Abstract
Novel synthesis of 2,5-bis[(1,3-thiazol-2-ylmethyl)amino]methyl-1,3,4-oxadiazole (LThiazole) is reported, along with the preparation of three new dinuclear Fe(II) complexes with different counterions [FeII2(μ2-LThiazole)2](BF4)4·2CH3CN (1), [FeII2(μ2-LThiazole)2](ClO4)4 (2) and [FeII2(μ2-LThiazole)2](CF3SO3)4·2CH3CN (3). The obtained complexes were characterized by single-crystal X-ray crystallography, SQUID magnetometry and IR spectroscopy. The structure of the crystalline material was determined at 120 K for 1 and 3. The magnetic properties of all three complexes (1–3) were measured between 2–300 K and clearly show that all three complexes stay in the high-spin state over the measured temperature range.
1. Introduction
The chemistry of first-row transition metals is fascinating because of their plethora of very interesting properties. The most interesting of all is possessing partially populated d-orbitals, which lead to their different oxidation and spin states [1,2]. The ambiguity around the spin states of the metals is not as thoroughly understood as the oxidation state. In theory, controlling the spin state of the transition metal is straightforward through ligand design. Molecular complexes containing d4 to d7 electronic configuration can have two different spin states which can be switched by applying an external stimulus. This is still fascinating and has potential for applications in sensors, displays and data storage devices [3,4,5,6,7]. In addition to this, spin state plays a very important role in metalloenzymatic reactions, biological activities and in catalysis involving organometallic compounds where different reactions take place via different spin states [8,9,10,11]. The phenomenon by which the compounds can switch their spin state from low spin (LS) to high spin (HS) with an external stimulus such as heat [12,13], pressure [14,15,16], light [17,18] or magnetic field [19] is called spin crossover (SCO). This phenomenon was first discovered in Fe(III) [20,21] but more commonly studied in Fe(II) [22,23,24,25] because the spin-state transition is between a diamagnetic low-spin (LS) state (S = 0) to a paramagnetic high-spin (HS) state (S = 2). This phenomenon is also rarely studied in Co(II) [26,27] and more rarely studied in Mn(III) [28,29,30,31] and Cr(II) [32,33,34]. In the case of the Fe(II) SCO complexes, the N-6 ligand donor set is commonly preferred [35]. The SCO phenomenon in solid state is sensitive to the effect of crystal packing [36,37,38], choice of counterion [39,40], presence of lattice solvents [41,42,43], and the choice of coligands [44,45]. In this quest of finding the right ligand field strength for the central metal ion to exhibit bistability of its spin state, various ligands have been explored and the ligands were designed to fine-tune the switching temperature.
Complexes based on 1,3,4 oxadiazole are seldom studied in SCO literature. Our recent advances have shown that the ligand design using (PMOD = 2,5-bis{[(2-pyridyl-methyl)amino]methyl}-1,3,4-oxadiazole) [46] together with Fe(II) leads to the formation of [Fe2(μ2-PMOD)2](X)4-type complexes where X = BF4, ClO4 and CF3SO3 [46]. The complex with the BF4− counterion stays in HS over the temperature range measured, while the complexes with the ClO4− and CF3SO3− show SCO behavior with a T1/2 = 150 K [46]. So far, to our best of our knowledge these are the only two 1,3,4-oxadiazole complexes reported that show SCO behavior. To explore this further, the ligand designed is altered by changing the axially coordinated six-membered pyridine rings with a five-membered thiazole ring with a sulfur as a heteroatom, to learn its influences on the SCO behavior of the dimeric Fe(II) complexes, which from the literature is known to reduce the ligand field. It is worthy to note that Fe(II) dimeric complexes with a very similar ligand based on 1,2,4-thiadiazole, reported earlier by some of us, was also SCO active [47]. Since the SCO phenomenon in solid state is highly dependent on the counterion, three different commonly used counterions (BF4−, ClO4− and CF3SO3−) have been selected and the results are shown here.
2. Materials and Methods
All chemicals were used without further purification and were purchased from Alfa Aesar, Acros Organics and Fisher Chemicals. The C, H and N elemental analyses were carried out on a Foss Heraeus Vario EL (Elementar Analysensysteme GmbH, Langenselbold, Germany) at the microanalytical laboratories of the department of chemistry at Johannes Gutenberg University Mainz. The infrared absorption spectra were collected at room temperature in a range of 3000–450 cm−1 on a Thermo Fisher NICOLET Nexus FT/IR-5700 spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) equipped with Smart Orbit ATR Diamond cell (Thermo Fisher Scientific, Waltham, MA, USA). X-ray diffraction data for the structure analysis were collected from suitable single crystals at 120 K with a STOE IPDS 2T at Johannes Gutenberg University Mainz. The structures were solved with ShelXT [48,49,50] and refined with ShelXL [49,50,51] implemented in the program Olex2 [52]. Magnetic data of a microcrystalline sample were collected using a Quantum Design SQUID magnetometer MPMSXL in a temperature range between 2–300 K with an applied field of 1 kOe (0.1 T). The magnetic contribution from the holder was experimentally determined and subtracted from the measured susceptibility data. The contribution from underlying diamagnetism was corrected using the estimation in equation χdia = kM∙10−6 cm3 mol−1 [53]. In this equation, M is the molecular weight of the compound and k is equal to 0.5.
2.1. Ligand Synthesis
- 1,2-Bis(Chloroacetyl)hydrazide
This was prepared by using a modified procedure from the literature [46]. Hydrazine hydrate (25.03 g, 0.50 mol, 1 eq) was dissolved in 200 mL diethyl ether. To this solution, chloroacetyl chloride (56.47 g, 0.50 mol, 1 eq), which was dissolved in 150 mL diethyl ether, was added under cooling and vigorous stirring, such that the temperature did not exceed 35 °C. A colorless suspension formed, which was stirred for 30 min after the complete addition of the chloroacetyl chloride, then 100 mL of deionized water was added followed by the slow addition of sodium carbonate (53.10 g, 0.50 mol, 1 eq). Afterwards, another equivalent of chloroacetyl chloride (56.47 g, 0.50 mol, 1 eq), again dissolved in 150 mL diethyl ether, was slowly added. After stirring at room temperature for 12 h, the colorless solid was filtered, washed with cold diethyl ether and recrystallized in ethyl acetate. 1,2-Bis(Chloroacetyl)hydrazide was isolated as a colorless powder in moderate yields (45.13 g, 0.24 mol, 48%). 1H-NMR (400 MHz, DMSO, δ (ppm)): 10.47 (s, 2H, NH), 4.15 (s, 4H, CH2). 13C-NMR (101 MHz, DMSO, δ (ppm)): 165.09 (O=C), 41.25 (CH2). FT-IR: (cm−1) = 3176, 3034, 2979, 2956, 2891, 1701, 1606, 1491, 1437, 1405, 1306, 1252, 1222, 1112, 1007, 934, 918, 792, 713, 639, 577, 538, 490, 460. EA (%): Calc. (found) C 25.97 (25.90), H 3.27 (3.01), N 15.14 (15.43).
