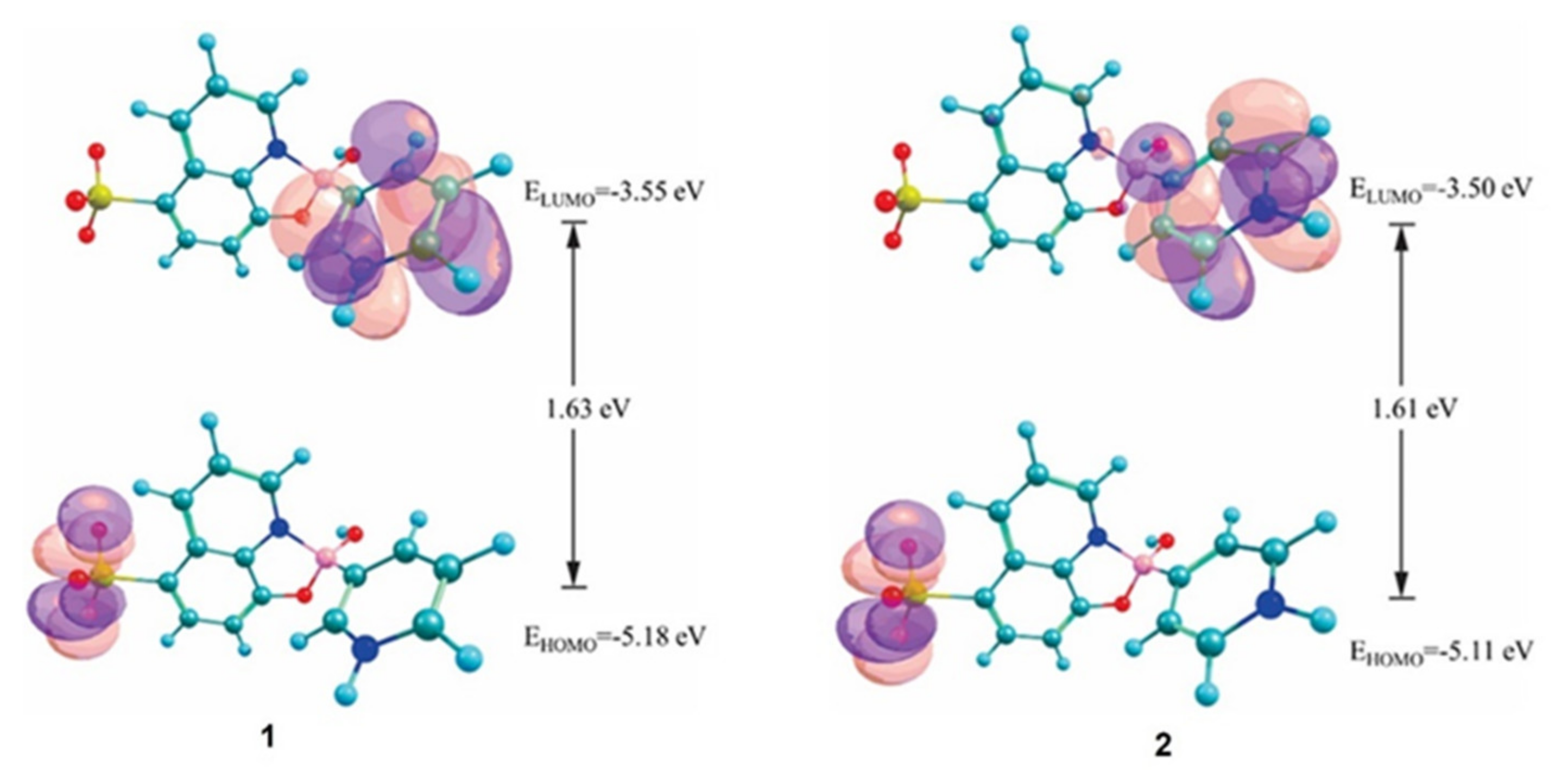
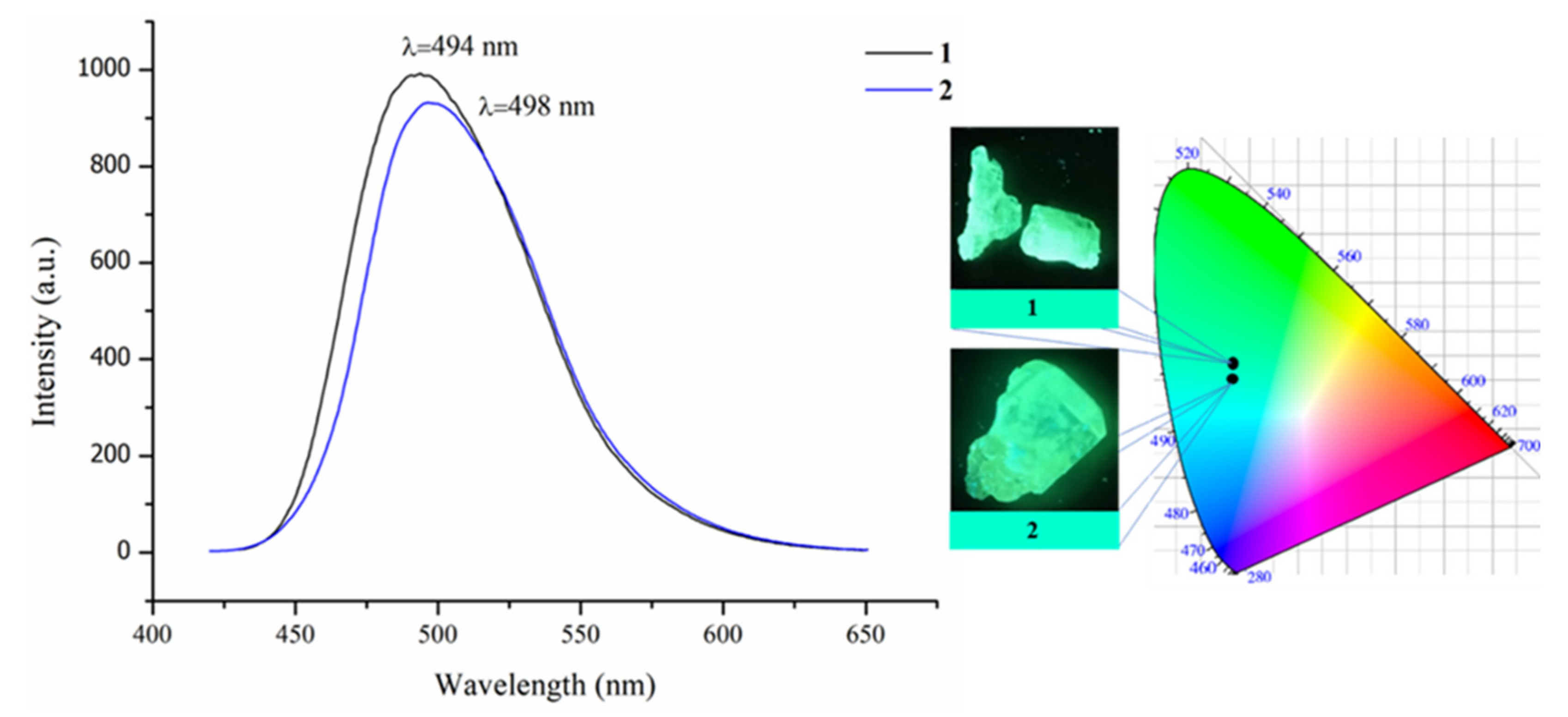
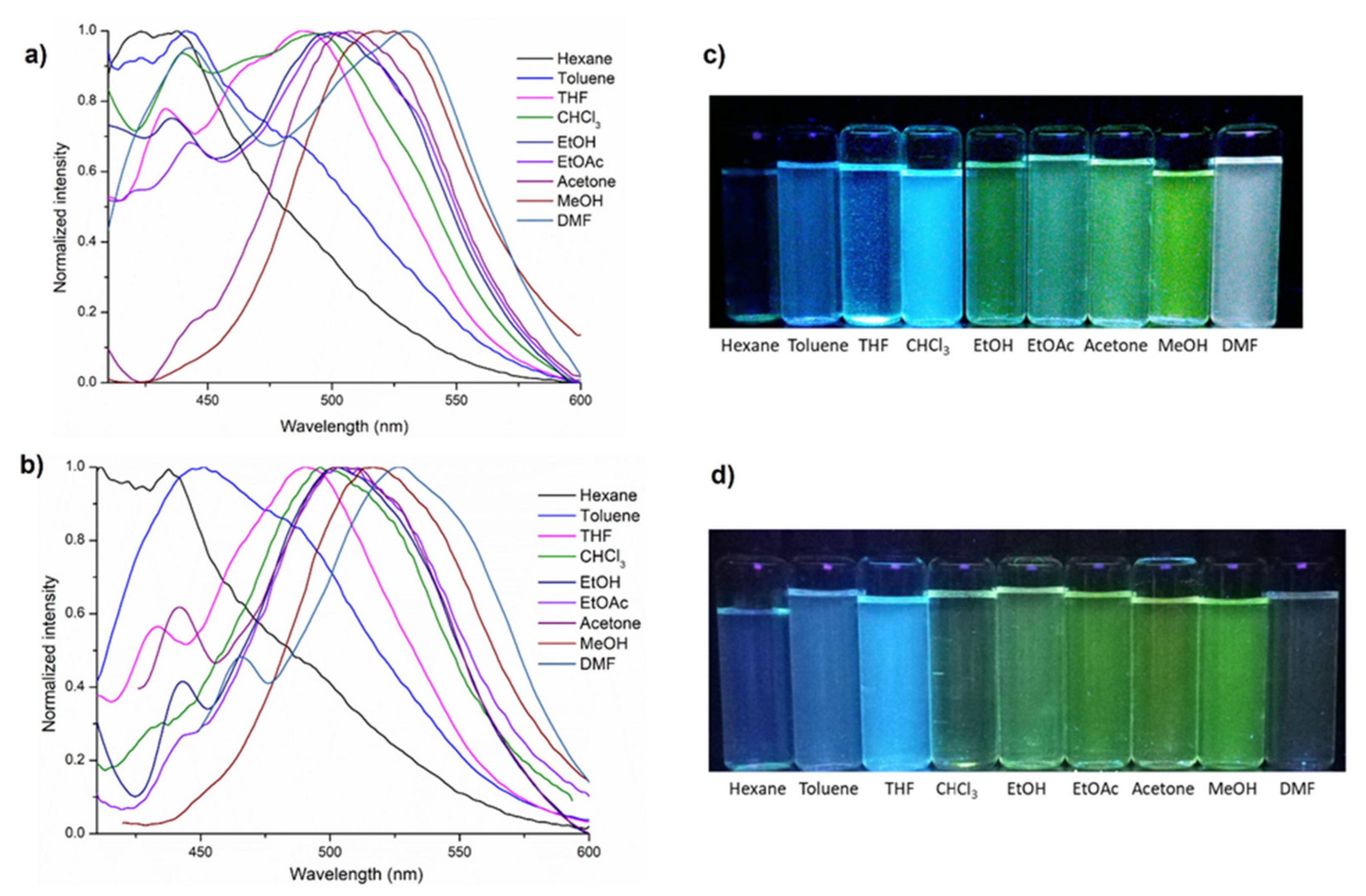
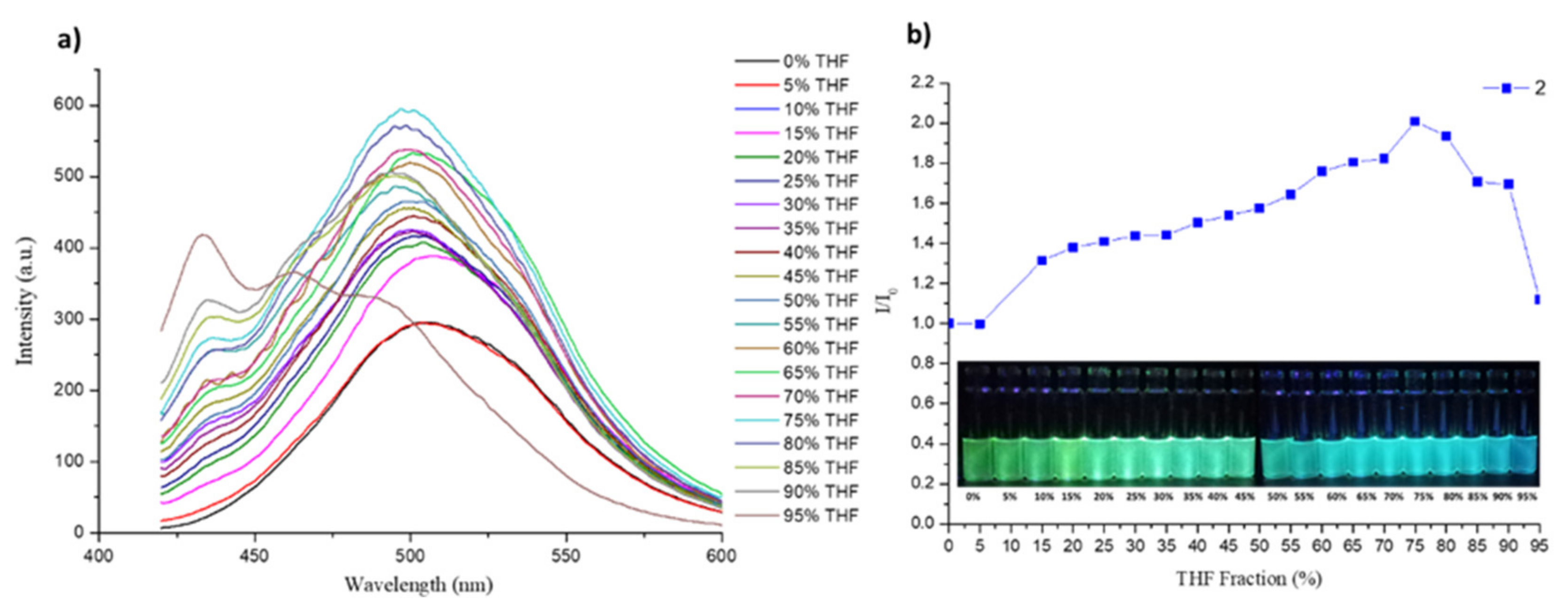
3.5. DFT Calculations and Evaluation of the Photophysical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Z.; He, W.; Guo, Z. Metal coordination in photoluminescent sensing. Chem. Soc. Rev. 2013, 42, 1568–1600. [Google Scholar] [CrossRef] [PubMed]
- Elgemeie, G.H.; Mohamed, R.A. Microwave synthesis of fluorescent and luminescent dyes (1990–2017). J. Mol. Struct. 2018, 1173, 707–742. [Google Scholar] [CrossRef]
- Wong, W.-Y.; Ho, C.-L. Functional metallophosphors for effective charge carrier injection/transport: New robust OLED materials with emerging applications. J. Mater. Chem. 2009, 19, 4457–4482. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, Y.Y.; Liu, X.; Lin, H.; Gao, K.; Lai, W.Y.; Huang, W. Organic solid-state lasers: A materials view and future development. Chem. Soc. Rev. 2020, 49, 5885–5944. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Q.; Feng, W.; Li, F. Luminescent Chemodosimeters for Bioimaging. Chem. Rev. 2013, 113, 192–270. [Google Scholar] [CrossRef]
- Benesperi, I.; Michaels, H.; Freitag, M. The researcher’s guide to solid-state dye-sensitized solar cells. J. Mater. Chem. C 2018, 6, 11903–11942. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Askim, J.R.; Suslick, K.S. The Optoelectronic Nose: Colorimetric and Fluorometric Sensor Arrays. Chem. Rev. 2019, 119, 231–292. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, J.; Fan, J.; Chao, H.; Peng, X. Recent progress in photosensitizers for overcoming the challenges of photodynamic therapy: From molecular design to application. Chem. Soc. Rev. 2021, 50, 4185–4219. [Google Scholar] [CrossRef]
- Entwistle, C.D.; Marder, T.B. Applications of Three-Coordinate Organoboron Compounds and Polymers in Optoelectronics. Chem. Mater. 2004, 16, 4574–4585. [Google Scholar] [CrossRef]
- Li, D.; Zhang, H.; Wang, Y. Four-coordinate organoboron compounds for organic light-emitting diodes (OLEDs). Chem. Soc. Rev. 2013, 42, 8416–8433. [Google Scholar] [CrossRef]
- Rodrigues, A.I.; Krishnamoorthy, P.; Gomes, C.S.B.; Carmona, N.; Di Paolo, R.E.; Pander, P.; Pina, J.; de Melo, J.S.S.; Días, F.B.; Calhorda, M.J.; et al. Luminescent halogen-substituted 2-(N-arylimino)pyrrolyl boron complexes: The internal heavy-atom effect. Dalton Trans. 2020, 49, 10185–10202. [Google Scholar] [CrossRef]
- Qi, Y.; Cao, X.; Zou, Y.; Yang, C. Color-tunable tetracoordinated organoboron complexes exhibiting aggregation-induced emission for the efficient turn-on detection of fluoride ions. Mater. Chem. Front. 2021, 5, 2353–2360. [Google Scholar] [CrossRef]
- Dolati, H.; Haufe, L.C.; Denker, L.; Lorbach, A.; Grotjahn, G.H.; Frank, R. Two π-Electrons Make the Difference: From BODIPY to BODIIM Switchable Fluorescent Dyes. Chem. Eur. J. 2020, 26, 1422–1428. [Google Scholar] [CrossRef]
- Boens, N.; Verbelen, B.; Ortiz, M.J.; Jiao, L.; Dehaen, W. Synthesis of BODIPY dyes through postfunctionalization of the boron dipyrromethene core. Coord. Chem. Rev. 2019, 399, 213024. [Google Scholar] [CrossRef]
