
Weakly Bound Dimer of a Diaryloxygermylene Derived from a tBuPh2Si-Substituted 2,2′-Methylenediphenol


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Procedures
2.2. Synthesis of 2-(tert-Butyldiphenylsilyl)-4-methylphenol (1a)
2.3. Synthesis of 2-(Methyldiphenylsilyl)-4-methylphenol (1b)
2.4. Synthesis of 2,2′-Methylenebis{6-(tert-butyldiphenylsilyl)-4-methylphenol} (2a)
2.5. Synthesis of 2,2′-Methylenebis{6-(methyldiphenylsilyl)-4-methylphenol} (2b)
2.6. Synthesis of Diaryloxygermylene (3a)
2.7. Synthesis of Diaryloxygermylene Dimer (3b)
2.8. Single-Crystal XRD Analysis
3. Results and Discussion
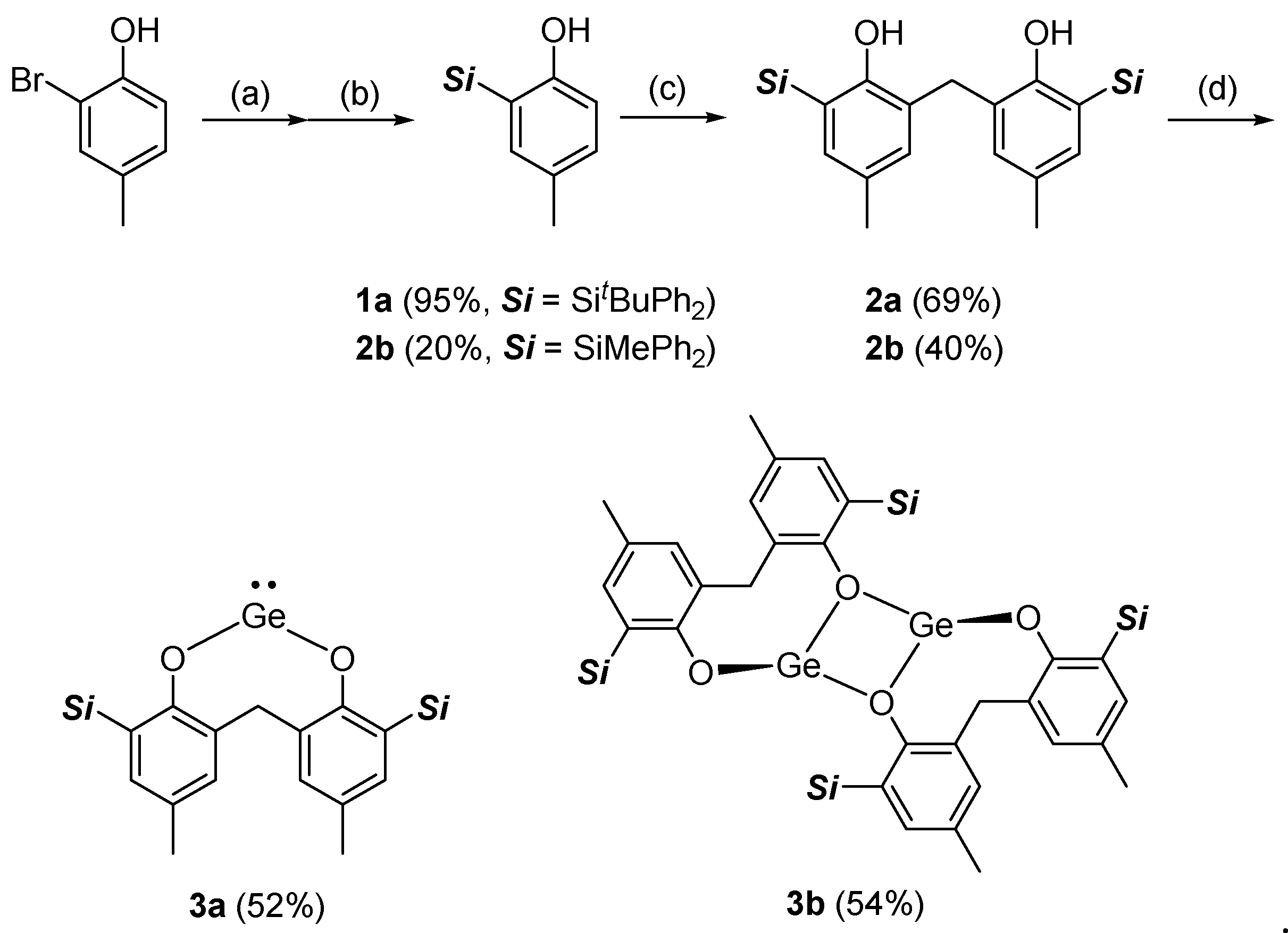
3.1. Synthesis
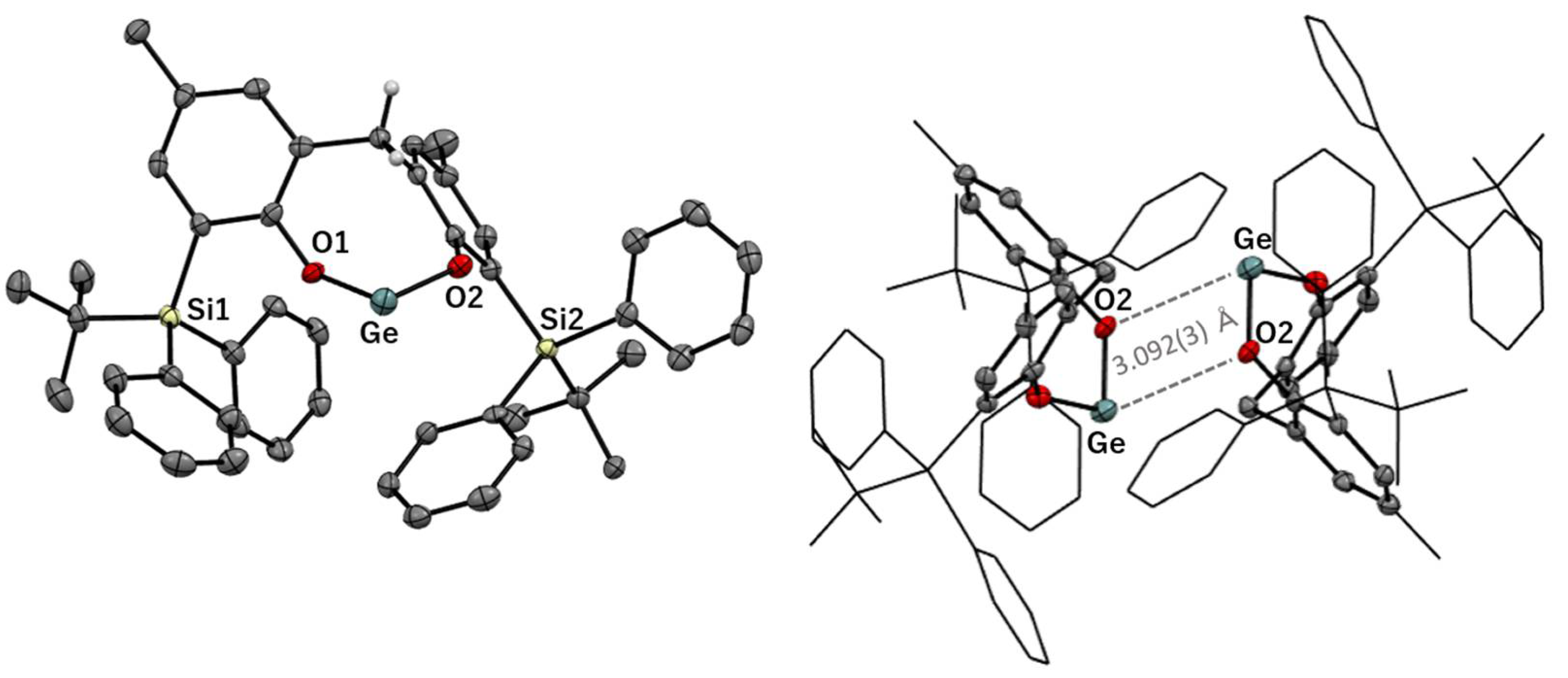
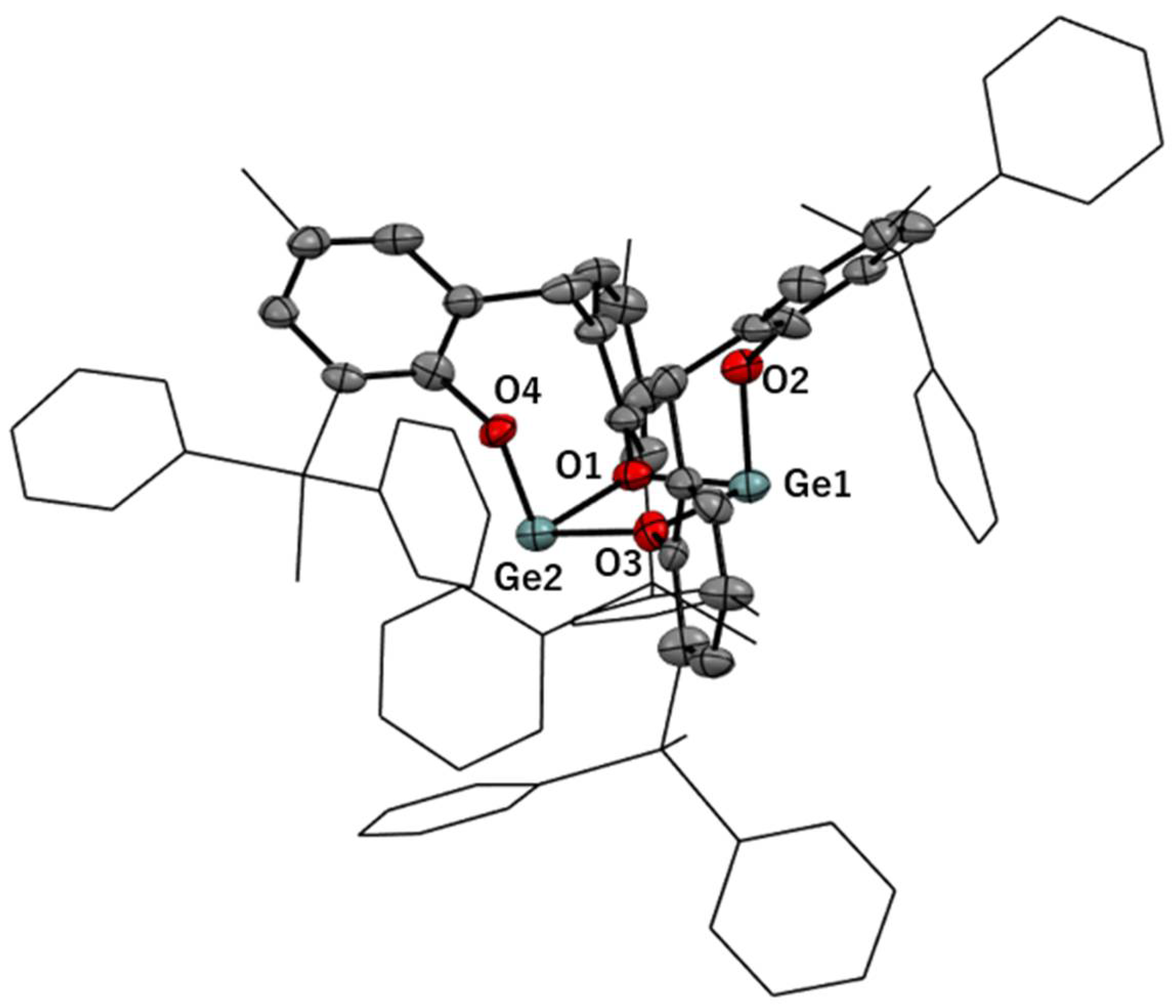
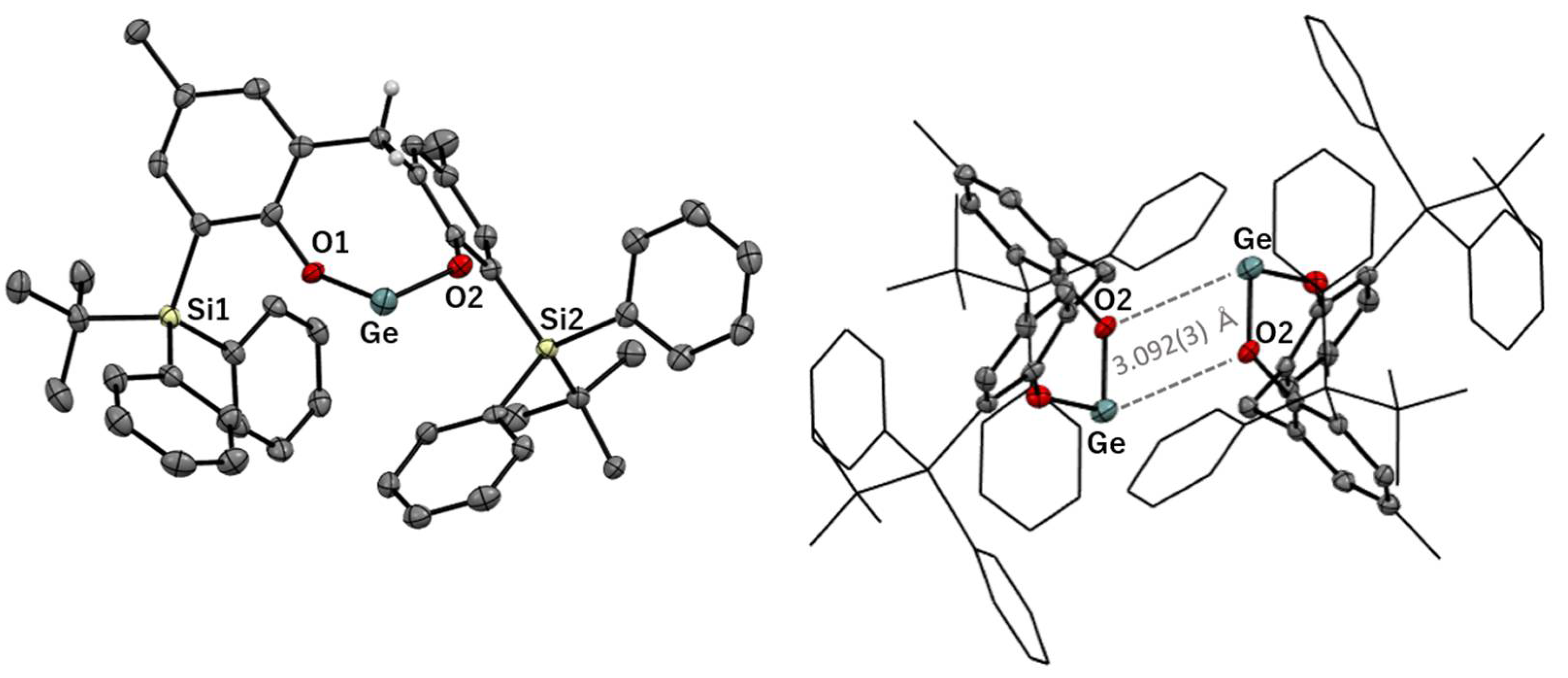
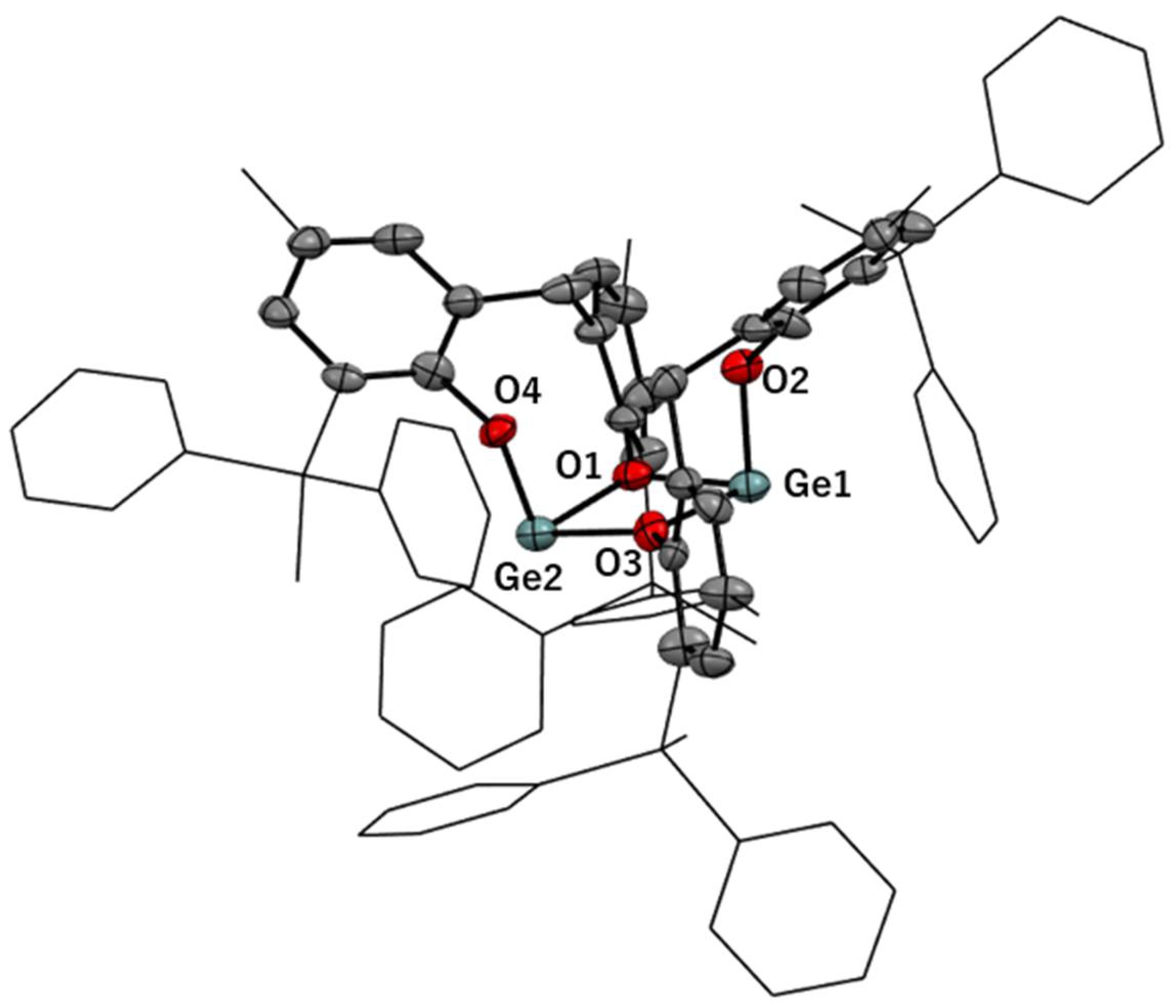
3.2. Solid-State Structures
3.3. NMR Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References and Note
- Mizuhata, Y.; Sasamori, T.; Tokitoh, N. Stable Heavier Carbene Analogues. Chem. Rev. 2009, 109, 3479–3511. [Google Scholar] [CrossRef]
- Power, P.P. Main-group elements as transition metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef]
- Hadlington, T.J.; Driess, M.; Jones, C. Low-valent group 14 element hydride chemistry: Towards catalysis. Chem. Soc. Rev. 2018, 47, 4176–4197. [Google Scholar] [CrossRef] [Green Version]
- Shan, C.; Yao, S.; Driess, M. Where silylene–silicon centres matter in the activation of small molecules. Chem. Soc. Rev. 2020, 49, 6733–6754. [Google Scholar] [CrossRef]
- Davidson, P.J.; Lappert, M.F. Stabilisation of metals in a low co-ordinative environment using the bis(trimethylsilyl)methyl ligand; coloured Sn and Pb alkyls, M[CH(SiMe3)2]2. J. Chem. Soc. Chem. Commun. 1973, 317a. [Google Scholar] [CrossRef]