- 2,5-Bis(chloromethyl)-1,3,4-oxadiazole
This was prepared by using a modified procedure from the literature [46]. Perchloric acid (60%, 8.75 g) and chloroacetyl chloride (30.00 g, 270.25 mmol, 5 eq) were added and heated at a temperature of 60 °C until a uniform phase was formed. 1,2-Bis(Chloroacetyl)hydrazide (10.00 g, 54.05 mmol, 1 eq) was then added in small portions with stirring; when it dissolved completely, the reaction mixture was allowed to stir vigorously for 24 h. The yellow solution was then added to a 150 mL ice-water mixture and the pH was adjusted to 9.0 with a saturated sodium-carbonate solution. The reaction mixture was extracted three times with 100 mL of diethyl ether. The combined extracts were washed with a 10% sodium-carbonate solution, dried over sodium sulfate and taken to dryness under reduced pressure, yielding a bright yellow oil. 2,5-Bis(chloromethyl)-1,3,4-oxadiazole was isolated from the crude product by column chromatography (SiO2, cyclohexane/ethyl acetate (3:1), Rf = 0.3) as a colorless oil (8.38 g, 50.18 mmol, 93%). 1H-NMR (400 MHz, CDCl3, δ (ppm)): 4.68 (s, 4H, CH2). 13C-NMR (101 MHz, CDCl3, δ (ppm)): 163.70 (CODA), 32.75 (CH2). FT-IR: (cm−1) = 3029 (w), 2969 (w), 1582 (s), 1565 (s), 1427 (s), 1382 (s), 1269 (m), 1233 (s), 1154 (m), 1038 (m), 989 (s), 968 (m), 918 (w), 886 (w), 780 (vs), 760 (vs), 719 (s), 654 (vs), 616 (s). EA (%): Calc. (found) C 28.77 (28.70), H 2.41 (2.79), N 16.78 (16.76).
- 2,5-Bis(azidomethyl)-1,3,4-oxadiazole
2,5-Bis(chloromethyl)-1,3,4-oxadiazole (19.68 g, 118.00 mmol, 1 eq), sodium azide (41.49 g, 638.00 mmol, 5.4 eq), and 18-crown-6 (2.81 g, 11.00 mmol, 0.09 eq) were suspended in 200 mL of acetone and stirred at room temperature for 18 h. The suspension was filtered and the solvent removed under reduced pressure, yielding a yellowish crude product. 2,5-Bis(azidomethyl)-1,3,4-oxadiazole was isolated as a colorless oil by column chromatography (SiO2, dichloromethane/diethyl ether (4:1), Rf = 0.6) in good yield (18.42 g, 102.26 mmol, 87%). 1H-NMR (400 MHz, CDCl3, δ (ppm)): 4.75 (s, 4H, CH2). 13C-NMR (101 MHz, CDCl3, δ (ppm)): 162.95 (CODA), 44.18 (CH2). FT-IR: (cm−1) = 3004 (w), 2941 (w), 2858 (vw), 2180 (sh, s), 2093 (vs), 1585 (m), 1566 (m), 1435 (m), 1394 (m), 1333 (m), 1257 (s), 1181 (s), 1043 (w), 980 (m), 944 (m), 889 (m), 789 (m), 687 (m), 649 (m), 555 (m). EA (%): Calc. (found) C 26.67 (26.71), H 2.24 (2.23), N 62.21 (61.82).
- 2,5-Bis(aminomethyl)-1,3,4-oxadiazole
2,5-Bis(azidomethyl)-1,3,4-oxadiazole (5.00 g, 27.76 mmol, 1 eq) was dissolved in 250 mL tetrahydrofuran and triphenylphosphine (17.47 g, 66.62 mmol, 2.4 eq) was added in portions while stirring. After completing addition and gas evolution, 17 mL of deionized water was added to the solution. The reaction was stirred at room temperature for 18 h. The solvent was then removed under reduced pressure to yield a yellow solid as crude product. 2,5-Bis(aminomethyl)-1,3,4-oxadiazole was isolated as a brown solid in yields (3.44 g, 26.85 mmol, 97%) by column chromatography with discontinuous gradient elution (SiO2, chloroform/methanol (4:1) → chloroform/methanol/triethylamine (80:20:1) → chloroform/methanol/triethylamine (67:33:1)). 1H-NMR (400 MHz, CDCl3, δ (ppm)): 4.05 (s, 4H, CH2), 1.64 (s, 4H, NH2). 13C-NMR (101 MHz, MeOD, δ (ppm)): 167.57 (CODA), 35.90 (CH2). FT-IR: (cm−1)= 3373 (s), 3304 (s), 3067 (m), 2967 (m), 2927 (m), 2848 (m), 2555 (w), 1663 (s), 1606 (s), 1572 (vs), 1550 (vs), 1438 (s), 1374 (m), 1339 (s), 1219 (m), 1176 (s), 1094 (s), 1005 (w), 980 (s), 960 (m), 899 (s), 871 (vs), 772 (m), 697 (w), 661 (w), 625 (w), 497 (w), 486 (w), 440 (w). EA (%): Calc. (found) C 37.49 (37.05), H 6.29 (7.21), N 43.73 (42.12).
- 2,5-Bis[(1,3-thiazol-2-ylmethyl)amino]methyl-1,3,4-oxadiazole (LThiazole)
To a solution of 2,5-Bis(aminomethyl)-1,3,4-oxadiazole (2.52 g, 19.7 mmol, 1 eq), thiazole-2-carbaldehyde (4.70 g, 41.55 mmol, 2.1 eq) and acetic acid (2.37 mL, 2.49 g, 41.55 mmol, 2.1 eq) in 1000 mL methanol was added sodium cyanoborohydride (6.19 g, 98.50 mmol, 5 eq) slowly in portions. Afterwards, the reaction mixture was refluxed for 24 h. The solvent was removed under reduced pressure, yielding a brown oil as crude product. LThiazole was isolated as brown oil in moderate yields (3.82 g, 11.84 mmol, 60%) by column chromatography (SiO2, chloroform/methanol (19:1)). 1H-NMR (400 MHz, CDCl3, COSY, δ (ppm)): 7.73 (d, 2H, HC=CH), 7.29 (d, 2H, HC=CH), 4.23 (s, 4H, TA-CH2), 4.12 (s, 4H, ODA-CH2). 13C-NMR (101 MHz, CDCl3, HSQC, HMBC, δ (ppm)): 169.84, 165.39, 142.6, 119.30, 49.97, 43.06. FT-IR: (cm−1) = 3277, 3109, 3079, 2913, 2838, 1670, 1587, 1502, 1468, 1427, 1337, 1315, 1238, 1185, 1138, 1054, 979, 875, 804, 771, 731, 677, 601, 499. FD-MS: m/z = 323.08 (100.00%), 324.08 (15.63%), 325.07 (9.37%).