- Kappaun, S.; Rentenberger, S.; Pogantsch, A.; Zojer, E.; Mereiter, K.; Trimmel, G.; Saf, R.; Möller, K.C.; Stelzer, F.; Slugovc, C. Organoboron Quinolinolates with Extended Conjugated Chromophores: Synthesis, Structure, and Electronic and Electroluminescent Properties. Chem. Mater. 2006, 18, 3539–3547. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Kamiński, R.; Durka, K.; Woźniak, K. Ground-State Charge-Density Distribution in a Crystal of the Luminescent ortho-Phenylenediboronic Acid Complex with 8-Hydroxyquinoline. J. Phys. Chem. A 2018, 122, 4508–4520. [Google Scholar] [CrossRef]
- Jarzembsk, K.N.; Kamiński, R.; Durka, K.; Kubsik, M. Engineering of Solvatomorphs of the Luminescent Complex of ortho-Phenylenediboronic Acid and 8-Hydroxyquinoline. Cryst. Growth Des. 2017, 17, 6836–6851. [Google Scholar] [CrossRef]
- Wesela-Bauman, G.; Ciećwierz, P.; Durka, K.; Luliński, S.; Serwatowski, J.; Woźniak, K. Heteroleptic (2-Fluoro-3-pyridyl)arylborinic 8-Oxyquinolinates for the Potential Application in Organic Light-Emitting Devices. Inorg. Chem. 2013, 52, 10846–10859. [Google Scholar] [CrossRef]
- Xu, S.; Evans, R.E.; Liu, T.; Zhang, G.; Demas, J.N.; Trindle, C.O.; Fraser, C.L. Aromatic Difluoroboron β-Diketonate Complexes: Effects of π-Conjugation and Media on Optical Properties. Inorg. Chem. 2013, 52, 3597–3610. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Han, Y.; Zhang, Z.; He, X.; Chen, Y. The effect of substituent number on mechanochromic luminescence of β-diketones and the corresponding boron complexes. Dye. Pigment. 2019, 166, 159–167. [Google Scholar] [CrossRef]
- Morse, G.E.; Bender, T.P. Boron Subphthalocyanines as Organic Electronic Materials. ACS Appl. Mater. Interfaces 2012, 4, 5055–5068. [Google Scholar] [CrossRef]
- Morse, G.E.; Helander, M.G.; Maka, J.F.; Lu, Z.H.; Bender, T.P. Fluorinated Phenoxy Boron Subphthalocyanines in Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces 2010, 2, 1934–1944. [Google Scholar] [CrossRef]
- Tamgho, I.S.; Hasheminasab, A.; Engle, J.T.; Nemykin, V.N.; Ziegler, C.J. A New Highly Fluorescent and Symmetric Pyrrole–BF2 Chromophore: BOPHY. J. Am. Chem. Soc. 2014, 136, 5623–5626. [Google Scholar] [CrossRef]
- Schrage, B.R.; Nemykin, V.N.; Ziegler, C.J. BOSHPY Fluorophores: BODIPY Analogues with Single Atom Controlled Aggregation. Org. Lett. 2021, 23, 5246–5250. [Google Scholar] [CrossRef]
- Rao, Y.L.; Wang, S. Four-Coordinate Organoboron Compounds with a π-Conjugated Chelate Ligand for Optoelectronic Applications. Inorg. Chem. 2011, 50, 12263–12274. [Google Scholar] [CrossRef]
- Rodríguez, A.I.; Figueira, C.A.; Gomes, C.S.B.; Suresh, D.; Ferreira, B.; Di Paolo, R.E.; de Sa Pereira, D.; Días, F.B.; Calhorda, M.J.; Morgado, J.; et al. Boron complexes of aromatic 5-substituted iminopyrrolyl ligands: Synthesis, structure, and luminescence properties. Dalton Trans. 2019, 48, 13337–13352. [Google Scholar] [CrossRef]
- Li, P.; Chan, H.; Lai, S.L.; Ng, M.; Chan, M.Y.; Yam, V.W.W. Four-Coordinate Boron Emitters with Tridentate Chelating Ligand for Efficient and Stable Thermally Activated Delayed Fluorescence Organic Light-Emitting Devices. Angew. Chem. Int. Ed. 2019, 58, 9088–9094. [Google Scholar] [CrossRef]
