
The Exchange-Correlation Effects on the Electronic Bands of Hybrid Armchair Single-Walled Carbon Boron Nitride Nanostructure



 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Computational Methods
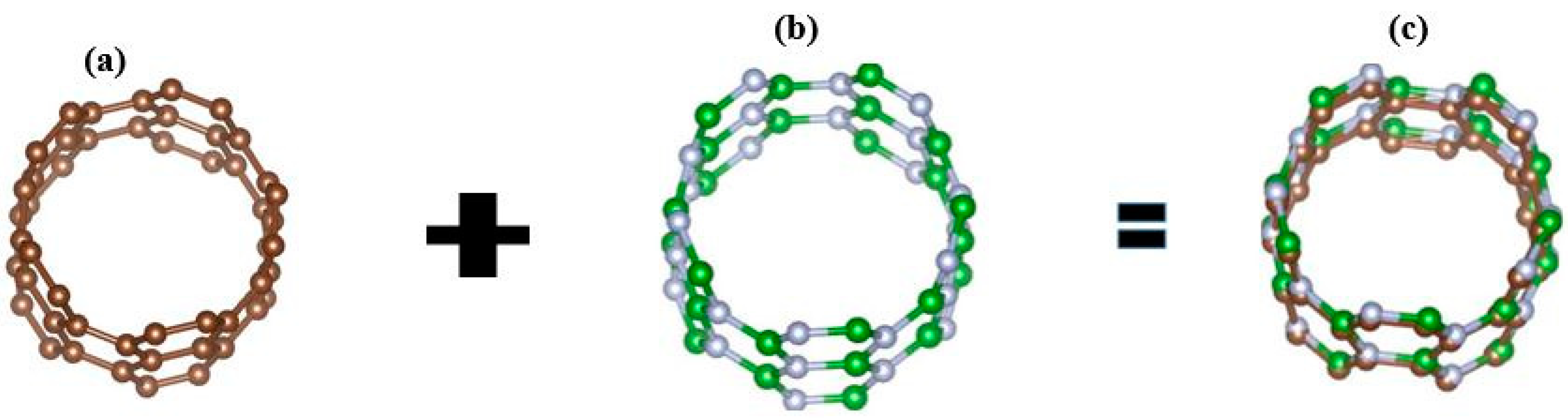
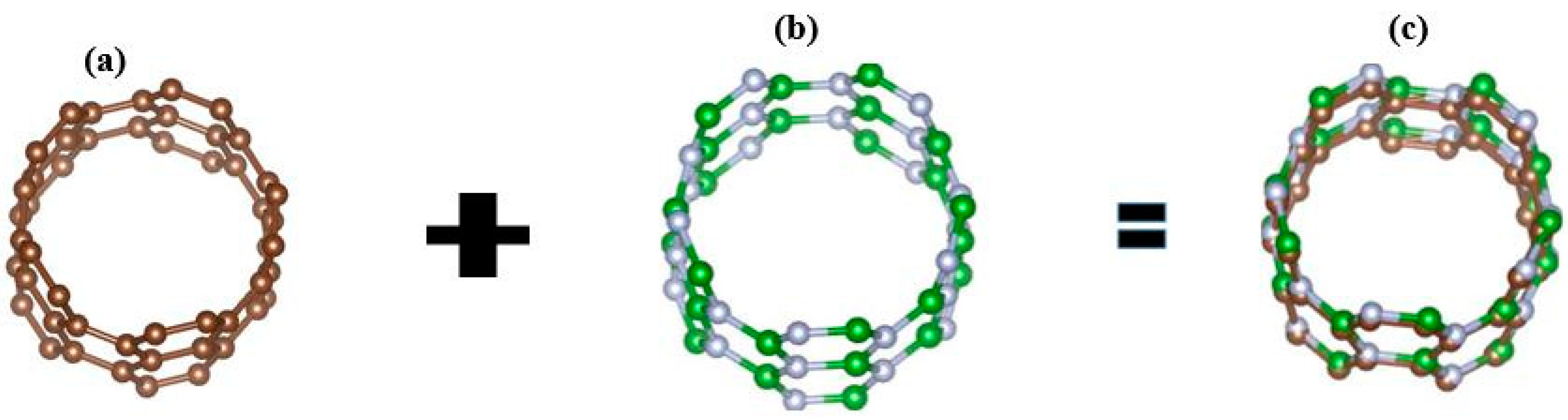
3. Geometry Optimization
4. Results and Discussion
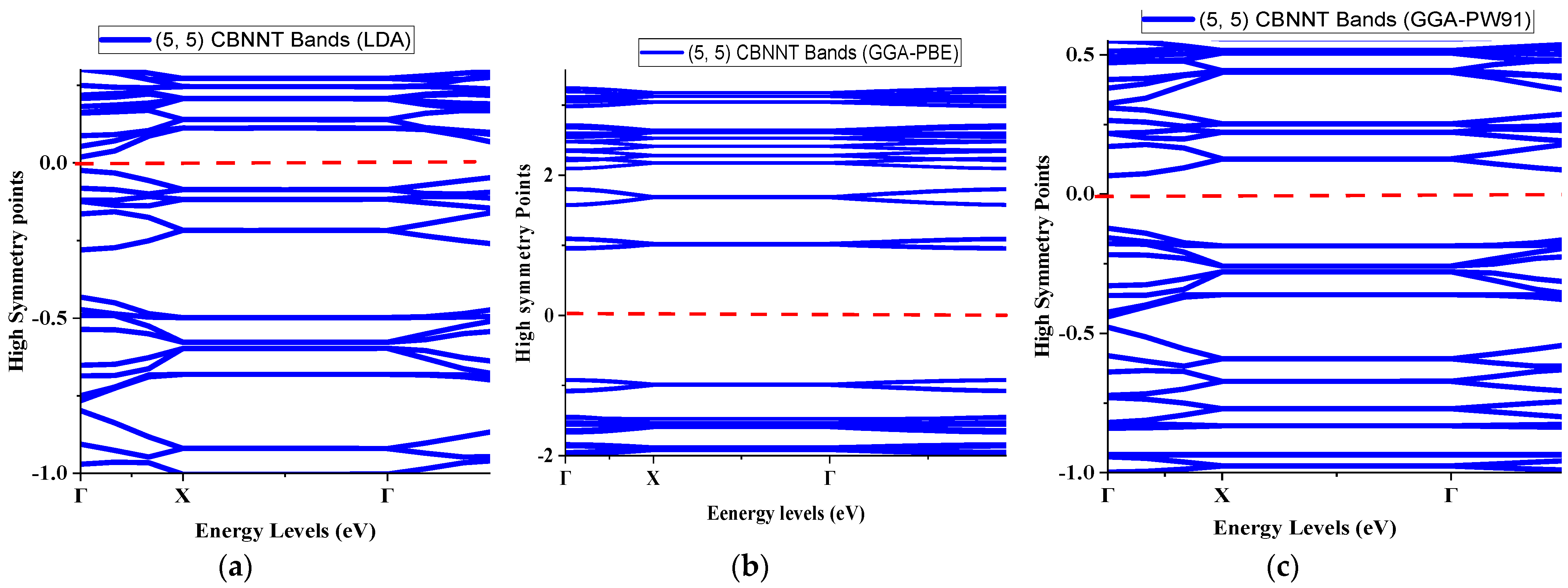
4.1. Bands Structures of (5, 5) CBNNT under Three Different Exchange-Correlation Functionals
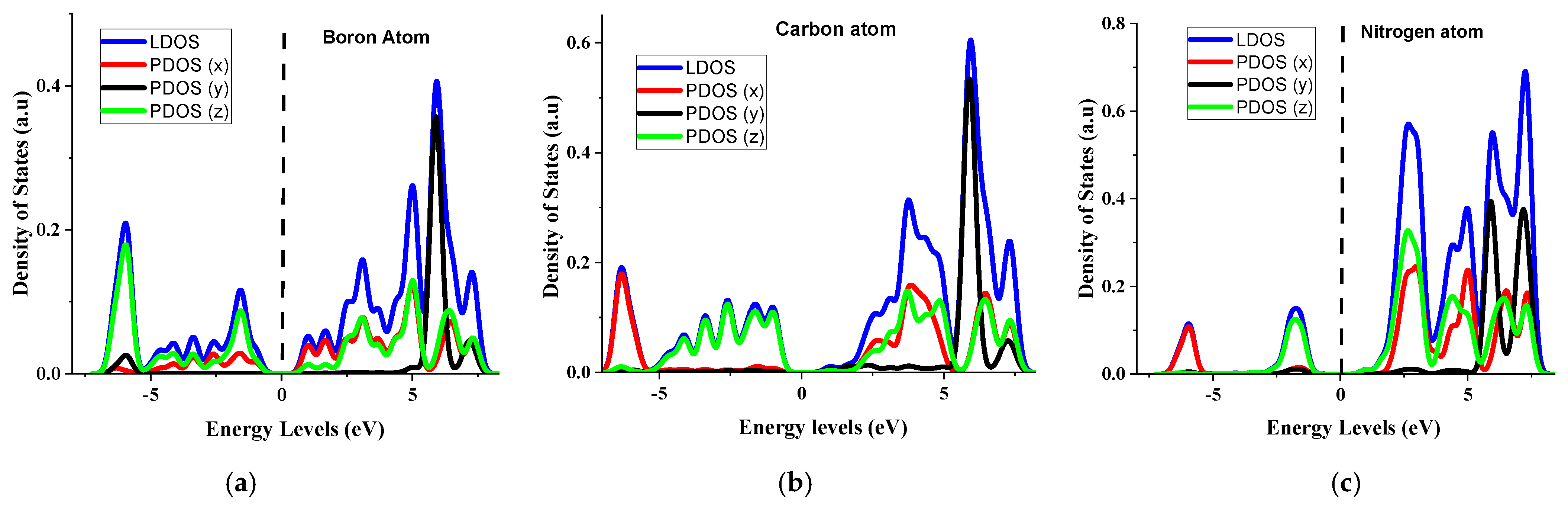
4.2. Analysis of the Density of States
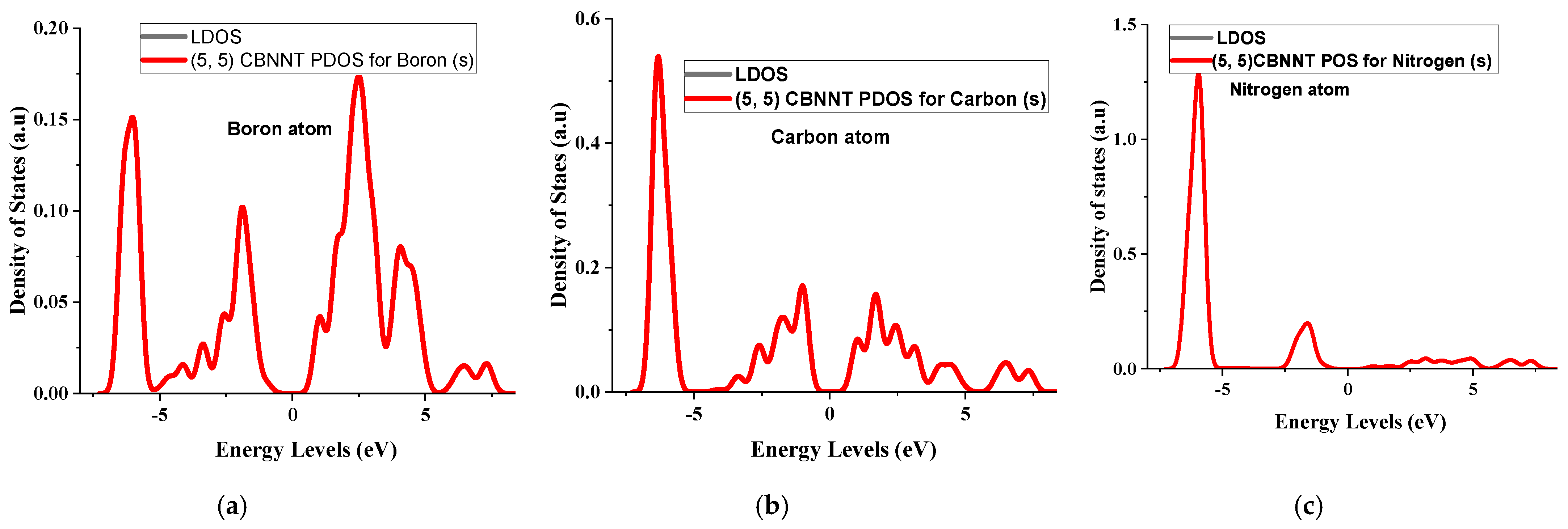
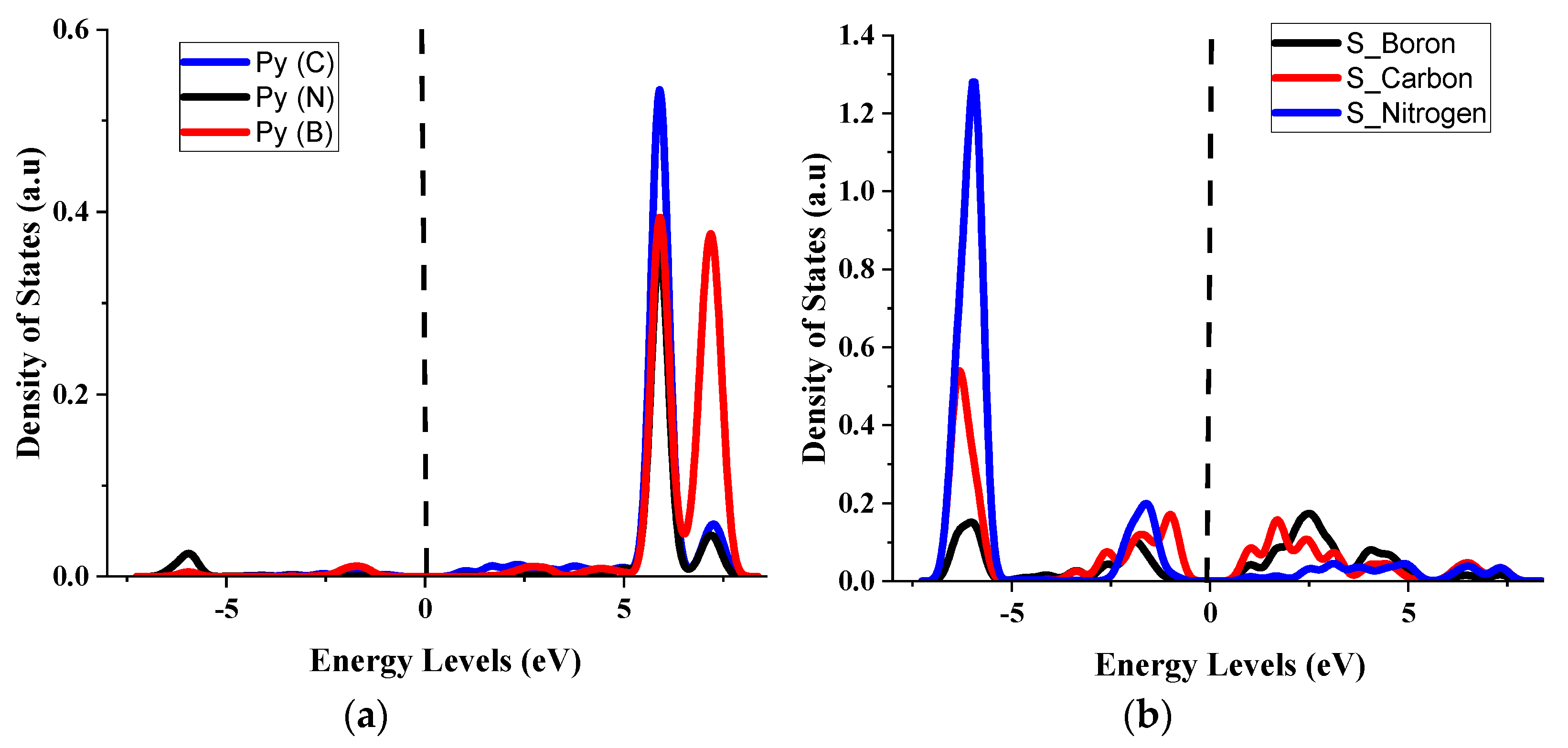
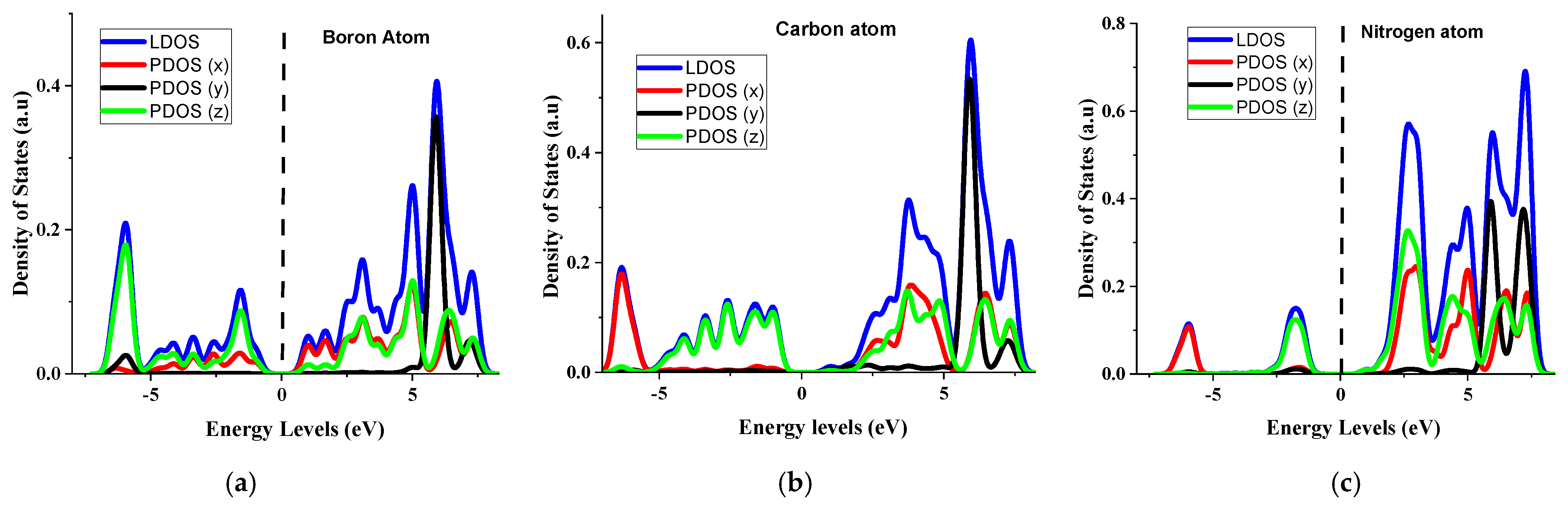
4.3. Partial Density of States for the (5, 5) CBNNT System
4.4. Effects of Py and S Orbitals in the Semiconductivity of (5, 5) CBNNT
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, M.; Evans, L. Carbon Nanotubes as Advanced Materials. J. Aust. Ceram. Soc. 2021, 36, 21–36. [Google Scholar]
- Powell, L.R.; Kim, M.; Wang, Y. Chirality-Selective Functionalization of Semiconducting Carbon Nanotubes with a Reactivity-Switchable Molecule. J. Am. Chem. Soc. 2017, 139, 12533–12540. [Google Scholar] [CrossRef] [PubMed]
- Melaibari, A.; Daikh, A.A.; Basha, M.; Abdalla, A.W.; Othman, R.; Almitani, K.H.; Hamed, M.A.; Abdelrahman, A.; Eltaher, M.A. Free Vibration of FG-CNTRCs Nano-Plates/Shells with Temperature-Dependent Properties. Material 2022, 10, 583. [Google Scholar] [CrossRef]
- Janas, D. Special Issue of Materials Focused on “Electrical, Thermal and Optical Properties of Nanocarbon Materials”. Materials 2022, 15, 1649. [Google Scholar] [CrossRef] [PubMed]