2.2. Complex Synthesis
- [Fe2(μ2-LThiazole)2](BF4)4·2CH3CN
LThiazole (32 mg, 0.1 mmol, 1 eq) was dissolved in 2 mL of acetonitrile. To this solution, Fe(BF4)2·6H2O (34 mg, 0.1 mmol, 1 eq), dissolved in 2 mL acetonitrile, was added dropwise and stirred for an hour. The reaction mixture was set for vapor diffusion against diethyl ether and kept undisturbed at room temperature. After three days, colorless block-shaped crystals suitable for X-ray structure analysis were obtained. The crystals were collected by filtration in moderate yields (23.79 mg, 0.022 mmol, 22%), washed with diethyl ether and air-dried. FT-IR: (cm−1) = 3442 (m), 3333 (s), 3101 (m), 2935 (w), 1619 (m), 1589 (m), 1516 (m), 1459 (w), 1421 (m), 1384 (w), 1296 (w), 1235 (w), 1159 (s), 1084 (vs), 1037 (vs), 901 (w), 884 (w), 786 (w), 762 (m), 655 (w), 617 (w), 594 (w), 522 (w). EA (%): Calc. (found) C 26.12 (26.18), H 2.56 (2.35), N 15.23 (15.33).
- [Fe2(μ2-LThiazole)2](ClO4)4
LThiazole (32 mg, 0.1 mmol, 1 eq) was dissolved in 2 mL of acetonitrile. To this solution, Fe(ClO4)2 × H2O (27 mg, 0.1 mmol, 1 eq), dissolved in 2 mL acetonitrile, was added dropwise and stirred for an hour. The reaction mixture was set for vapor diffusion against diethyl ether and kept undisturbed at room temperature. After three days, colorless microcrystalline needles were obtained. The compound was collected by filtration in low yields (17.76 mg, 0.015 mmol, 15%) and washed with diethyl ether and air-dried. FT-IR: (cm−1) = 3422 (s), 3307 (vs), 3125 (m), 3097 (m), 2938 (w), 1617 (m), 1513 (m), 1451 (m), 1429 (m), 1384 (m), 1282 (m), 1233 (m), 1146 (vs), 1115 (vs), 1088 (vs), 974 (w), 897 (m), 884 (m), 637 (s), 626 (vs), 593 (w). EA (%): Calc. (found) C 24.97 (25.01), H 2.45 (2.40), N 14.56 (14.63).
- [Fe2(μ2-LThiazole)2](CF3SO3)4· 2CH3CN
LThiazole (32 mg, 0.1 mmol, 1 eq) was dissolved in 2 mL of acetonitrile. To this solution, Fe(CF3SO3)2 (35 mg, 0.1 mmol, 1 eq), dissolved in 2 mL acetonitrile, was added dropwise and stirred for an hour. The reaction mixture was set for vapor diffusion against diethyl ether and kept undisturbed at room temperature. After one week, colorless block-shaped crystals suitable for X-ray analysis were obtained. The crystals were collected by filtration in low yields (22.54 mg, 0.017 mmol, 17%), washed with diethyl ether and air-dried. FT-IR: (cm−1) = 3509 (m), 3277 (s), 3138 (w), 3106 (m), 2967 (w), 2930 (w), 2850 (w), 2359 (w), 2258 (w), 1618 (s), 1513 (m), 1454 (m), 1429 (s), 1384 (w), 1277 (vs), 1255 (vs), 1227 (vs), 1158 (vs), 1086 (s), 1029 (vs), 979 (w), 886 (m), 786 (w), 770 (m), 759 (m), 7435 (m), 639 (vs), 594 (w), 574 (m), 518 (s). EA (%): Calc. (found) C 26.79 (26.42), H 2.39 (2.05), N 13.67 (13.54).
3. Results and Discussion
3.1. Synthesis
The first two steps of the ligand synthesis include the condensation reaction of hydrazine monohydrate and chloroacetyl chloride to 1,2-bis(chloracetyl)hydrazide, followed by the cyclization reaction to 2,5-bis(chloromethyl)-1,3,4-oxadiazole with slide modifications to the literature. [46] This is followed by the nucleophilic substitution reaction of 2,5-bis(chloromethyl)-1,3,4-oxadiazole with sodium azide, resulting in 2,5-bis(azidomethyl)-1,3,4-oxadiazole in good yields (87%). In the last step, the Staudinger reduction of 2,5-bis(azidomethyl)-1,3,4-oxadiazole with triphenylphosphine and water afforded 2,5-bis(aminomethyl)-1,3,4-oxadiazole in a very good yield (97%), which was reacted with thiazole-2-carboxaldehyde to form 2,5-bis[(1,3-thiazol-2-ylmethyl)amino]methyl-1,3,4-oxadiazole (LThiazole), in moderate yield (60%). The in situ-formed imine compound was reduced with sodium cyanoborohydride (LThiazole) to the desired amine. Reductive amination of 2,5-bis(aminomethyl)-1,3,4-oxadiazole with thiazole-2-carbaldehyde to yield LThiazole (Scheme 1), using sodium cyanoborohydride with catalytic amounts of acetic acid. The successful formation of the desired ligand was characterized by 1H-NMR, 13C-NMR and IR spectroscopy (Supplementary Materials, Figures S1–S9). Complexation reaction of LThiazole with three different appropriate iron salts in acetonitrile at inert atmosphere yielded pure crystalline complexes 1–3, which were characterized by elemental analysis, infrared spectroscopy, SQUID magnetometry and X-ray crystallography in the case of 1 and 3 at 120 K. Attempts to obtain crystals in the case of 2 suitable for X-ray was not successful, even after several attempts of recrystallization yielding colorless pure microcrystalline powder in all cases.
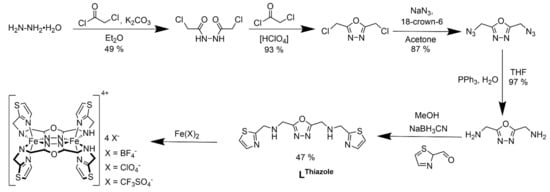
Scheme 1.
Scheme showing multistep synthesis of LThiazole and three corresponding dinuclear Fe(II) complexes 1–3.
3.2. Crystallographic Discussion of 1 and 3
The X-ray crystal data collected for 1 and 3 at 120 K show that the two complexes are isostructural to each other and crystallize in a triclinic P space group (Appendix A). The crystallographic data and the selected bond lengths are tabulated in Table 1 and Table 2. The two iron(II) ions are bridged by the 1,3,4-oxadiazole moieties of two opposing ligands (Figure 1). The secondary amine functions of both ligands are also coordinating in the equatorial position, thus saturating the horizontal vertices of both octahedral coordination spheres. The axial position is filled by the thiazole moiety using the nitrogen donor. Both ligands coordinate in a cis-axial manner (Figure 1, left).

Table 1.
Selected bond lengths for complexes 1 and 3 at 120 K.
Table 2.
Crystallography data for complexes 1 and 3.