- Murali, A.C.; Nayak, P.; Venkatasubbaiah, K. Recent advances in the synthesis of luminescent tetra-coordinated boron compounds. Dalton Trans. 2022, 51, 5751. [Google Scholar] [CrossRef]
- Kothavale, S.S.; Lee, J.Y. Three- and Four-Coordinate, Boron-Based, Thermally Activated Delayed Fluorescent Emitters. Adv. Opt. Mater. 2020, 8, 2000922. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Jiao, C.; Ye, K.; Zhang, H.; Zhang, J.; Wang, Y. 2-(2-Hydroxyphenyl)benzimidazole-Based Four-Coordinate Boron-Containing Materials with Highly Efficient Deep-Blue Photoluminescence and Electroluminescence. Inorg. Chem. 2015, 54, 2652–2659. [Google Scholar] [CrossRef]
- Suresh, D.; Ferreira, B.; Lopes, P.S.; Gomes, C.S.B.; Krishnamoorthy, P.; Charas, A.; Vila-Viçosa, D.; Morgado, J.; Calhorda, M.J.; Maçanita, A.L.; et al. Boron complexes of aromatic ring fused iminopyrrolyl ligands: Synthesis, structure, and luminescence properties. Dalton Trans. 2016, 45, 15603–15620. [Google Scholar] [CrossRef] [PubMed]
- Bismillah, A.N.; Aprahamian, I. Fundamental studies to emerging applications of pyrrole-BF2 (BOPHY) fluorophores. Chem. Soc. Rev. 2021, 50, 5631–5649. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Liu, Q.D.; Bai, D.R.; Jia, W.L.; Tao, Y.; Wang, S. Organoboron Compounds with an 8-Hydroxyquinolato Chelate and Its Derivatives: Substituent Effects on Structures and Luminescence. Inorg. Chem. 2005, 44, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, S.; Gon, M.; Tanaka, K.; Chujo, Y. The Design Strategy for an Aggregation- and Crystallization-Induced Emission-Active Molecule Based on the Introduction of Skeletal Distortion by Boron Complexation with a Tridentate Ligand. Crystals 2020, 10, 615. [Google Scholar] [CrossRef]
- Yarnton, O. Crystallizer; Version 1.171.37.35; Agilent Technologies: Oxford, UK, 2014. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. ShelXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0-New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Brandenburg, K. Diamond; Version 4.3.2; Crystal Impact GbR: Bonn, Germany, 2017. [Google Scholar]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer; Version 2.1; University of Western Australia: Perth, Australia, 2007. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Mennucci, V.B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision A. 02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J.; Eppinnett, K.; Hovell, W.L.; Gilliland, R. GaussView, version 5.0.9; Semichem, Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Burke, K.; Werschnik, J.; Gross, E.K.U. Time-dependent density functional theory: Past, present, and future. J. Chem. Phys. 2005, 123, 062206. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; DeFrees, D.J.; People, J.A.; Gordon, M.S. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Scrocco, E.; Tomasi, J. Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Euristic Interpretation by Means of Electrostatic Molecular Potentials. Adv. Quantum Chem. 1978, 11, 115–193. [Google Scholar]