- Harris, D.H.; Lappert, M.F. Monomeric, volatile bivalent amides of group IV elements, M(NR12)2 and M(NR1R2)2(M = Ge, Sn, or Pb; R1 = Me3Si, R2=Me3C). J. Chem. Soc. Chem. Commun. 1974, 895–896. [Google Scholar] [CrossRef]
- McCrea-Hendrick, M.L.; Bursch, M.; Gullett, K.L.; Maurer, L.R.; Fettinger, J.C.; Grimme, S.; Power, P.P. Counterintuitive Interligand Angles in the Diaryls E{C6H3-2,6-(C6H2-2,4,6-iPr3)2}2 (E = Ge, Sn, or Pb) and Related Species: The Role of London Dispersion Forces. Organometallics 2018, 37, 2075–2085. [Google Scholar] [CrossRef]
- Perla, L.G.; Kulenkampff, J.M.; Fettinger, J.C.; Power, P.P. Steric and Electronic Properties of the Bulky Terphenyl Ligand ArtBu6 (ArtBu6 = C6H3-2,6-(C6H2-2,4,6-tBu3)2) and Synthesis of Its Tin Derivatives ArtBu6SnCl, ArtBu6SnSn(H)2ArtBu6, and ArtBu6SnSnArtBu6: A New Route to a Distannyne via Thermolysis of the Asymmetric Hydride ArtBu6SnSn(H)2ArtBu6. Organometallics 2018, 37, 4048–4054. [Google Scholar]
- Matsuo, T.; Tamao, K. Fused-Ring Bulky “Rind” Groups Producing New Possibilities in Elemento-Organic Chemistry. Bull. Chem. Soc. Jpn. 2015, 88, 1201–1220. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, F.; Nishino, R.; Yukimoto, M.; Sugamata, K.; Minoura, M. Synthesis, Structure, and Reactivity of a Thermally Stable Dialkylgermylene. Bull. Chem. Soc. Jpn. 2020, 93, 249–251. [Google Scholar] [CrossRef]
- Protchenko, A.V.; Birjkumar, K.H.; Dange, D.; Schwarz, A.D.; Vidovic, D.; Jones, C.; Kaltsoyannis, N.; Mountford, P.; Aldridge, S. A Stable Two-Coordinate Acyclic Silylene. J. Am. Chem. Soc. 2012, 134, 6500–6503. [Google Scholar] [CrossRef]
- Protchenko, A.V.; Dange, D.; Schwarz, A.D.; Tang, C.Y.; Phillips, N.; Mountford, P.; Jones, C.; Aldridge, S. Heavy metal boryl chemistry: Complexes of cadmium, mercury and lead. Chem. Commun. 2014, 50, 3841–3844. [Google Scholar] [CrossRef]
- Weinert, C.S.; Fenwick, A.E.; Fanwick, P.E.; Rothwell, I.P. Synthesis, structures and reactivity of novel germanium(ii) aryloxide and arylsulfide complexes. Dalton Trans. 2003, 532–539. [Google Scholar] [CrossRef]