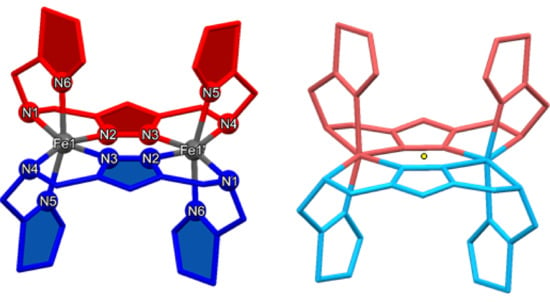
Figure 1.
(Left): Labeling scheme for [Fe2(-LThiazole)2]4+ complex cations. Cis-axial coordination mode of the ligands is displayed by the colors red and blue. (Right): Symmetry equivalent moieties in all [Fe2(-LThiazole)2]4+ complex cations due to an intrinsic inversion center (yellow dot). The color scheme was adapted to the symmetry-equivalent units.
The asymmetric unit consists of one ligand coordinated to an iron(II) center, two perchlorate anions and one acetonitrile in the case of 1 (Figure 2). The complex cation is located on top of a center of inversion in the middle of both ab-planes of the unit cell. Thus, half of the complex cation points into the unit cell while the other half formally lies outside of the unit cell. One tetrafluoroborate and its symmetry-generated pendant lie between the two halves of the two complex molecules. The acetonitrile molecules are located between the thiazole rings above an oxadiazole unit (Figure S11). This BF4− anion is involved in hydrogen bonding, connecting the complex cations in a one-dimensional way (Figure S13). The amine function of one ligand and a fluorine atom of the tetrafluoroborate shows that N1-H1⋯F1 distance is equal to 2.01 Å, with an N1-H1-F1 angle of 165.5°. Two other fluorine atoms of the same BF4− anion form additional hydrogen bonds to a neighboring complex cation. The N4-H4⋯F4 distance is found to be 1.95 Å with an enclosed N4-H4-F4 angle of 165.5°, while the N4-H4⋯F3 distance is equal to 2.65 Å. The N4-H4-F3 angle appears to be 129.9°. Due to the bridging nature of the BF4− counter ion, a one-dimensional chain of complex cation is formed via hydrogen bonds (Figure S13). Since the complex cation is inverse-symmetrical, the coordination spheres of both iron(II) centers are equivalent (Figure 2, right). Thus, only the coordination environment of Fe1 will be discussed. The mean distance between Fe1 and a secondary amine function [(Fe-NAmine) = (d(Fe1-N1) + d(Fe1-N4))/2] is found to be 2.34 Å, which represents the longest Fe-N bond type. The next smaller average bond length can be found between the iron(II) ion and the oxadiazole nitrogen atom [(Fe-NODA) = (d(Fe1-N2) + d(Fe1-N3))/2 = 2.14 Å]. With an average value of (Fe-NTA) = (d(Fe1-N5) + d(Fe1-N6))/2 = 2.13 Å, the bonds between the metal center and the thiazole ring are the shortest. While the amine function only offers σ-bonding, the metal ligand bonds to the oxadiazol and thiazole moieties can have further π-back bonding character [54]. Therefore, the bond distances between Fe1 and the heteroaromatic compounds are shorter than those to the amine groups.
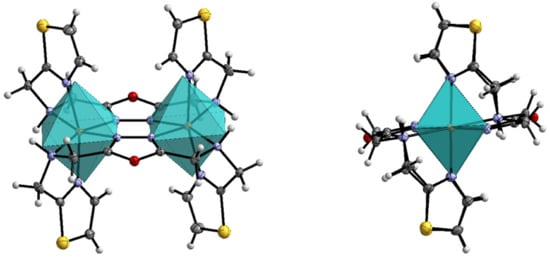
Figure 2.
(Left): Molecular structure of 1 and its ideal coordination octahedron with front view and view onto the Fe-Fe axis (right), calculated with SHAPE [55]. Scheme: dark grey—C, grey—H, violet—N, red—O, yellow—S, orange—Fe. ORTEP representation with atomic displacement parameters set to 50% probability.
The average Fe-N distance in the complex 1 is found to be (Fe-N) = 2.21 Å. This value suggests a [HS-HS] state of the complex at 120 K. The octahedral distortion parameter (FeN6) is equal to 123.9°, displaying a very strong deviation of the ideal coordination octahedron. This is further confirmed by continuous shape measurement, resulting in a value of CShM(Oh) = 4.09 (Figure 3). Selected bond lengths are tabulated in Table 1. It is known by literature that high-spin iron(II) complexes have a larger distortion than low-spin complexes. Thus, the octahedral distortion parameter for LS complexes is usually less than 80°, while it is greater than 100° for HS complexes [54]. This can be explained by the asymmetric electron configuration of the iron(II) ion in the high-spin state, compared to the symmetric population of the t2g-orbitals of the low-spin species.
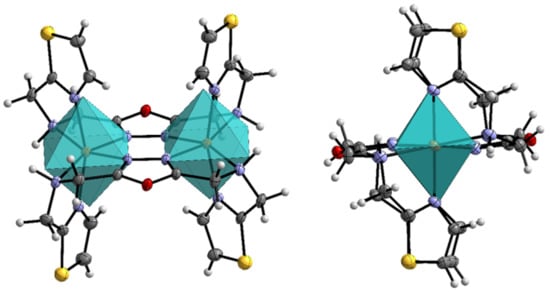
Figure 3.
Molecular structure of 3 and its ideal coordination octahedron with front view (left) and view onto the Fe-Fe axis (right), calculated with SHAPE [55]. Color scheme: dark grey—C, grey—H, violet—N, red—O, yellow—S, orange—Fe. ORTEP representation with atomic displacement parameters set to 50% probability.
In the case of complex 3, the asymmetric unit consists of one LThaizole coordinated to an iron(II) ion, one acetonitrile and two trifluoromethanesulfonate anions (Figure S15). Thus, one half of the unit cell is symmetry generated. The distance d(O1-N7) between the oxadiazole (O1) and the acetonitrile (N7) equals to 2.90 Å. On the other side of the oxadiazole motif a triflate anion is located with d(O1-S3) = 3.60 Å (Figure S15). Two distinguishable hydrogen bonds between an amine function of the ligand and a triflate anion are found in complex 3. The first one can be found between N3-H3⋯O4 with a distance of 1.97 Å and an N3-H3-O4 angle of 161°. Next to that, a second hydrogen bond N4-H4⋯O7 (2.05 Å) is formed, showing an N4-H4-O7 angle of 153° (Figure S16).
The bond lengths of the complex cation look very similar to 1, where the longest average Fe-N bond type is displayed by (Fe-NAmine) (2.36 Å), followed by (Fe-NODA) (2.14 Å) and (Fe-NTA) (2.11 Å). These bond distances result in a mean Fe-N bond distance of 2.21 Å. The octahedral distortion parameter ((FeN6) = 128 °) shows a larger distortion of the coordination sphere than found for 1, which is confirmed by continuous shape measurement (CShM(Oh) = 4.57).The bond lengths indicate that both iron centers stay in [HS-HS] state at the given temperature of 120 K. Selected bond lengths are tabulated in Table 1.