- Wu, Q.; Esteghamatian, M.; Hu, N.X.; Popovic, Z.; Enright, G.; Tao, Y.; D’Iorio, M.; Wang, S. Synthesis, Structure, and Electroluminescence of BR2q (R = Et, Ph, 2-Naphthyl and q = 8-Hydroxyquinolato). Chem. Mater. 2000, 12, 79–83. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Kamiński, R.; Durka, K.; Kubsik, M.; Nawara, K.; Witkowska, E.; Wiloch, M.; Luliński, S.; Waluk, J.; Głowacki, I.; et al. New class of easily-synthesisable and modifiable organic materials for applications in luminescent devices. Dye. Pigment. 2017, 138, 267–277. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(i) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 9, 955–964. [Google Scholar] [CrossRef]
- Höpfl, H.J. The tetrahedral character of the boron atom newly defined—A useful tool to evaluate the N→B bond. J. Organomet. Chem. 1999, 581, 129–149. [Google Scholar] [CrossRef]
- Dar, A.A.; Ganie, A.A. Irreversible Thermochromism in Organic Salts of Sulfonated Anils. Cryst. Growth Des. 2020, 20, 3888–3897. [Google Scholar] [CrossRef]
- Duan, W.; Liu, B.; Gong, N.; Famulari, A.; Guo, F. Polymorphs and Transformations of the Solid Forms of Organic Salts of 5-Sulfosalicylic Acid and Isonicotinamide. Cryst. Growth Des. 2020, 20, 7606–7614. [Google Scholar] [CrossRef]
- Schmidtmann, M.; Wilson, C.C. Hydrogen transfer in pentachlorophenol–Dimethylpyridine complexes. CrystEngComm 2008, 10, 177–183. [Google Scholar] [CrossRef]
- Campos-Gaxiola, J.J.; García-Grajeda, B.A.; Hernández-Ahuactzi, I.F.; Guerrero-Álvarez, J.A.; Höpfl, H.; Cruz-Enríquez, A. Supramolecular networks in molecular complexes of pyridine boronic acids and polycarboxylic acids: Synthesis, structural characterization and fluorescence properties. CrystEngComm 2017, 19, 3760. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Garcia-Grajeda, B.A.; Höpfl, H.; Guerrero-Alvarez, J.A.; Campos-Gaxiola, J.J.; Cruz-Enríquez, A. 2,6-Dihy dr oxy-4-oxo-2-(pyridin-1-ium-3-yl)-4H-1,3,2-benzodioxaborinin-2-ide 0.67-hydrate. Acta Cryst. 2014, E70, o388–o389. [Google Scholar]
- Ruelas-Álvarez, G.Y.; Cárdenas-Valenzuela, A.J.; Cruz-Enríquez, A.; Höpfl, H.; Campos-Gaxiola, J.J.; Rodríguez-Rivera, M.A.; Rodríguez-Molina, B. Exploration of the Luminescence Properties of Organic Phosphate Salts of 3-Quinoline- and 5-Isoquinolineboronic Acid. Eur. J. Inorg. Chem. 2019, 2019, 2707–2724. [Google Scholar] [CrossRef]
- Główka, M.L.; Martynowski, D.; Kozłowska, K. Stacking of six-membered aromatic rings in crystals. J. Mol. Struct. 1999, 474, 81–89. [Google Scholar] [CrossRef]
- Thakuria, R.; Nath, N.K.; Saha, B.K. The Nature and Applications of π–π Interactions: A Perspective. Cryst. Growth Des. 2019, 19, 523–528. [Google Scholar] [CrossRef]
- Ganie, A.A.; Ahangar, A.A.; Dar, A.A. Sulfonate…Pyridinium Supramolecular Synthon: A Robust Interaction Utilized to Design Molecular Assemblies. Cryst. Growth Des. 2019, 19, 4650–4660. [Google Scholar] [CrossRef]
- Ahmad, I.; Ganie, A.A.; Dar, A.A. Achievement of enhanced solubility and improved optics in molecular complexes based on a sulfonate–Pyridinium supramolecular synthon. CrystEngComm 2020, 22, 3933–3942. [Google Scholar] [CrossRef]