- Goel, S.C.; Chiang, M.Y.; Buhro, W.E. Preparation of six lead(II) dialkoxides, X-ray crystal structures of [Pb(μ,η1-OCH2CH2OMe)2]∞ and [Pb3(μ-O-tert-Bu)6], and hydrolysis studies. Inorg. Chem. 1990, 29, 4640–4646. [Google Scholar] [CrossRef]
- Kitschke, P.; Rüffer, T.; Korb, M.; Lang, H.; Schneider, W.B.; Auer, A.A.; Mehring, M. Intramolecular C–O Insertion of a Germanium(II) Salicyl Alcoholate: A Combined Experimental and Theoretical Study. Eur. J. Inorg. Chem. 2015, 2015, 5467–5479. [Google Scholar] [CrossRef]
- Kitschke, P.; Walter, M.; Rüffer, T.; Seifert, A.; Speck, F.; Seyller, T.; Spange, S.; Lang, H.; Auer, A.A.; Kovalenko, M.V.; et al. Porous Ge@C materials via twin polymerization of germanium(ii) salicyl alcoholates for Li-ion batteries. J. Mater. Chem. A 2016, 4, 2705–2719. [Google Scholar] [CrossRef] [Green Version]
- Boyle, T.J.; Doan, T.Q.; Steele, L.A.M.; Apblett, C.; Hoppe, S.M.; Hawthorne, K.; Kalinich, R.M.; Sigmund, W.M. Tin(ii) amide/alkoxide coordination compounds for production of Sn-based nanowires for lithium ion battery anode materials. Dalton Trans. 2012, 41, 9349–9364. [Google Scholar] [CrossRef]
- Stanciu, C.; Richards, A.F.; Stender, M.; Olmstead, M.M.; Power, P.P. New terphenylphenoxides of group 13 and 14 elements. Polyhedron 2006, 25, 477–483. [Google Scholar] [CrossRef]
- Dickie, D.A.; MacIntosh, I.S.; Ino, D.D.; He, Q.; Labeodan, O.A.; Jennings, M.C.; Schatte, G.; Walsby, C.J.; Clyburne, J.A.C. Synthesis of the bulky m-terphenyl phenol Ar*OH (Ar* = C6H3-2,6-Mes2, Mes=2,4,6-trimethylphenyl) and the preparation and structural characterization of several of its metal complexes. Can. J. Chem. 2008, 86, 20–31. [Google Scholar] [CrossRef]