- Foygel, M.; Morris, R.D.; Anez, D.; French, S.; Sobolev, V.L. Theoretical and computational studies of carbon nanotube composites and suspensions: Electrical and thermal conductivity. Phys. Rev. B 2015, 71, 1–6. [Google Scholar] [CrossRef]
- Zhigilei, L.V.; Salaway, R.N.; Wittmaack, B.K.; Volkov, A.N. Computational Studies of Thermal Transport Properties of Carbon Nanotube Materials. In Carbon Nanotubes for Interconnects; Springer: Cham, Switzerland, 2017; pp. 129–161. [Google Scholar] [CrossRef]
- Trivedi, M. Recent Development and Applications of Carbon Nanotubes. Chem. Sci. Rev. Lett. 2020, 9, 502–510. [Google Scholar] [CrossRef]
- Sundaram, R.M.; Sekiguchi, A.; Sekiya, M.; Yamada, T.; Hata, K. Copper/carbon nanotube composites: Research trends and outlook. R. Soc. Open Sci. 2018, 5, 180814. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Kim, M.J.; Park, C.; Fay, C.C.; Chu, S.-H.; Kingston, C.T.; Simard, B. Scalable manufacturing of boron nitride nanotubes and their assemblies: A review. Semicond. Sci. Technol. 2016, 32, 3–13. [Google Scholar] [CrossRef]
- Saifuddin, N.; Raziah, A.Z.; Junizah, A.R. Carbon Nanotubes: A Review on Structure and Their Interaction with Proteins. J. Chem. 2013, 2013, 676815. [Google Scholar] [CrossRef]
- Yanar, N.; Yang, E.; Park, H.; Son, M.; Choi, H. Boron Nitride Nanotube (BNNT) Membranes for Energy and Environmental Applications. Membranes 2020, 10, 430. [Google Scholar] [CrossRef]
- Nematollahi, P.; Esrafili, M.D.; Bagheri, A. Functionalization of single-walled (n, 0) carbon and boron nitride nanotubes by carbonyl derivatives (n = 5, 6): A DFT Study. Can. J. Chem. 2016, 94, 2–21. [Google Scholar] [CrossRef] [Green Version]
- Gharbavi, K.; Badehian, H. Structural and electronic properties of armchair (7, 7) carbon nanotubes using DFT. Comput. Mater. Sci. 2014, 82, 159–164. [Google Scholar] [CrossRef]
