
A Puzzling Protein from Variovorax paradoxus Has a PLP Fold Type IV Transaminase Structure and Binds PLP without Catalytic Lysine

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. VP5454 Expression, Purification and Characterization
2.2. Protein Crystallization
2.3. Data Collection, Processing, Structure Solution and Refinement
2.4. Structure Analysis and Validation
2.5. Molecular Modeling
3. Results
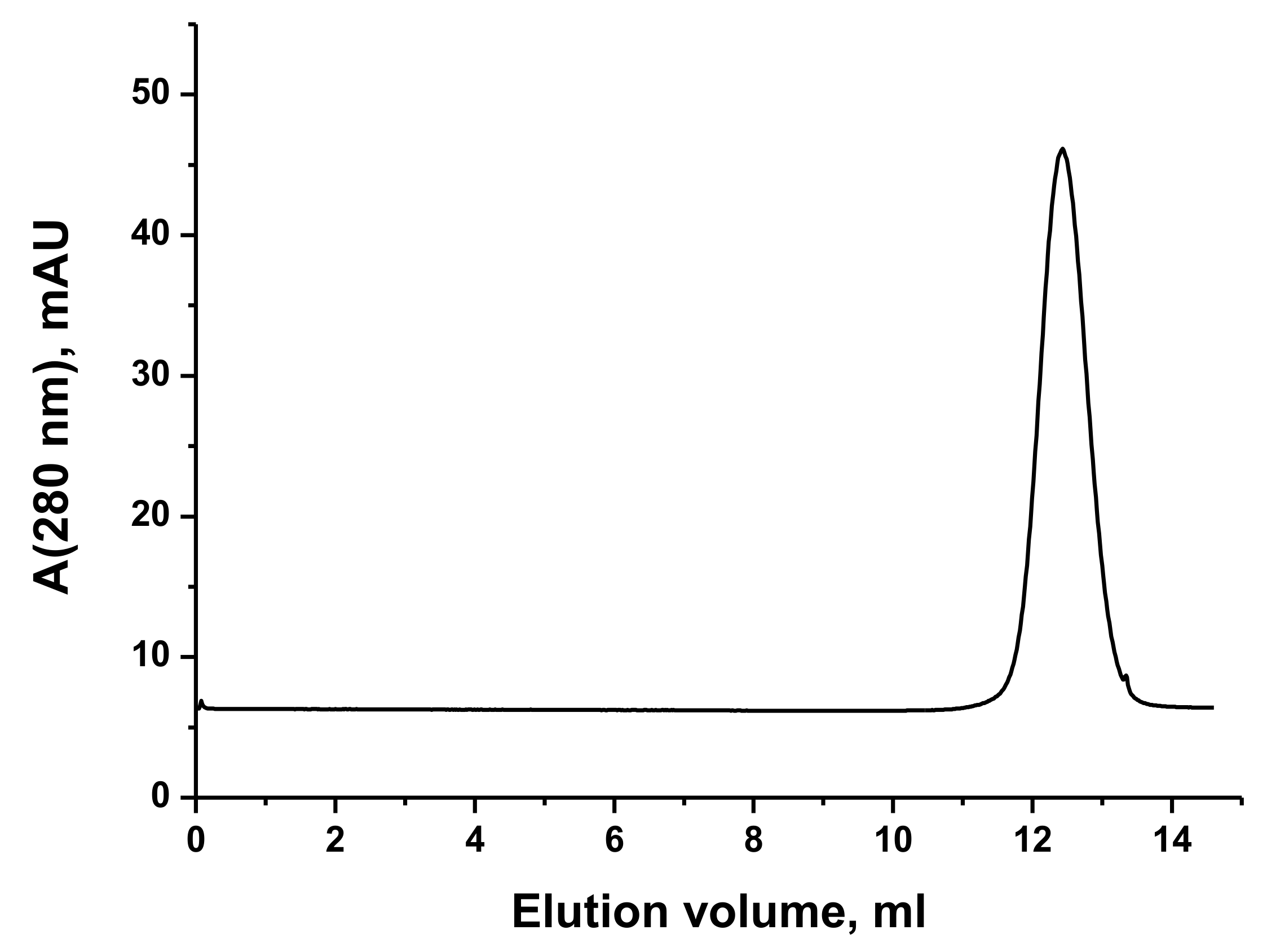
3.1. VP5454 Purification and Biochemical Analysis
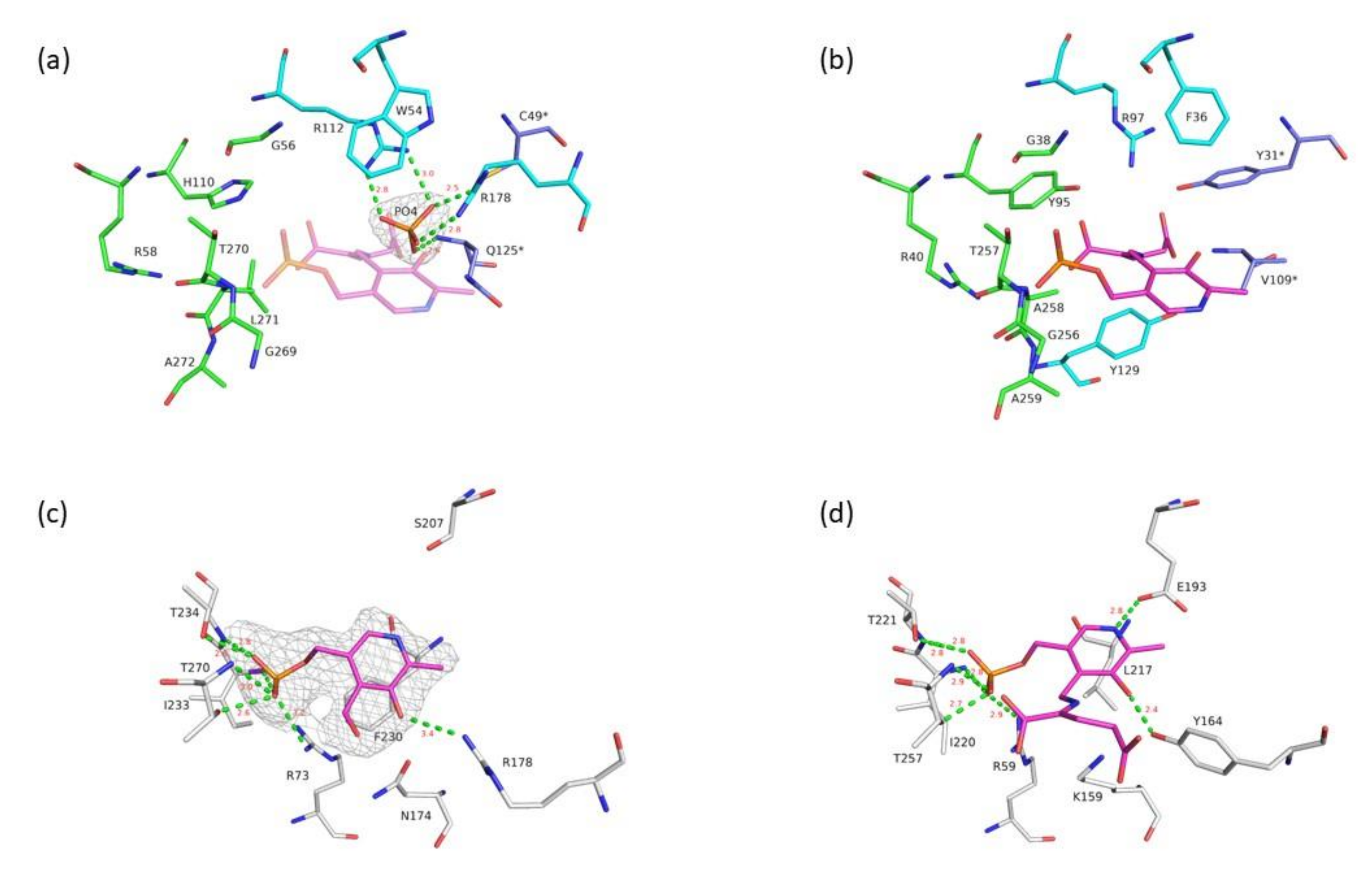
3.2. VP5454 Has Typical TAs Fold and Is Mostly Similar to BCAT TAs
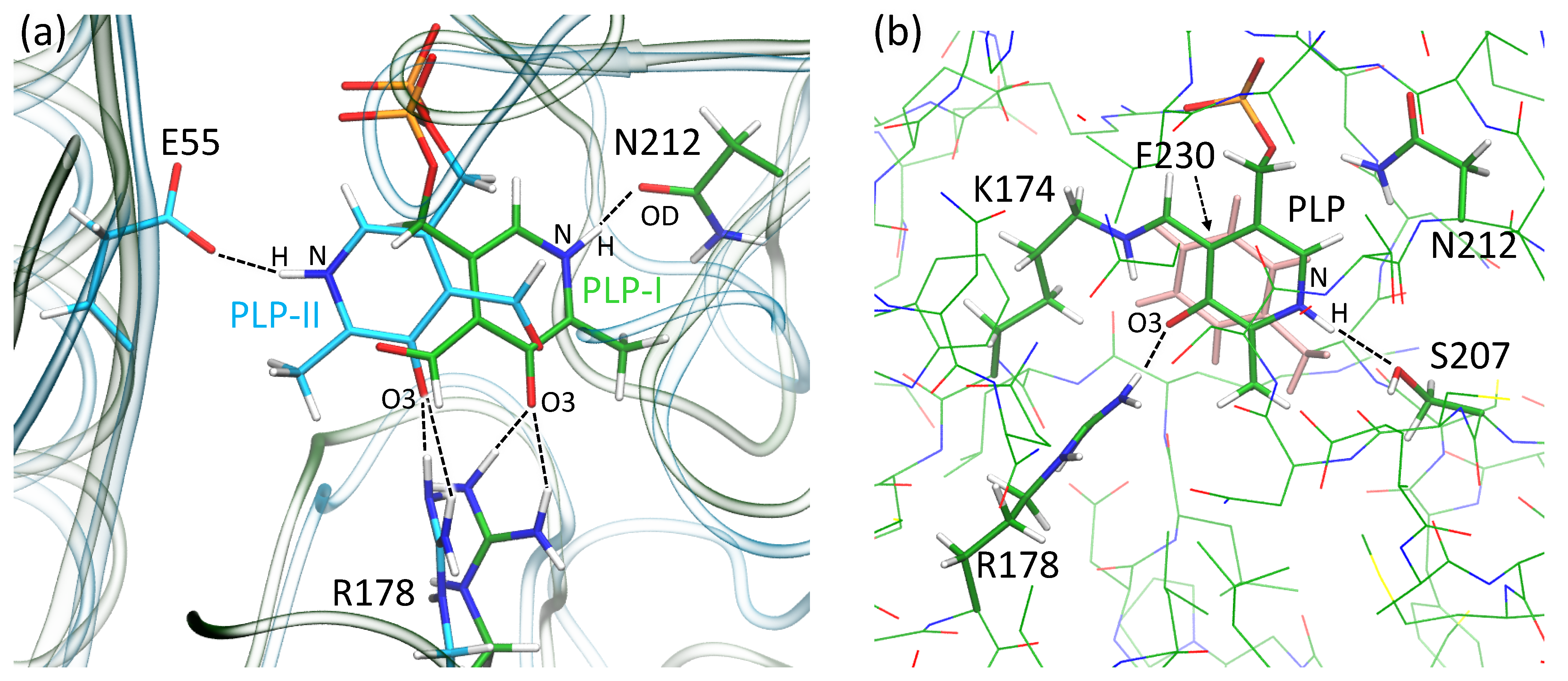
3.3. Active Site of VP5454 Can Accommodate PLP Molecule
3.4. Molecular Modeling Supports the Ability of VP5454 to Non-Covalently Bind PLP
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gu, X.; Zhao, J.; Chen, L.; Li, Y.; Yu, B.; Tian, X.; Min, Z.; Xu, S.; Gu, H.; Sun, J.; et al. Application of Transition-Metal Catalysis, Biocatalysis, and Flow Chemistry as State-of-the-Art Technologies in the Synthesis of LCZ696. J. Org. Chem. 2020, 85, 6844–6853. [Google Scholar] [CrossRef]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 2020, 60, 88–119. [Google Scholar] [CrossRef] [PubMed]
- Eliot, A.C.; Kirsch, J.F. Pyridoxal Phosphate Enzymes: Mechanistic, Structural, and Evolutionary Considerations. Annu. Rev. Biochem. 2004, 73, 383–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toney, M.D. Controlling reaction specificity in pyridoxal phosphate enzymes. Biochim. et Biophys. Acta (BBA) Proteins Proteom. 2011, 1814, 1407–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höhne, M.; Schätzle, S.; Jochens, H.; Robins, K.; Bornscheuer, U.T. Rational assignment of key motifs for function guides in silico enzyme identification. Nat. Chem. Biol. 2010, 6, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Grishin, N.V.; Phillips, M.A.; Goldsmith, E.J. Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein Sci. 1995, 4, 1291–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezsudnova, E.Y.; Popov, V.; Boyko, K.M. Structural insight into the substrate specificity of PLP fold type IV transaminases. Appl. Microbiol. Biotechnol. 2020, 104, 2343–2357. [Google Scholar] [CrossRef]
- Steffen-Munsberg, F.; Vickers, C.; Kohls, H.; Land, H.; Mallin, H.; Nobili, A.; Skalden, L.; Bergh, T.V.D.; Joosten, H.-J.; Berglund, P.; et al. Bioinformatic analysis of a PLP-dependent enzyme superfamily suitable for biocatalytic applications. Biotechnol. Adv. 2015, 33, 566–604. [Google Scholar] [CrossRef]
- Iwasaki, A.; Matsumoto, K.; Hasegawa, J.; Yasohara, Y. A novel transaminase, (R)-amine:pyruvate aminotransferase, from Arthrobacter sp. KNK168 (FERM BP-5228): Purification, characterization, and gene cloning. Appl. Microbiol. Biotechnol. 2011, 93, 1563–1573. [Google Scholar] [CrossRef]
- Stekhanova, T.N.; Rakitin, A.L.; Mardanov, A.V.; Bezsudnova, E.Y.; Popov, V.O. A Novel highly thermostable branched-chain amino acid aminotransferase from the crenarchaeon Vulcanisaeta moutnovskia. Enzym. Microb. Technol. 2017, 96, 127–134. [Google Scholar] [CrossRef]
- Bezsudnova, E.Y.; Boyko, K.M.; Nikolaeva, A.Y.; Zeifman, Y.S.; Rakitina, T.V.; Suplatov, D.A.; Popov, V.O. Biochemical and structural insights into PLP fold type IV transaminase from Thermobaculum terrenum. Biochimie 2018, 158, 130–138. [Google Scholar] [CrossRef]
- Boyko, K.M.; Stekhanova, T.N.; Nikolaeva, A.Y.; Mardanov, A.V.; Rakitin, A.L.; Ravin, N.V.; Bezsudnova, E.Y.; Popov, V.O. First structure of archaeal branched-chain amino acid aminotransferase from Thermoproteus uzoniensis specific for l-amino acids and R-amines. Extremophiles 2016, 20, 215–225. [Google Scholar] [CrossRef]