- Rekken, B.D.; Brown, T.M.; Olmstead, M.M.; Fettinger, J.C.; Power, P.P. Stable Plumbylene Dichalcogenolate Monomers with Large Differences in Their Interligand Angles and the Synthesis and Characterization of a Monothiolato Pb(II) Bromide and Lithium Trithiolato Plumbate. Inorg. Chem. 2013, 52, 3054–3062. [Google Scholar] [CrossRef]
- Rekken, B.D.; Brown, T.M.; Fettinger, J.C.; Lips, F.; Tuononen, H.M.; Herber, R.H.; Power, P.P. Dispersion Forces and Counterintuitive Steric Effects in Main Group Molecules: Heavier Group 14 (Si–Pb) Dichalcogenolate Carbene Analogues with Sub-90° Interligand Bond Angles. J. Am. Chem. Soc. 2013, 135, 10134–10148. [Google Scholar] [CrossRef]
- Cetinkaya, B.; Gumrukcu, I.; Lappert, M.F.; Atwood, J.L.; Rogers, R.D.; Zaworotko, M.J. Bivalent germanium, tin, and lead 2,6-di-tert-butylphenoxides and the crystal and molecular structures of M(OC6H2Me-4-But2-2,6)2 (M = Ge or Sn). J. Am. Chem. Soc. 1980, 102, 2088–2089. [Google Scholar] [CrossRef]
- Gerung, H.; Boyle, T.J.; Tribby, L.J.; Bunge, S.D.; Brinker, C.J.; Han, S.M. Solution Synthesis of Germanium Nanowires Using a Ge2+ Alkoxide Precursor. J. Am. Chem. Soc. 2006, 128, 5244–5250. [Google Scholar] [CrossRef] [Green Version]
- Weinert, C.S.; Fanwick, P.E.; Rothwell, I.P. Novel germanium(ii) binaphthoxide complexes: Synthesis and crystal structure of (R,R)-[Ge{OC20H10(OSiMe3)-2′-(SiMe3)2-3,3′}2] and (R)-[Ge{O2C20H10(SiMe2Ph)2-3,3′}{NH3}]; catalytic function of Ge[N(SiMe3)2]2 for the mono-silylation of 3,3′-disubstituted-1,1′-bi-2,2′-naphthols. J. Chem. Soc. Dalton Trans. 2002, 2948–2950. [Google Scholar] [CrossRef]
- Someşan, A.-A.; Le Coz, E.; Roisnel, T.; Silvestru, C.; Sarazin, Y. Stable lead(ii) boroxides. Chem. Commun. 2018, 54, 5299–5302. [Google Scholar] [CrossRef]
- Loh, Y.K.; Ying, L.; Fuentes, M.Á.; Do, D.C.H.; Aldridge, S. An N-Heterocyclic Boryloxy Ligand Isoelectronic with N-Heterocyclic Imines: Access to an Acyclic Dioxysilylene and its Heavier Congeners. Angew. Chem., Int. Ed. 2019, 58, 4847–4851. [Google Scholar] [CrossRef] [PubMed]