- Itas, Y.S.; Ndikilar, C.E.; Zangina, T.; Hafeez, H.Y.; Safana, A.A.; Khandaker, M.U.; Ahmad, P.; Abdullahi, I.; Olawumi, B.K.; Babaji, M.A.; et al. Synthesis of Thermally Stable h-BN-CNT Hetero-Structures via Microwave Heating of Ethylene under Nickel, Iron, and Silver Catalysts. Crystal 2021, 11, 1097. [Google Scholar] [CrossRef]
- Ahmadi, S.; Raeisi, M.; Eslami, L.; Rajabpour, A. Thermoelectric Characteristics of Two-Dimensional Structures for Three Different Lattice Compounds of B-C-N and Graphene Counterpart BX (X = P, As, and Sb) Systems. J. Phys. Chem. 2021, 125, 14525–14537. [Google Scholar] [CrossRef]
- Shao, J.; Beaufils, C.; Kolmogorov, A.N. Ab initio engineering of materials with stacked hexagonal tin frameworks. Sci. Rep. 2016, 6, 28369. [Google Scholar] [CrossRef]
- Omidvar, A.; Hadipour, N. Density functional theory studies of carbon nanotube-Graphene nanoribbon hybrids. J. Iran. Chem. Soc. 2013, 10, 1239–1246. [Google Scholar] [CrossRef]
- An, W.; Turner, H. Linking Carbon and Boron-Nitride Nanotubes: Heterojunction Energetics and Band Gap Tuning. J. Phys. Chem. Lett. 2010, 1, 2269–2273. [Google Scholar] [CrossRef]
- Kostoglou, N. Boron Nitride Nanotubes Versus Carbon Nanotubes: A Thermal Stability and Oxidation Behavior Study. Nanomaterials 2020, 10, 2435. [Google Scholar] [CrossRef]
- An, Y.; Sun, Y.; Jiao, J.; Zhang, M.; Wang, K.; Chen, X.; Wu, D.; Wang, T.; Fu, Z.; Jiao, Z. The rectifying effect of heterojunctions composed of carbon and boron nitride nanotubes. Org. Electron. 2017, 50, 43–47. [Google Scholar] [CrossRef]
- El-Barbary, A.A.; Eid, K.M.; Kamel, M.A.; Taha, H.O.; Ismail, G.H. Adsorption of CO, CO2, NO and NO2 on Carbon Boron Nitride Hetero Junction: DFT Study. J. Surf. Eng. Mater. Adv. Technol. 2015, 5, 57979. [Google Scholar] [CrossRef] [Green Version]