3.3. Infrared Spectroscopy
IR spectra of the isolated complexes are shown in Figure 4. All three compounds 1–3 show a blueshift of the symmetric N-H stretching frequency [νs(N-H)] compared to the free ligand LThiazole (3277 cm−1). Here, the biggest difference is found in 1 (3333 cm−1), followed by 2 (3307 cm−1) and 3 (3278 cm−1). In most cases, the coordination of a NHR2 group to transition metals results in a redshift of the symmetric N-H stretching frequency [56]. However, there are also reported cases of a shift towards higher wavenumbers [57]. The different shifts of the symmetric N-H stretching frequency can be caused by varying hydrogen bonds to the secondary NHR2 group of the ligand. The coordination of the oxadiazole-N results in a red shift of the corresponding C=N stretching mode, moving to lower wave numbers from a strong ν(C=N) stretching at 1667 cm−1 for the free ligand to less pronounced weak stretching around 1640 cm−1 as expected due to the weakening of the C=N bond when binding to the transition metal [58]. In addition to the νs(N-H) band, the counter ion-specific IR bands are also clearly visible. Hence, 1 shows very pronounced asymmetric B-F stretching bands νas(BF4) at 1084 cm−1, 1052 cm−1 and 1036 cm−1. The IR band at 762 cm−1 can be assigned to the symmetric stretching mode νs(BF4), while the bands at 522 cm−1, 506 cm−1 and 500 cm−1 display deformation vibrations δ(BF4) [58].
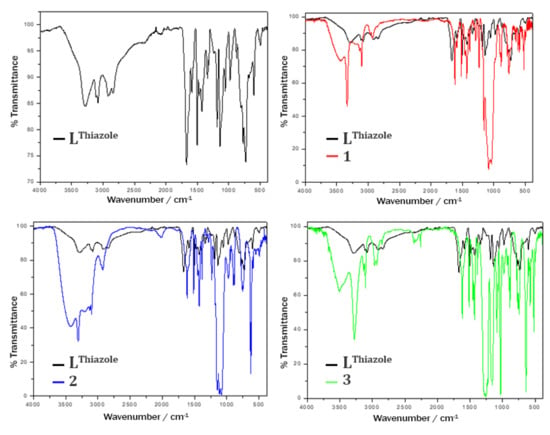
Figure 4.
Comparison of IR spectra of LThiazole (black, top left) with 1 (red line, top right), 2 (blue, bottom left) and 3 (green, bottom right). All complex spectra are overlaid onto the ligand spectra (black line).
Complex 2 shows vibration bands at 1147 cm−1, 1115 cm−1 and 1086 cm−1, which result from asymmetric stretching vibrations νas(ClO4). In addition, an asymmetric bending mode δas(ClO4) at 625 cm−1 can be observed [58]. Based on the distinct N-H stretching vibration, the perchlorate-specific bands and the ligand-like fingerprint region in the IR spectrum of 2, a successful synthesis of 2 in the desired form of [Fe2(-LThiazole)2](ClO4)4 is verified. The IR spectrum of 3 clearly shows symmetric and asymmetric stretching vibrations of the free triflate anion in the region of approx. 1020–1300 cm−1 (νas(SO3): 1278 cm−1, νs(CF3): 1253 cm−1, νas(CF3): 1227 cm−1 and νs(SO3): 1029 cm−1). In addition, bending bands can be found at 639 cm−1 [δs(SO3)], 573 cm−1 [δas(CF3)] and 517 cm−1 [δas(SO3)] [58].
3.4. Magnetic Measurement of 1–3
Magnetic measurements of 1–3 were carried out in a temperature range from 300–2 K at 0.1 T. Therefore, the magnetic susceptibility was determined and plotted as χMT product versus the temperature (T) for each complex (Figure 5). [Fe2(μ2-LThiazole)2](BF4)4 ∙ 2CH3CN (1) and [Fe2(μ2-LThiazole)2](CF3SO3)4·2CH3CN (3) were measured as colorless block-shaped crystals, while [Fe2(μ2-LThiazole)2](ClO4)4 (2) was isolated and studied as microcrystalline powder. The χMT value of 2 at 300 K is equal to 7.28 cm3 mol−1 K. This value is higher than the spin-only value, which is 6 cm3 mol−1 K for two S = 2 spin centers with g = 2 (Figure 5). Below 50 K, the χMT product for 2 drops significantly to 0.62 cm3 mol−1 K at 2 K. The decline of χMT from high temperatures towards 2 K is caused by antiferromagnetic coupling between the oxadiazole-bridged iron(II) ions. Simulation with the spin-exchange Hamiltonian H = −2Jij ∑Si · Sj leads to a weak antiferromagnetic-exchange interaction parameter J = −0.66 (2) cm−1 with g = 2.217 (4), (Figure 5, blue solid line). The data were fitted with the PHI program [59]. High crystallographic anisotropy can lead to pronounced magnetic anisotropy, which in turn can cause the orientation of the crystals within the applied magnetic field. Since the crystals were measured in a nonblocked way, an alignment of the crystals with the magnetic field can occur. The resulting measured magnetic moment can therefore be formally higher than expected. For 1 and 3, in contrast, the χMT product increases with decreasing temperature, which is due to orientation effects of the single crystals. Both complexes show a constant χMT value of approx. 7 cm3 mol−1 K in the range of 300–150 K. Regardless of the magnetic coupling of both iron(II) centers, complexes 1–3 do not exhibit SCO activity. Thus, all three complexes 1–3 remain in the [HS-HS] state. The change of the six-membered pyridine group in the sidearms to the five-membered thiazole did not lead to the desired SCO-active dinuclear iron(II) compounds.
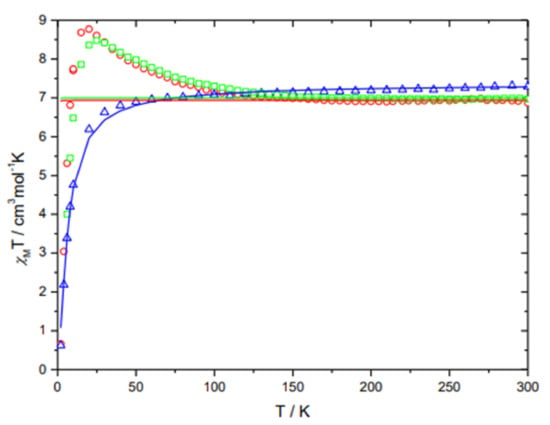
Figure 5.
Temperature-dependent behavior of 1 (red open circles), 2 (blue open triangles) and 3 (green open squares) in form of the combined χMT vs. T plots.