- Kuriki, R.; Kuwabara, T.; Ishii, Y. Synthesis and structures of diaryloxystannylenes and -plumbylenes embedded in 1,3-diethers of thiacalix [4]arene. Dalton Trans. 2020, 49, 12234–12241. [Google Scholar] [CrossRef] [PubMed]
- Hascall, T.; Parkin, G.; Hascall, T.; Rheingold, A.L.; Guzei, I. Subvalent germanium and tin complexes supported by a dianionic calixarene ligand: Structural characterization of exo and endo isomers of [Butcalix(TMS)2]Ge. Chem. Commun. 1998, 101–102. [Google Scholar] [CrossRef]
- Hascall, T.; Pang, K.; Parkin, G. exo and endo Isomerism of subvalent tin and germanium complexes derived from 1,3-diethers of p-tert-butylcalix [4]arene. Tetrahedron 2007, 63, 10826–10833. [Google Scholar] [CrossRef]
- McBurnett, B.G.; Cowley, A.H. Binuclear tin and germanium calix [4]arenes. Chem. Commun. 1999, 17–18. [Google Scholar] [CrossRef]
- Wetherby, A.E.; Goeller, L.R.; DiPasquale, A.G.; Rheingold, A.L.; Weinert, C.S. Synthesis and Structures of an Unusual Germanium(II) Calix [4]arene Complex and the First Germanium(II) Calix [8]arene Complex and Their Reactivity with Diiron Nonacarbonyl. Inorg. Chem. 2007, 46, 7579–7586. [Google Scholar] [CrossRef] [PubMed]
- Schrick, A.C.; Rheingold, A.L.; Weinert, C.S. A divalent germanium complex of calix [5]arene. Dalton Trans. 2011, 40, 6629–6631. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.J.; Balsells, J.; Carroll, P.J.; Walsh, P.J. Optimization of Asymmetric Catalysts Using Achiral Ligands: Metal Geometry-Induced Ligand Asymmetry. Org. Lett. 2001, 3, 2161–2164. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H.; Matsuo, T. Aryloxide-based multidentate ligands for early transition metals and f-element metals. J. Organomet. Chem. 2004, 689, 4228–4243. [Google Scholar] [CrossRef]