- Desiraju, G.R. The C−H⋯O Hydrogen Bond: Structural Implications and Supramolecular Design. Acc. Chem. Res. 1996, 29, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Ganie, A.A.; Ismail, T.M.; Sajith, P.K.; Dar, A.A. Validation of the supramolecular synthon preference through DFT and physicochemical property investigations of pyridyl salts of organo-sulfonates. New J. Chem. 2021, 45, 4780–4790. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- Loots, L.; Barbour, L.J. A simple and robust method for the identification of π–π packing motifs of aromatic compounds. CrystEngComm 2012, 14, 300–304. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 0, 3814–3816. [Google Scholar] [CrossRef]
- Wood, P.A.; McKinnon, J.J.; Parsons, S.; Pidcock, E.; Spackman, M.A. Analysis of the compression of molecular crystal structures using Hirshfeld surfaces. CrystEngComm 2008, 10, 368–376. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Cryst. 2004, B60, 627–668. [Google Scholar] [CrossRef]
- Capelli, S.C.; Bürgi, H.B.; Dittrich, B.; Grabowsky, S.; Jayatilaka, D. Hirshfeld atom refinement. IUCrJ 2014, 1, 361–379. [Google Scholar] [CrossRef] [Green Version]
- Woinska, M.; Jayatilaka, D.; Spackman, M.A.; Edwards, A.J.; Dominiak, P.M.; Wozniak, K.; Nishibori, E.; Sugimoto, K.; Grabowsky, S. Hirshfeld atom refinement for modelling strong hydrogen bonds. Acta Cryst. 2014, A70, 483–498. [Google Scholar] [CrossRef]
- Politzer, P.; Truhlar, D.G. Introduction: The role of the electrostatic potential in chemistry. In Chemical Applications of Atomic and Molecular Electrostatic Potentials; Politzer, P., Truhlar, D.G., Eds.; Springer: Boston, MA, USA, 1981. [Google Scholar] [CrossRef]
- Murray, J.S.; Sen, K. Molecular Electrostatic Potentials, Concepts and Applications, 1st ed.; Elsevier: Amsterdam, The Netherlands, 1996. [Google Scholar]
- Barba, V.; Vázquez, J.; López, F.; Santillan, R.; Farfán, N. Mono- and diboronates derived from tridentate ONO ligands and arylboronic acids. J. Organomet. Chem. 2005, 690, 2351–2357. [Google Scholar] [CrossRef]
- Gonzalez-Hernández, A.; León-Negrete, A.; Galván-Hidalgo, J.M.; Gómez, E.; Barba, V. Hexacyclic monomeric boronates derived from tridentate shiff-base ligands fused by dative N → B bond. J. Mol. Struct. 2020, 1207, 127779. [Google Scholar] [CrossRef]
- Smith, M.K.; Northrop, B.H. Vibrational Properties of Boroxine Anhydride and Boronate Ester Materials: Model Systems for the Diagnostic Characterization of Covalent Organic Frameworks. Chem. Mater. 2014, 26, 3781–3795. [Google Scholar] [CrossRef]
- Videnova-Adrabinska, V.; Nowak, A.M.; Wilk, M.; Janczak, J.; Baran, J. Crystal polymorphism of sodium benzene-1,3-dicarboxy-5-sulfonate monohydrate. J. Mol. Struct. 2011, 996, 53–63. [Google Scholar] [CrossRef]
- Furer, V.L.; Vandyukov, A.E.; Popova, E.V.; Solovieva, S.E.; Antipin, I.S.; Kovalenko, V.I. Vibrational spectra study of p-sulfonatocalix[4]arene containing azobenzene groups. J. Mol. Struct. 2020, 1200, 127058. [Google Scholar] [CrossRef]
- Sandhu, B.; Fonari, M.S.; Sawyer, K.; Timofeeva, T.V. A series of crystalline solids composed of aminopyridines and succinic, fumaric, and sebacic acids. J. Mol. Struct. 2013, 1052, 125–134. [Google Scholar] [CrossRef]