- Hong-Xia, L.; He-Ming, Z.; Jiu-Xu, S.; Zhi-Yong, Z. Electronic transport properties of an (8, 0) carbon/boron nitride nanotube heterojunction. Chin. Phys. B 2010, 19, 037104. [Google Scholar] [CrossRef]
- Liu, H.; Turner, H. Oxygen Adsorption Characteristics on Hybrid Carbon and Boron-Nitride Nanotubes. Comput. Chem. 2014, 35, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Yap, Y.K. Hetero-Junctions of Boron Nitride and Carbon Nanotubes: Synthesis and Characterization; Technical Report No. DOE-MTU-ER46294; Michigan Technological University: Houghton, MI, USA, 2013; pp. 1–7. [Google Scholar]
- Ahmad, P.; Khandaker, M.U.; Khan, Z.R.; Amin, Y.M. Synthesis of boron nitride nanotubes via chemical vapour deposition: A comprehensive review. RSC Adv. 2015, 5, 35116–35137. [Google Scholar] [CrossRef]
- Nafiu, S.; Apalangya, V.A.; Yaya, A.; Sabi, E.B. Boron Nitride Nanotubes for Curcumin Delivery as an Anticancer Drug: A DFT Investigation. Appl. Phys. 2022, 12, 879. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Song, J.; Zhang, Z. Electronic structures of an (8, 0) boron nitride/carbon nanotube heterojunction. J. Semicond. 2010, 31, 013001. [Google Scholar] [CrossRef]
- Dhungana, K.; Pati, R. Boron nitride nanotubes for spintronics. Sensors 2014, 14, 17655–17685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, L.; Xie, Y.; Zhang, S.; Zhang, J. Band Engineering of Carbon Nanotubes for Device Applications. Matter 2020, 3, 664–694. [Google Scholar] [CrossRef]
- Chen, C.-W.; Lee, M.-H.; Clark, S. Band gap modification of single-walled carbon nanotube and boron nitride nanotube under a transverse electric field. Nanotechnology 2004, 15, 1837–1843. [Google Scholar] [CrossRef]