- Watanabe, T.; Ishida, Y.; Matsuo, T.; Kawaguchi, H. Reductive Coupling of Six Carbon Monoxides by a Ditantalum Hydride Complex. J. Am. Chem. Soc. 2009, 131, 3474–3475. [Google Scholar] [CrossRef]
- Buccella, D.; Tanski, J.M.; Parkin, G. Factors Influencing Coordination versus Oxidative Addition of C−H Bonds to Molybdenum and Tungsten: Structural and Spectroscopic Evidence That the Calixarene Framework Promotes C−H Bond Activation. Organometallics 2007, 26, 3275–3278. [Google Scholar] [CrossRef]
- Ishida, Y.; Kawaguchi, H. Methylene-Linked Anilide—Bis(aryloxide) Ligands: Lithium, Sodium, Potassium, Chromium(III), and Vanadium(III) Ligation. Inorg. Chem. 2014, 53, 6775–6787. [Google Scholar] [CrossRef]
- Kuwabara, T.; Toriumi, T.; Suzuki, M.; Ishii, Y. Selective Double CH Activation at a Methylene Carbon in Methylenediphenol Derivatives to Generate Carbene-Bridged Dinuclear Iridium Complexes. Organometallics 2020, 39, 4500–4509. [Google Scholar] [CrossRef]
- Jacobson, R.A. Private Communication to Rigaku Corp.; Rigaku Corp.: Tokyo, Japan, 1998. [Google Scholar]
- Crystal Structure 4.0: Single Crystal Structure Analysis Package; Rigaku Corp.: Tokyo, Japan, 2000–2010.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment-Olex2 dissected. Acta Crystallogr. Sec. A 2015, 71, 59–75. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sec. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Simchen, G.; Pfletschinger, J. Anionic O → C Rearrangements in Trialkylsiloxy-benzenes and Trialkylsiloxypyridines. Angew. Chem. Int. Ed. Engl. 1976, 15, 428–429. [Google Scholar] [CrossRef]
- Ikawa, T.; Nishiyama, T.; Nosaki, T.; Takagi, A.; Akai, S. A Domino Process for Benzyne Preparation: Dual Activation of o-(Trimethylsilyl)phenols by Nonafluorobutanesulfonyl Fluoride. Org. Lett. 2011, 13, 1730–1733. [Google Scholar] [CrossRef]