- Okada, K.; Hirotsu, K.; Hayashi, H.; Kagamiyama, H. Structures of Escherichia coli Branched-Chain Amino Acid Aminotransferase and Its Complexes with 4-Methylvalerate and 2-Methylleucine: Induced Fit and Substrate Recognition of the Enzyme. Biochemistry 2001, 40, 7453–7463. [Google Scholar] [CrossRef]
- Sugio, S.; Petsko, G.A.; Manning, J.M.; Soda, K.; Ringe, D. Crystal Structure of a D-Amino Acid Aminotransferase: How the Protein Controls Stereoselectivity. Biochemistry 1995, 34, 9661–9669. [Google Scholar] [CrossRef]
- Thomsen, M.; Skalden, L.; Palm, G.J.; Höhne, M.; Bornscheuer, U.T.; Hinrichs, W. Crystallographic characterization of the (R)-selective amine transaminase fromAspergillus fumigatus. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 1086–1093. [Google Scholar] [CrossRef]
- Wybenga, G.G.; Crismaru, C.G.; Janssen, D.B.; Dijkstra, B.W. Structural Determinants of the β-Selectivity of a Bacterial Aminotransferase. J. Biol. Chem. 2012, 287, 28495–28502. [Google Scholar] [CrossRef] [Green Version]
- Bakunova, A.K.; Nikolaeva, A.Y.; Rakitina, T.V.; Isaikina, T.Y.; Khrenova, M.G.; Boyko, K.M.; Popov, V.O.; Bezsudnova, E.Y. The Uncommon Active Site of D-Amino Acid Transaminase from Haliscomenobacter hydrossis: Biochemical and Structural Insights into the New Enzyme. Molecules 2021, 26, 5053. [Google Scholar] [CrossRef]
- Boyko, K.; Gorbacheva, M.; Rakitina, T.; Korzhenevskiy, D.; Vanyushkina, A.; Kamashev, D.; Lipkin, A.; Popov, V. Expression, purification, crystallization and preliminary X-ray crystallographic analysis of the histone-like HU protein fromSpiroplasma melliferumKC3. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 24–27. [Google Scholar] [CrossRef] [Green Version]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 62, 72–82. [Google Scholar] [CrossRef]