4. Conclusions
We report the synthesis and magnetic properties of three novel dinuclear Fe(II) complexes based on a new bridging 1,3,4-oxadiazole ligand bearing coordinating thiadiazole side arms. All three compounds are locked in the high-spin state [HS-HS]. The reason for this can be assigned to a combination of geometrical and electronic effects. From our earlier work where ligand field strength in imidazole-arm-containing thiadiazole-based [Fe2(μ2-I2MTD)2](X)4 complexes (I2MTD = 2,5-bis[(1H-imidazol-2-ylmethyl)amino]methyl1,3,4-thiadiazole) is lower than pyridyl-arm-containing thiadiazole-based complexes of type [Fe2(μ2-PMTD)2](X)4 (PMTD = 2,5-bis[(2-pyridyl-methyl)amino]methyl-1,3,4-thiadiazole), (X = BF4−, ClO4− and CF3SO3−) [46]. While the [Fe2(μ2-I2MTD)2](X)4-type complexes remain in the [HS-HS] state between 2–300 K, the compounds of type [Fe2(μ2-PMTD)2](X)4 stabilize the diamagnetic [LS-LS] state, showing only the start of an incomplete SCO above room temperatures. This is because of the different angles between the heteroaromatic ring and the adjacent methylene group. The angle α, formed by the pyridine ring, is approximately 120°, whereas the angle β, formed by a five-membered ring, is approximately 125°. Due to the steric effect of such ligand systems, the bigger angle β results in longer Fe-N-bonds favoring the high-spin state. The same arguments seem to apply for the analogous oxadiazole-based ligands LThiazole and PMOD. In addition to that, thiazole is considered to generate a weaker ligand field than pyridine, which we understand from the basic ligand-field arguments and literature.
Interestingly, the coordination of a 5-membered thiazole heteroaromatic ring in the apical positions (1–3) does not lead to longer axial bond lengths compared to the [Fe2(μ2-PMOD)2] (X)4-type complexes [46]. In contrast, all three complexes show smaller average Fe-N bond lengths to the axially coordinated aromatic heterocycle than the analogous iron(II) complexes of PMOD. It appears that the increased angle β (Figure 6) has more effect on the Fe-NAmine bonds. The fact that complexes 1–3 have shorter axial bond lengths and significantly longer Fe-NAmine distances than the [Fe2(μ2-PMOD)2] (X)4 complexes clearly shows the modification in the steric effect of the ligand. Most likely, stronger π-back bonding to the thiazole ring in 1–3 still cannot compensate for the sterically unfavorable constraint of the five-membered ring in the ligand side arm. Thus, the increased steric tension and the lower ligand-field strength of LThiazole lead to dinuclear iron(II) [HS-HS] complexes without any SCO behavior in all three cases.
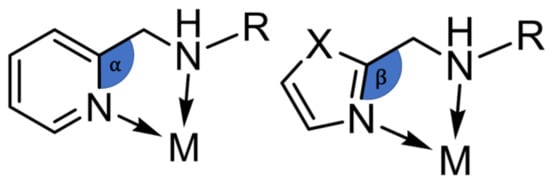
Figure 6.
Scheme of different coordination angles between a 6-membered ring (left) and 5-membered ring (right) in the ligand side arm.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst12030404/s1. S1. Infrared Spectra: Figure S1: IR spectrum of 2,5-Bis[(1,3-thiazol-2-ylmethyl)amino]methyl-1,3,4-oxadiazole (LThiazole), Figure S2: IR spectrum of [Fe2(μ2-LThiazole)2](BF4)4∙2MeCN (1), Figure S3: IR spectrum of [Fe2(μ2-LThiazole)2](ClO4)4 (2), Figure S4: IR spectrum of [Fe2(μ2-LThiazole)2](CF3SO3)4∙2MeCN (3). S2. NMR Spectroscopy: Figure S5: 1H-NMR spectrum of 2,5-Bis[(1,3-thiazol-2-ylmethyl)amino]methyl-1,3,4-oxadiazole (LThiazole) in CDCl3; Figure S6: 13C-NMR spectrum of 2,5-Bis[(1,3-thiazol-2-ylmethyl)amino]methyl-1,3,4-oxadiazole (LThiazole) in CDCl3; Figure S7: 1H-1H-COSY spectrum of 2,5-Bis[(1,3-thiazol-2-ylmethyl)amino]methyl-1,3,4-oxadiazole (LThiazole) in CDCl3; Figure S8: 1H-13C-HSQC spectrum of 2,5-Bis[(1,3-thiazol-2-ylmethyl)amino]methyl-1,3,4-oxadiazole (L5,TA) in CDCl3; Figure S9: 1H-13C-HMBC spectrum of 2,5-Bis[(1,3-thiazol-2-ylmethyl)amino]methyl-1,3,4-oxadiazole (L5,TA) in CDCl3. S3. FD Mass spectroscopy: Figure S10: FD mass spectrum of 2,5-Bis[(1,3-thiazol-2-ylmethyl)amino]methyl-1,3,4-oxadiazole (LThiazole). S4. Crystallography: Figure S11: Asymmetric Unit of 1 with front and side view; Figure S12: Unique hydrogen bonds in 1 between complex cations and tetrafluoroborate anions; Figure S13: One-dimensional network of complex cations via hydrogen bonds in 1; Figure S14: Unit cell of 1 with view along the bisector of γ and perspective; Figure S15: Asymmetric Unit of 3 with front and side view; Figure S16: Unique hydrogen bonds in 3 between complex cations and tetrafluoroborate anions; Figure S17: Unit cell of 3 with view along the a-axis and perspective; Table S1: Selected angles of complex 1 and 3.
Author Contributions
Synthesis, investigations, experimental, J.E.; methodology, J.E. and E.R.; spectroscopy, spectrometry and magnetic data analysis, J.E. and S.S.; crystal structure analysis, L.M.C.; writing—original draft preparation, S.S. and J.E.; writing—review and editing, S.S. and E.R.; supervision, E.R.; funding acquisition, E.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the JGU.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The magnetic data presented in this study are available on request from the corresponding author.
Acknowledgments
Dieter Schollmeyer is kindly acknowledged for X-ray data acquisition.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
CCDC files with deposition numbers 2152968–2152969 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif (accessed on 18 February 2022), or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033.