- Casiraghi, G.; Casnati, G.; Cornia, M.; Pochini, A.; Puglia, G.; Sartori, G.; Ungaro, R. Selective reactions using metal phenoxides. Part 1. Reactions with formaldehyde. J. Chem. Soc. Perkin Trans. 1 1978, 318–321. [Google Scholar] [CrossRef]
- Fjeldberg, T.; Hitchcock, P.B.; Lappert, M.F.; Smith, S.J.; Thorne, A.J. Chemistry of bulky alkoxides of bivalent germanium and tin; structures of gaseous [Sn(OBut)2]2 and crystalline Ge(OCBut3)2. J. Chem. Soc. Chem. Commun. 1985, 939–941. [Google Scholar] [CrossRef]
- Boyle, T.J.; Tribby, L.J.; Ottley, L.A.M.; Han, S.M. Synthesis and Characterization of Germanium Coordination Compounds for Production of Germanium Nanomaterials. Eur. J. Inorg. Chem. 2009, 2009, 5550–5560. [Google Scholar] [CrossRef] [Green Version]
- Khoury, N.; Pastor, S.D.; Rahni, D.; Richardson, C.F.; Syed, N.A.; Shum, S.P.; Chandrasekaran, A. The Conformation of Sterically Congested Eight-membereed Rings Containing Germanium: First X-ray Crystallographic Characterization of the Boat Conformation. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 483–497. [Google Scholar] [CrossRef]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef] [Green Version]
- Veith, M.; Belot, C.; Huch, V.; Zimmer, M. Influence of the Solvent on the Formation of New Tin(II) Methoxides Containing Thienyl Substituents: Crystal Structure and NMR Investigations. Z. Anorg. Allg. Chem. 2009, 635, 942–948. [Google Scholar] [CrossRef]
- Hill, M.S.; Johnson, A.L.; Lowe, J.P.; Molloy, K.C.; Parish, J.D.; Wildsmith, T.; Kingsley, A.L. Aerosol-assisted CVD of SnO from stannous alkoxide precursors. Dalton Trans. 2016, 45, 18252–18258. [Google Scholar] [CrossRef]
- Desiraju, G.R. The C-H···O hydrogen bond in crystals: What is it? Acc. Chem. Res. 1991, 24, 290–296. [Google Scholar] [CrossRef]
- Steiner, T. Unrolling the hydrogen bond properties of C–H···O interactions. Chem. Commun. 1997, 727–734. [Google Scholar] [CrossRef]