- Vagin, A.A.; Teplyakov, A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Isupov, M.N.; Boyko, K.M.; Sutter, J.-M.; James, P.; Sayer, C.; Schmidt, M.; Schönheit, P.; Nikolaeva, A.Y.; Stekhanova, T.N.; Mardanov, A.V.; et al. Thermostable Branched-Chain Amino Acid Transaminases From the Archaea Geoglobus acetivorans and Archaeoglobus fulgidus: Biochemical and Structural Characterization. Front. Bioeng. Biotechnol. 2019, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Computational Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Struct. Biol. Crystallogr. 2004, D60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, J.A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; Mackerell, A.D.M. Impact of 2′-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Liu, H.; Hou, T. CaFE: A tool for binding affinity prediction using end-point free energy methods. Bioinformatics 2016, 32, 2216–2218. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-D.; Lin, C.-H.; Chuankhayan, P.; Huang, Y.-C.; Hsieh, Y.-C.; Huang, T.-F.; Guan, H.-H.; Liu, M.-Y.; Chang, W.-C.; Chen, C.-J. Crystal Structures of Complexes of the Branched-Chain Aminotransferase from Deinococcus radiodurans with α-Ketoisocaproate and l- Glutamate Suggest the Radiation Resistance of This Enzyme for Catalysis. J. Bacteriol. 2012, 194, 6206–6216. [Google Scholar] [CrossRef] [Green Version]
- Mistry, D.; Powles, N. The relative hydrolytic reactivities of pyrophosphites and pyrophosphates. Org. Biomol. Chem. 2013, 11, 5727–5733. [Google Scholar] [CrossRef] [Green Version]
- Goto, M.; Miyahara, I.; Hayashi, H.; Kagamiyama, H.; Hirotsu, K. Crystal Structures of Branched-Chain Amino Acid Aminotransferase Complexed with Glutamate and Glutarate: True Reaction Intermediate and Double Substrate Recognition of the Enzyme. Biochemistry 2003, 42, 3725–3733. [Google Scholar] [CrossRef]