References
- Shriver, D.F.; Atkins, P.W.; Langford, C.H. Inorganic Chemistry, 2nd ed.; Oxford University Press: Oxford, UK; Melbourne, Australia; Tokyo, Japan, 1994. [Google Scholar]
- Melson, G. Coordination Chemistry of Macrocyclic Compounds; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Bousseksou, A.; Molnár, G.; Salmon, L.; Nicolazzi, W. Molecular spin crossover phenomenon: Recent achievements and prospects. Chem. Soc. Rev. 2011, 40, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Halcrow, M.A. Spin-Crossover Materials—Properties and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Létard, J.-F.; Guionneau, P.; Goux-Capes, L. Towards Spin Crossover Applications. Top. Curr. Chem. 2004, 235, 221–249. [Google Scholar]
- Molnár, G.; Rat, S.; Salmon, L.; Nicolazzi, W.; Bousseksou, A. Spin crossover Nanomaterials: From fundamental concepts to devices. Adv. Mater. 2018, 30, 1703862. [Google Scholar] [CrossRef] [PubMed]
- Jeon, I.R.; Park, J.G.; Haney, C.R.; Harris, T.D. Spin crossover iron(II) complexes as PARACEST MRI thermometers. Chem. Sci. 2014, 5, 2461. [Google Scholar] [CrossRef]
- Gural’skiy, I.Y.A.; Shylin, S.I.; Ksenofontov, V.; Tremel, W. Cooperative High-Temperature Spin Crossover accompanied by a highly anisotropic structural distortion. Eur. J. Inorg. Chem. 2017, 2017, 3125–3131. [Google Scholar] [CrossRef]
- Halcrow, M. The effect of ligand design on metal ion spin state-Lessons from spin crossover complexes. Crystals 2016, 6, 58. [Google Scholar] [CrossRef]
- Krewald, V.; Retegan, M.; Neese, F.; Lubitz, W.; Pantazis, D.A.; Cox, N. Spin state as a marker for the structural evolution of nature’s water-spilling catalyst. Inorg. Chem. 2016, 55, 488–501. [Google Scholar] [CrossRef]
- Swart, M.; Gruden, M. Spinning around in transition-metal chemistry. Acc. Chem. Res. 2016, 49, 2690–2697. [Google Scholar] [CrossRef]
- Hayami, S.; Gu, Z.; Yoshiki, H.; Fujishima, A.; Sato, O. Iron(III) spin-crossover compounds with a wide apparent thermal hysteresis around room temperature. J. Am. Chem. Soc. 2001, 123, 11644–11650. [Google Scholar] [CrossRef]
- Kahn, O.; Codjovi, E. Iron(II)-1,2,4-triazole spin transition molecular materials. Phil. Trans. R. Soc. Lond. A 1996, 354, 359–379. [Google Scholar]
- Gaspar, A.B.; Molnár, G.; Rotaru, A.; Shepherd, H.J. Pressure effect investigations on spin crossover coordination compounds. C. R. Chim. 2018, 21, 1095–1120. [Google Scholar] [CrossRef]
- Hammack, W.S.; Conti, A.J.; Hendrickson, D.N.; Drickamer, H.G. Pressure-induced spin-state interconversion of [Fe(6-Me-py)3tren] (ClO4)2. J. Am. Chem. Soc. 1989, 111, 1738–1741. [Google Scholar] [CrossRef]
- Wang, Y.; Ying, J.; Zhou, Z.; Sun, J.; Wen, T.; Zhou, Y.; Li, N.; Zhang, Q.; Han, F.; Xiao, Y.; et al. Emergent superconductivity in an iron-based honeycomb lattice initiated by pressure-driven spin crossover. Nat. Commun. 2018, 9, 1914. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S. Distortion pathways of transition metal coordination polyhedral induced by chelating topology. Chem. Rev. 2015, 115, 13447–13483. [Google Scholar] [CrossRef]
- Bousseksou, A.; Molnár, G.; Real, J.A.; Tanaka, K. Spin crossover and photomagnetism in dinuclear iron(II) compounds. Coord. Chem. Rev. 2007, 251, 1822–1833. [Google Scholar] [CrossRef]
- Jakobsen, V.B.; Chikara, S.; Yu, J.-X.; Dobbelaar, E.; Kelly, C.T.; Ding, X.; Weickert, F.; Trzop, E.; Collet, E.; Cheng, H.-P.; et al. Giant magnetoelectric coupling and magnetic-field-induced permanent switching in a spin crossover Mn(III) complex. Inorg. Chem. 2021, 60, 6167–6175. [Google Scholar] [CrossRef]
- Cambi, L.; Szegö, L. über die magnetische susceptibilität der komplexen verbindungen. Ber. Dtsch. Chem. Ges. B 1931, 64, 2591–2598. [Google Scholar] [CrossRef]
- Sundaresan, S.; Kühne, I.A.; Kelly, C.T.; Barker, A.; Salley, D.; Müller-Bunz, H.; Powell, A.K.; Morgan, G.G. Anion influence on spin state in two novel Fe(III) compounds: [Fe(5F-sal2333)]. Crystals 2018, 9, 19. [Google Scholar] [CrossRef]
- Craig, G.A.; Roubeau, O.; Aromí, G. Spin state switching in 2,6-bis(pyrazol-3-yl)pyridine (3-bpp) based Fe(II) complexes. Coord. Chem. Rev. 2014, 269, 13–31. [Google Scholar] [CrossRef]
- Feltham, H.L.C.; Barltrop, A.S.; Brooker, S. Spin crossover in Iron(II) complexes of 3,4,5-tri-substituted-1,2,4-triazole (Rdpt), 3,5-di-substituted-1,2,4-triazolate (dpt-), and related ligands. Coord. Chem. Rev. 2017, 344, 26–53. [Google Scholar] [CrossRef]
- Gamez, P.; Costa, J.S.; Quesada, M.; Aromí, G. Iron spin-crossover compounds: From fundamental studies to practical applications. Dalton Trans. 2009, 38, 7845–7853. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, S.; Kitchen, J.A.; Brooker, S. Hydrophobic tail length in spin crossover active iron(II) complexes predictably tunes T1/2 in solution and enables surface immobilization. Inorg. Chem. Front. 2020, 7, 2050–2059. [Google Scholar] [CrossRef]
- Fürmeyer, F.; Münzberg, D.; Carrella, L.M.; Rentschler, E. First cobalt(II) Spin crossover compound with N4S2-donorset. Molecules 2020, 25, 855. [Google Scholar] [CrossRef]
- Goodwin, H.A. Spin crossover in cobalt(II) systems. Top. Curr. Chem. 2004, 234, 23–47. [Google Scholar]
- Jakobsen, V.B.; Trzop, E.; Gavin, L.C.; Dobbelaar, E.; Chikara, S.; Ding, X.; Esien, K.; Müller-Bunz, H.; Felton, S.; Zapf, V.S.; et al. Stress-induced domain wall motion in a ferroelastic Mn3+ spin crossover complex. Angew. Chem. Int. Ed. 2020, 59, 13305–13312. [Google Scholar] [CrossRef]
- Morgan, G.G.; Murnaghan, K.D.; Müller-Bunz, H.; McKee, V.; Harding, C.J. A manganese(III) complex that exhibits spin crossover triggered by geometric tuning. Angew. Chem. Int. Ed. 2006, 45, 7192–7195. [Google Scholar] [CrossRef]
- Sim, P.G.; Sinn, E. First manganese(III) spin crossover, first d4 crossover. Comment on cytochrome oxidase. J. Am. Chem. Soc. 1981, 103, 241–243. [Google Scholar] [CrossRef]