- Faizan, M.; Alam, M.J.; Afroz, Z.; Nunes Rodrigues, V.H.; Ahmad, S. Growth, structure, Hirshfeld surface and spectroscopic properties of 2-amino-4-hydroxy-6-methylpyrimidinium-2,3-pyrazinedicorboxylate single cristal. J. Mol. Struct. 2018, 1155, 695–710. [Google Scholar] [CrossRef]
- Zhou, Z.; Parr, R. G Activation hardness: New index for describing the orientation of electrophilic aromatic substitution. J. Am. Chem. Soc. 1990, 112, 5720–5724. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Ciofini, L.; Adamo, C.; Valero, R.; Zhao, Y.; Truhlar, D.G. On the Performances of the M06 Family of Density Functionals for Electronic Excitation Energies. J. Chem. Theory Comput. 2010, 6, 2071–2085. [Google Scholar] [CrossRef]
- Insuasty, D.; Cabrera, L.; Ortiz, A.; Insuasty, B.; Quiroga, J.; Abonia, R. Synthesis, photophysical properties and theoretical studies of new bis-quinolin curcuminoid BF2-complexes and their decomplexed derivatives. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 230, 118065. [Google Scholar] [CrossRef]
- Frath, D.; Massue, J.; Ulrich, G.; Ziessel, R. Luminescent Materials: Locking π-Conjugated and Heterocyclic Ligands with Boron(III). Angew. Chem. Int. Ed. 2014, 53, 2290–2310. [Google Scholar] [CrossRef]
- Urban, M.; Durka, K.; Górka, P.; Wiosna-Sałyga, G.; Nawara, K.; Jankowski, P.; Luliński, S. The effect of locking π-conjugation in organoboron moieties in the structures of luminescent tetracoordinate boron complexes. Dalton Trans. 2019, 48, 8642–8663. [Google Scholar] [CrossRef]
- Más-Montoya, M.; Montenegro, M.F.; Espinosa Ferao, A.; Tárraga, A.; Rodríguez-López, J.N.; Curiel, D. Rigid π-Extended Boron Difluoride Complex with Mega-Stokes Shift for Bioimaging. Org. Lett. 2020, 22, 3356–3360. [Google Scholar] [CrossRef]
- Sharma, S.; Wei, Z.; Grozema, F.C.; Sengupta, S. Structure–Property relationships in multi-stimuli responsive BODIPY-biphenyl-benzodithiophene TICT rigidochromic rotors exhibiting (pseudo-)Stokes shifts up to 221 nm. Phys. Chem. Chem. Phys. 2020, 22, 25514–25521. [Google Scholar] [CrossRef]
- Massue, J.; Jacquemin, D.; Ulrich, G. Boranils: Versatile Multifunctional Organic Fluorophores for Innovative Applications. Organics 2021, 2, 20. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.L.; Luo, D.; Xiao, P.; Xiao, W.; Song, Y.; Ang, Q.; Liu, B. Strategies to Achieve High-Performance White Organic Light-Emitting Diodes. Materials 2017, 10, 1378. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Kiburu, I.; Shah, S.; Jäkle, F. Luminescence tuning of organoboron quinolates through substituent variation at the 5-position of the quinolato moiety. Org. Lett. 2006, 8, 5227–5230. [Google Scholar] [CrossRef]
- Zhang, Z.; Bi, H.; Zhang, Y.; Yao, D.; Gao, H.; Fan, Y.; Zhang, H.; Wang, Y.; Wang, Y.; Chen, Z.; et al. Luminescent Boron-Contained Ladder-Type π-Conjugated Compounds. Inorg. Chem. 2009, 48, 7230–7236. [Google Scholar] [CrossRef]