- Mizushina, Y.; Xu, X.; Matsubara, K.; Murakami, C.; Kuriyama, I.; Oshige, M.; Takemura, M.; Kato, N.; Yoshida, H.; Sakaguchi, K. Pyridoxal 5′-phosphate is a selective inhibitor in vivo of DNA polymerase α and ε. Biochem. Biophys. Res. Commun. 2003, 312, 1025–1032. [Google Scholar] [CrossRef]
- Vega, D.E.; Margolin, W. Suppression of a Thermosensitive zipA Cell Division Mutant by Altering Amino Acid Metabolism. J. Bacteriol. 2018, 200, e00535-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerma-Ortiz, C.; Jeffryes, J.G.; Cooper, A.J.; Niehaus, T.D.; Thamm, A.M.; Frelin, O.; Aunins, T.; Fiehn, O.; de Crécy-Lagard, V.; Henry, C.S.; et al. ‘Nothing of chemistry disappears in biology’: The Top 30 damage-prone endogenous metabolites. Biochem. Soc. Trans. 2016, 44, 961–971. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | |
| Diffraction source | BL41XU, Spring8 |
| Wavelength (Å) | 1.0 |
| Temperature (K) | 100 |
| Detector | EIGER |
| Crystal-to-detector distance (mm) | 250.00 |
| Rotation range per image (°) | 0.5 |
| Total rotation range (°) | 180 |
| Space group | P3221 |
| a, b, c (Å) | 164.30, 164.30, 190.42 |
| α, β, γ (°) | 90, 90, 120 |
| Unique reflections | 131283 (6431) |
| Resolution range (Å) | 190.42–2.30 (2.34–2.30) |
| Completeness (%) | 99.7 (99.5) |
| Average redundancy | 20.5 (21.3) |
| 〈I/σ(I)〉 | 7.5 (0.8) |
| Rpim (%) | 1.7 (65.9) |
| CC1/2 | 99.6 (63.5) |
| Refinement | |
| Rfact (%) | 20.5 |
| Rfree (%) | 22.8 |
| RMS deviations | |
| Bonds (Å) | 0.02 |
| Angles (°) | 2.21 |
| Ramachandran plot | |
| Most favored (%) | 97.5 |
| Allowed (%) | 2.4 |
| No. atoms | |
| Protein | 13,632 |
| Water | 611 |
| PO4 | 30 |
| PLP | 45 |
| Other ligands | 97 |
| B-factors (Å2) | |
| Protein | 42.8 |
| Water | 37.0 |
| PLP | 32.9 |
| PO4 | 42.6 |
| Other ligands | 75.1 |
| Protein (PDB Code) | Aligned Residues, % | RMSD, Å | Sequence Identity, % |