- Liu, X.; Han, M.; Zhang, X.; Hou, H.; Pang, S.; Wu, Q. Tuning Electronic Structures of BN and C Double-Wall Hetero-Nanotubes. J. Nanomater. 2015, 2015, 326294. [Google Scholar] [CrossRef] [Green Version]
- Akter, N.; Mawardi Ayob, M.T.; Radiman, S.; Khandaker, M.U.; Osman, H.; Alamri, S. Bio-Surfactant Assisted Aqueous Exfoliation of High-Quality Few-Layered Graphene. Crystals 2021, 11, 944. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, F. A comparison of three DFT exchange-correlation functionals and two basis sets for the prediction of the conformation distribution of hydrated polyglycine. J. Chem. Phys. 2021, 155, 094104. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Zabel, T.; Sigg, H. Group IV direct band gap photonics: Methods, challenges, and opportunities. Front. Mater. 2015, 2, 52. [Google Scholar] [CrossRef] [Green Version]
- Crowley, J.M.; Tahir-Kheli, J., III. Resolution of the Band Gap Prediction Problem for Materials Design. J. Phys. 2016, 7, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Zviagin, V.; Grundmann, M.; Schmidt-Grund, R. Impact of Defects on Magnetic Properties of Spinel Zinc Ferrite Thin Films. Phys. Status Solidi B 2020, 257, 1900630. [Google Scholar] [CrossRef]
- Wang, G. First-Principles Studies of Group IV and Group V Related Two Dimensional Materials. Ph.D. Thesis, Michigan Technological University, Houghton, MI, USA, 2016. [Google Scholar] [CrossRef]
- Punter, A.; Nava, P.; Carissan, Y. Atomic pseudopotentials for reproducing π-orbital electron behavior in sp2 carbon atoms. Int. J. Quantum Chem. 2019, 119, 25914. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Cao, H.; Dong, J. Electronic properties of group-IV monochalcogenide nanoribbons: Studied from first-principles calculations. Phys. Lett. A 2017, 381, 3747–3753. [Google Scholar] [CrossRef]