- Guieu, S.; Esteves, C.I.; Rocha, J.; Silva, A.M. Multicomponent Synthesis of Luminescent Iminoboronates. Molecules 2020, 25, 6039. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.Y.; Shen, Q.J.; Zhao, X.R.; Yan, X.Q.; Pang, X.; Jin, W.J. Phosphorescent co-crystal assembled by 1,4-diiodotetrafluorobenzene with carbazole based on C–I⋯π halogen bonding. J. Mater. Chem. 2012, 22, 5336–5343. [Google Scholar] [CrossRef]
- Bolton, O.; Lee, K.; Kim, H.J.; Lin, K.Y.; Kim, J. Activating efficient phosphorescence from purely organic materials by crystal design. Nat. Chem. 2011, 3, 415. [Google Scholar] [CrossRef]
- Li, Q.; Tang, Y.; Hu, W.; Li, Z. Fluorescence of Nonaromatic Organic Systems and Room Temperature Phosphorescence of Organic Luminogens: The Intrinsic Principle and Recent Progress. Small 2018, 14, 1801560. [Google Scholar] [CrossRef] [PubMed]
- Galer, V.; Korosec, R.C.; Vidmar, M.; Šket, B. Crystal Structures and Emission Properties of the BF2 Complex 1-Phenyl-3-(3,5-dimethoxyphenyl)-propane-1,3-dione: Multiple Chromisms, Aggregation- or Crystallization-Induced Emission, and the Self-Assembly Effect. J. Am. Chem. Soc. 2014, 136, 7383–7739. [Google Scholar] [CrossRef] [PubMed]
- Chai, Z.; Wang, C.; Wang, J.; Liu, F.; Xie, Y.; Zhang, Y.Z.; Li, J.R.; Li, Q.; Li, Z. Abnormal room temperature phosphorescence of purely organic boron-containing compounds: The relationship between the emissive behaviorand the molecular packing, and the potential related applications. Chem. Sci. 2017, 8, 8336–8344. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Xu, D.; Wang, Y.; Zhang, C.; Yang, Y.; Liu, X.; Han, A.; Wang, Y. Aggregation-induced emission and mechanofluorochromism of tetraphenylbutadiene modified β-ketoiminate boron complexes. Dye. Pigment. 2018, 150, 165–173. [Google Scholar] [CrossRef]
- Potopnyk, M.A.; Volyniuk, D.; Luboradzki, R.; Lazauskas, A.; Grazulevicius, J.V. Aggregation-Induced Emission-Active Carbazolyl-Modified Benzo[4,5]thiazolo[3,2-c]oxadiazaborinines as Mechanochromic Fluorescent Materials. Eur. J. Org. Chem. 2021, 2021, 2772–2781. [Google Scholar] [CrossRef]
- Hong, Y.; Lam, J.W.Y.; Tang, B.Z. Aggregation-induced emission. Chem. Soc. Rev. 2011, 40, 5361–5388. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lam, J.W.Y.; Kwok, R.T.K.; Liu, B.; Tang, B.Z. Aggregation-induced emission: Fundamental understanding and future developments. Mater. Horiz. 2019, 6, 428–433. [Google Scholar] [CrossRef]
- Yin, P.A.; Wan, Q.; Niu, Y.; Peng, Q.; Wang, Z.; Li, Y.; Qin, A.; Shuai, Z.; Tang, B.Z. Theoretical and Experimental Investigations on the Aggregation-Enhanced Emission from Dark State: Vibronic Coupling Effect. Adv. Electron. Mater. 2020, 6, 2000255. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, S. Diboron and Triboron Compounds Based on Linear and Star-Shaped Conjugated Ligands with 8-Hydroxyquinolate Functionality: Impact of Intermolecular Interaction and Boron Coordination on Luminescence. J. Org. Chem. 2006, 71, 6485–6496. [Google Scholar] [CrossRef]
- Chen, S.; Qiu, R.; Yu, Q.; Zhang, X.; Wei, M.; Dai, Z. Boranil dyes bearing tetraphenylethene: Synthesis, AIE/AIEE effect properties, pH sensitive properties and application in live cell imaging. Tetrahedron Lett. 2018, 59, 2671–2678. [Google Scholar] [CrossRef]