|---|---|---|---|
| Branched-chain amino acid transaminases from Archaeoglobus fulgidus (5MQZ) | 91 | 1.18 | 36 |
| Branched-chain amino acid transaminases from Geoglobus acetivorans (5CM0) | 91 | 1.19 | 38 |
| Branched-chain amino acid aminotransferase from Thermus thermophilus (1WRV) | 87 | 1.51 | 29 |
| Branched-chain amino acid transaminases from Pseudomonas aeruginosa (6NST) | 83 | 1.58 | 37 |
| Branched-chain amino acid transaminases from Escherichia coli (1I1K, 1IYE) | 83 | 1.57 | 26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyko, K.M.; Matyuta, I.O.; Nikolaeva, A.Y.; Rakitina, T.V.; Popov, V.O.; Bezsudnova, E.Y.; Khrenova, M.G. A Puzzling Protein from Variovorax paradoxus Has a PLP Fold Type IV Transaminase Structure and Binds PLP without Catalytic Lysine. Crystals 2022, 12, 619. https://doi.org/10.3390/cryst12050619
Boyko KM, Matyuta IO, Nikolaeva AY, Rakitina TV, Popov VO, Bezsudnova EY, Khrenova MG. A Puzzling Protein from Variovorax paradoxus Has a PLP Fold Type IV Transaminase Structure and Binds PLP without Catalytic Lysine. Crystals. 2022; 12(5):619. https://doi.org/10.3390/cryst12050619
Chicago/Turabian StyleBoyko, Konstantin M., Ilya O. Matyuta, Alena Y. Nikolaeva, Tatiana V. Rakitina, Vladimir O. Popov, Ekaterina Yu. Bezsudnova, and Maria G. Khrenova. 2022. "A Puzzling Protein from Variovorax paradoxus Has a PLP Fold Type IV Transaminase Structure and Binds PLP without Catalytic Lysine" Crystals 12, no. 5: 619. https://doi.org/10.3390/cryst12050619
APA StyleBoyko, K. M., Matyuta, I. O., Nikolaeva, A. Y., Rakitina, T. V., Popov, V. O., Bezsudnova, E. Y., & Khrenova, M. G. (2022). A Puzzling Protein from Variovorax paradoxus Has a PLP Fold Type IV Transaminase Structure and Binds PLP without Catalytic Lysine. Crystals, 12(5), 619. https://doi.org/10.3390/cryst12050619