- Sundaresan, S.; Kühne, I.A.; Evesson, C.; Harris, M.M.; Fitzpatrick, A.J.; Ahmed, A.; Müller-Bunz, H.; Morgan, G.G. Compressed Jahn-Teller octahedra and spin quintet-triplet switching in coordinatively elastic manganese(III) complexes. Polyhedron 2021, 208, 115386. [Google Scholar] [CrossRef]
- Garcia, Y.; Gutlich, P. Thermal spin crossover in Mn(II), Mn(III), Cr(II) and Co(III) coordination compounds. Top. Curr. Chem. 2004, 234, 49–62. [Google Scholar]
- Halepoto, D.M.; Holt, D.G.L.; Larkworthy, L.F.; Leigh, G.J.; Povey, D.C.; Smith, G.W. Spin crossover in Chromium(II) complexes and the crystal and molecular structure of the high spin form of bis[1,2-bis(diethylphosphino)ethane]di-iodochromium(II). Chem. Comm. 1989, 18, 1322–1323. [Google Scholar] [CrossRef]
- Navarro, L.; Rodriguez, F.; Cirera, J. Controlling the spin-crossover behavior of the [Cr(indenyl)2] family via ligand functionalization. Dalton Trans. 2021, 50, 8704–8710. [Google Scholar] [CrossRef] [PubMed]
- Nihei, M.; Shiga, T.; Maeda, Y.; Oshio, H. Spin crossover iron(III) complexes. Coord. Chem. Rev. 2007, 251, 2606–2621. [Google Scholar] [CrossRef]
- Barrios, L.A.; Peyrecave-Lleixà, E.; Craig, G.A.; Roubeau, O.; Teat, S.J.; Aromí, G. Unusual crystal packing in a family of [Fe{2,6-bis(pyrazol-3-yl)pyridine}2]2+ compounds and the effect on the occurrence of spin crossover and its cooperative character. Eur. J. Inorg. Chem. 2014, 2014, 6013–6021. [Google Scholar] [CrossRef]
- Schlamp, S.; Dankhoff, K.; Weber, B. Amphiphilic iron(II) complexes with short alkyl chains-crystal packing and spin transition properties. New J. Chem. 2014, 38, 1965–1972. [Google Scholar] [CrossRef]
- Vela, S.; Novoa, J.J.; Ribas-Arino, J. Insights into the crystal-packing effects on the spin crossover of [FeII(1-bpp)]2+-based materials. Phys. Chem. Chem. Phys. 2014, 16, 27012–27024. [Google Scholar] [CrossRef]
- Galet, A.; Gaspar, A.B.; Muñoz, M.C.; Real, J.A. Influence of the counterion and the solvent molecules in the spin crossover system [Co(4-terpyridone)2]Xp·nH2O. Inorg. Chem. 2006, 45, 4413–4422. [Google Scholar] [CrossRef] [PubMed]
- Quesada, M.; Prins, F.; Bill, E.; Kooijman, H.; Gamez, P.; Roubeau, O.; Spek, A.L.; Haasnoot, J.G.; Reedijk, J. Counterion effect on the spin-transition properties of the cation [Fe(btzx)3]2+ (btzx=m-Xylylenebis(tetrazole)). Chem. Eur. J. 2008, 14, 8486–8499. [Google Scholar] [CrossRef]
- Alhassanat, A.; Völker, L.; Gamer, C.; Rauguth, A.; Kredel, A.; Luo, C.; Radu, F.; Sapozhnik, A.A.; Mashoff, T.; Rentschler, E.; et al. Solvent-iduced high-spin transition in double-decker 3d-4f mettacrowns. Phys. Rev. B 2019, 99, 104404. [Google Scholar] [CrossRef]
- Barrett, S.A.; Kilner, C.A.; Halcrow, M.A. Spin-crossover in [Fe(3-bpp)2][BF4]2 in different solvents-A dramatic stabilization of the low-spin state in water. Dalton Trans. 2011, 40, 12021–12024. [Google Scholar] [CrossRef]
- Bartel, M.; Absmeier, A.; Jameson, G.N.L.; Werner, F.; Kato, K.; Takata, M.; Boča, R.; Hasegawa, M.; Mereiter, K.; Caneschi, A.; et al. Modification of spin crossover behavior through solvent assisted formation and solvent inclusion in a triply interpenetrating three-dimensional network. Inorg. Chem. 2007, 46, 4220–4229. [Google Scholar] [CrossRef]
- Fei, B.; Chen, X.Q.; Cai, Y.D.; Fang, J.-K.; Tong, M.L.; Tucek, J.; Bao, X. Substituent effects on the fluorescent spin-crossover Fe(II) complexes of rhodamine 6G hydrazones. Inorg. Chem. Front. 2018, 6, 1671–1676. [Google Scholar] [CrossRef]
- Hogue, R.W.; Miller, R.G.; White, N.G.; Feltham, H.L.; Jameson, G.N.; Brooker, S. Hysteretic spin crossover in iron(II) complexes of a new pyridine-triazole-pyrazine ligand is tuned by choice of NCE co-ligand. Chem. Comm. 2014, 50, 1435–1437. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.; Rentschler, E. The first 1,3,4-oxadiazole based di-nuclear iron(II) complexes showing spin crossover behavior with hysteresis. Eur. J. Inorg. Chem. 2016, 1955–1960. [Google Scholar] [CrossRef]
- Fürmeyer, F.; Carrella, L.M.; Ksenofontov, V.; Möller, A.; Rentschler, E. Phase trapping in multistep spin crossover compound. Inorg. Chem. 2020, 59, 2843–2852. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar]
- Sheldrick, G.M.; Schneider, T.R. SHELXL: High-resolution refinement. Methods Enzymol. 1997, 277, 319–343. [Google Scholar] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Tateiwa, N.; Haga, Y.; Fisk, Z.; Ōnuki, Y. Miniature ceramic-anvil high-pressure cell for magnetic measurements in a commercial superconducting quantum interference device magnetometer. Rev. Sci. Instrum. 2011, 82, 053906. [Google Scholar] [CrossRef]
- Sundberg, R.J.; Martin, R.B. Interactions of histidine and other imidazole derivatives with transition metal ions in chemical and biological systems. Chem. Rev. 1974, 74, 471–517. [Google Scholar] [CrossRef]
- Alvarez, S.; Alemany, P.; Casanova, D.; Cirera, J.; Llunell, M.; Avnir, D. Continuous symmetry measures: A new tool in quantum chemistry. Coord. Chem. Rev. 2005, 249, 1693–1708. [Google Scholar] [CrossRef]
- Svatos, G.F.; Curran, C.; Quagliano, J.V. Infrared absorption spectra of inorganic coordination complexes, the N-H Stretching vibration in coördination compounds. J. Am. Chem. Soc. 1955, 77, 6159–6163. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, V.; Upadhyay, N.; Sharma, S. Interactions of atrazine with transition metal ions in aqueous media: Experimental and computational approach. 3 Biotech 2015, 5, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Banwell, C.N.; McCash, E.M. Fundamentals of Molecular Spectroscopy; McGraw-Hill: New York, NY, USA, 1994. [Google Scholar]
- Chilton, N.F.; Anderson, R.P.; Turner, L.D.; Soncini, A.; Murray, K.S. PHI: A powerful new program for the analysis of anistropic monomeric and exchange-coupled polynuclear d- and f-block complexes. J. Comput. Chem. 2013, 34, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).