- Vaz, P.A.A.M.; Rocha, J.; Silva, A.M.S.; Guieu, S. Aggregation-induced emission enhancement of chiral boranils. New J. Chem. 2018, 42, 18166–18171. [Google Scholar] [CrossRef]
- Varghese, S.; Das, S. Role of Molecular Packing in Determining Solid-State Optical Properties of π-Conjugated Materials. J. Phys. Chem. Lett. 2011, 2, 863–873. [Google Scholar] [CrossRef]
- Li, Q.; Li, Z. The Strong Light-Emission Materials in the Aggregated State: What Happens from a Single Molecule to the Collective Group. Adv. Sci. 2017, 4, 1600484. [Google Scholar] [CrossRef]
- Yan, X.; Zhu, P.; Zhou, Z.; Yang, H.; Lana, H.; Xiao, S. Aggregation-induced emission enhancement (AIEE)-active boron-difluoride dyes with reversible mechanochromic fluorescence. RSC Adv. 2019, 9, 35872–35877. [Google Scholar] [CrossRef] [Green Version]
- Chattapadhyay, D.; Mondal, S.; Kumar, S.; Haldar, D. Topology-Controlled AIEE of Iminocoumarin Luminophores. Chem. Asian J. 2021, 16, 2723–2728. [Google Scholar] [CrossRef]
- Gu, L.; Liu, R.; Shi, H.; Wang, Q.; Song, G.; Zhu, X.; Yuan, S.; Zhu, H. Synthesis, Luminescent Properties of aza-Boron-Diquinomethene Difluoride Complexes and Their Application for Fluorescent Security Inks. J. Fluoresc. 2016, 26, 407–412. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, H.; Yu, J. Luminescence anti-counterfeiting: From elementary to advanced. Aggregate 2021, 2, 20–34. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruelas-Álvarez, G.Y.; Cárdenas-Valenzuela, A.J.; Galaviz-Moreno, L.L.; Cruz-Enríquez, A.; Campos-Gaxiola, J.J.; Höpfl, H.; Baldenebro-López, J.; Vargas-Olvera, E.C.; Miranda-Soto, V.; García Grajeda, B.A.; et al. Four-Coordinate Monoboron Complexes with 8-Hydroxyquinolin-5-Sulfonate: Synthesis, Crystal Structures, Theoretical Studies, and Luminescence Properties. Crystals 2022, 12, 783. https://doi.org/10.3390/cryst12060783
Ruelas-Álvarez GY, Cárdenas-Valenzuela AJ, Galaviz-Moreno LL, Cruz-Enríquez A, Campos-Gaxiola JJ, Höpfl H, Baldenebro-López J, Vargas-Olvera EC, Miranda-Soto V, García Grajeda BA, et al. Four-Coordinate Monoboron Complexes with 8-Hydroxyquinolin-5-Sulfonate: Synthesis, Crystal Structures, Theoretical Studies, and Luminescence Properties. Crystals. 2022; 12(6):783. https://doi.org/10.3390/cryst12060783
Chicago/Turabian StyleRuelas-Álvarez, Glenda Y., A. Jaquelin Cárdenas-Valenzuela, Luis L. Galaviz-Moreno, Adriana Cruz-Enríquez, José J. Campos-Gaxiola, Herbert Höpfl, Jesús Baldenebro-López, Eva C. Vargas-Olvera, Valentín Miranda-Soto, Blanca A. García Grajeda, and et al. 2022. "Four-Coordinate Monoboron Complexes with 8-Hydroxyquinolin-5-Sulfonate: Synthesis, Crystal Structures, Theoretical Studies, and Luminescence Properties" Crystals 12, no. 6: 783. https://doi.org/10.3390/cryst12060783
APA StyleRuelas-Álvarez, G. Y., Cárdenas-Valenzuela, A. J., Galaviz-Moreno, L. L., Cruz-Enríquez, A., Campos-Gaxiola, J. J., Höpfl, H., Baldenebro-López, J., Vargas-Olvera, E. C., Miranda-Soto, V., García Grajeda, B. A., & Glossman-Mitnik, D. (2022). Four-Coordinate Monoboron Complexes with 8-Hydroxyquinolin-5-Sulfonate: Synthesis, Crystal Structures, Theoretical Studies, and Luminescence Properties. Crystals, 12(6), 783. https://doi.org/10.3390/cryst12060783