- Sun, Y.; Nishida, T. Band Structures of Strained Semiconductors; Springer: Boston, MA, USA, 2009. [Google Scholar] [CrossRef]
- Landau, A.; Khistyaev, K.; Dolgikh, S.; Krylov, A.I. Frozen natural orbitals for ionized states within equation-of-motion coupled-cluster formalism. J. Chem. Phys. 2010, 132, 014109. [Google Scholar] [CrossRef] [Green Version]
- Steeve, C.; Salahub, D.R. Density Functional Theory, Methods, Techniques, and Applications. In Atomic Clusters and Nanoparticles. Agregats Atomiques et Nanoparticules; Springer: Berlin/Heidelberg, Germany, 2000; Volume 73, pp. 105–160. [Google Scholar]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into Current Limitations of Density Functional Theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef] [Green Version]
- Garza, J.; Vargas, R.; Nichols, J.; Dixon, D.A. Orbital energy analysis with respect to LDA and self-interaction corrected exchange-only potentials. J. Chem. Phys. 2001, 114, 639–651. [Google Scholar] [CrossRef]
- Giarusso, S.; Gori-Giorgi, P. Exchange-Correlation Energy Densities and Response Potentials: Two Definitions and Analytical Model for the Strong-Coupling Limit of a Stretched Bond. J. Phys. Chem. 2020, 124, 2473–2482. [Google Scholar] [CrossRef]
- Grüning, M.; Gritsenko, O.V.; Baerends, E.J. Exchange-correlation energy and potential as approximate functionals of occupied and virtual Kohn-Sham orbitals: Application to dissociating H2. J. Chem. Phys. 2003, 118, 7183. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DFT Method | Theoretical Results | Experimental Results | References |
|---|---|---|---|