- Steiner, T. C–H···O hydrogen bonding in crystals. Crystallogr. Rev. 2003, 9, 177–228. [Google Scholar] [CrossRef]
- Ash, E.L.; Sudmeier, J.L.; Day, R.M.; Vincent, M.; Torchilin, E.V.; Haddad, K.C.; Bradshaw, E.M.; Sanford, D.G.; Bachovchin, W.W. Unusual 1H NMR chemical shifts support (His) Cε1–H···O=C H-bond: Proposal for reaction-driven ring flip mechanism in serine protease catalysis. Proc. Natl. Acad. Sci. USA 2000, 97, 10371–10376. [Google Scholar] [CrossRef] [Green Version]
- Vibhute, A.M.; Deva Priyakumar, U.; Ravi, A.; Sureshan, K.M. Model molecules to classify CH···O hydrogen-bonds. Chem. Commun. 2018, 54, 4629–4632. [Google Scholar] [CrossRef]
- Since such downfield shifts have also been observed for a related tin compound having SiiPr3 substituents instead of SiMePh2 and SitBuPh2, we exclude the possibility that the downfield shifts are caused by ring currents of the Ph rings.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamazaki, R.; Kuriki, R.; Sugihara, A.; Ishii, Y.; Kuwabara, T. Weakly Bound Dimer of a Diaryloxygermylene Derived from a tBuPh2Si-Substituted 2,2′-Methylenediphenol. Crystals 2022, 12, 605. https://doi.org/10.3390/cryst12050605
Yamazaki R, Kuriki R, Sugihara A, Ishii Y, Kuwabara T. Weakly Bound Dimer of a Diaryloxygermylene Derived from a tBuPh2Si-Substituted 2,2′-Methylenediphenol. Crystals. 2022; 12(5):605. https://doi.org/10.3390/cryst12050605
Chicago/Turabian StyleYamazaki, Ryo, Ryunosuke Kuriki, Asuka Sugihara, Youichi Ishii, and Takuya Kuwabara. 2022. "Weakly Bound Dimer of a Diaryloxygermylene Derived from a tBuPh2Si-Substituted 2,2′-Methylenediphenol" Crystals 12, no. 5: 605. https://doi.org/10.3390/cryst12050605
APA StyleYamazaki, R., Kuriki, R., Sugihara, A., Ishii, Y., & Kuwabara, T. (2022). Weakly Bound Dimer of a Diaryloxygermylene Derived from a tBuPh2Si-Substituted 2,2′-Methylenediphenol. Crystals, 12(5), 605. https://doi.org/10.3390/cryst12050605