| DFT-theoretical | Reported the possibility of obtaining band gap by combining CNT segments and BNNT segments. | None | [18] |
| DFT-Theoretical | It has been reported that the CNT-BNNT can be competitive in thermodynamical stability for sufficiently large segments of building blocks in the axial direction. | [19] | |
| Non-equilibrium Green’s function method combined with the density functional theory | Carbon and boron nitride nanotubes were obtained with semiconducting properties of 2.2 eV; results predicted that CBNNT could become potential candidates in the field of nano rectifiers. | [20] | |
| Density functional theory and using basis set 6–31 g (d,p) | Reported the band gap of 1.21 and 2.52 eV which is close with our results of PW91. | [21] | |
| Geometry optimization implemented in the CASTEP package | Reported 2.3 eV band gap in hetero nanotubes with the lowest unoccupied molecular orbital and the highest occupied molecular orbital mainly located on the carbon nanotube section. | [22] | |
| Vienna ab initio simulation package | The electrical conductivity of CBNNT is increased by oxygen absorption. | [23] | |
| CVD | 2.41 eV band gap was reported. Recommended for theoretical and computational confirmatory tests. | [24] | |
| VASP code | Reported 1.06 eV band gap, also reported that the highest occupied and lowest unoccupied orbital gap of carbon-boron-nitride hetero nanotubes can be significantly tuned by modifying the CNT and BNNT general geometry. | [25] | |
| LDA | Reported 5.6 eV band gap in BNNT. Furthermore, analysis of the HOMO–LUMO gap after the adsorption process showed that the HOMO value increased marginally while the LUMO value decreased dramatically in the curcumin-BNNT complexes | [26] | |
| GGA-PBE | Reported 1.83 eV band gap, which agrees with this current research. Also, reported that the band gap of the CBNNT system is greatly influenced by the nanotube aspect ratio. | [27] | |
| GGA-PW91 | Reported 2.52 eV band gap, highlighted the potentials of CBNNT for the next generation spintronics. | [28] |
| Iterations | Delta-h | Delta-r | K-Point (Gamma) | Iterations | ||||
|---|---|---|---|---|---|---|---|---|
| a1 | a2 | a3 | ||||||
| 0 | 1.000000 | 1.000000 | 1.000000 | 41 | 41 | 0 | ||
| 1 | 1.7171 × 10−3 | 7.5733 × 10−2 | 1.001208 | 1.001208 | 1.000000 | 20 | 20 | 0 |
| 2 | −4.1446 × 10−5 | −1.8236 × 10−4 | 1.001205 | 1.001205 | 1.000000 | 0 | 0 | 0 |
| 3 | 0.0000e + 00 | 0.0000e + 00 | 1.001205 | 1.001205 | 1.000000 | |||
| S/No | Pseudopotential | Total Energy Achieved (Ry) | The Calculated Band Gap (eV) | SCF Iterations |
|---|---|---|---|---|
| 1 | LDA | −1299.17 | 0.043 | 72 |
| 2 | PBE | −1444.79 | 1.87 | 72 |
| 3 | PW91 | −1314.23 | 0.19 | 74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itas, Y.S.; Suleiman, A.B.; Ndikilar, C.E.; Lawal, A.; Razali, R.; Khandaker, M.U.; Ahmad, P.; Tamam, N.; Sulieman, A. The Exchange-Correlation Effects on the Electronic Bands of Hybrid Armchair Single-Walled Carbon Boron Nitride Nanostructure. Crystals 2022, 12, 394. https://doi.org/10.3390/cryst12030394
Itas YS, Suleiman AB, Ndikilar CE, Lawal A, Razali R, Khandaker MU, Ahmad P, Tamam N, Sulieman A. The Exchange-Correlation Effects on the Electronic Bands of Hybrid Armchair Single-Walled Carbon Boron Nitride Nanostructure. Crystals. 2022; 12(3):394. https://doi.org/10.3390/cryst12030394
Chicago/Turabian StyleItas, Yahaya Saadu, Abdussalam Balarabe Suleiman, Chifu E. Ndikilar, Abdullahi Lawal, Razif Razali, Mayeen Uddin Khandaker, Pervaiz Ahmad, Nissren Tamam, and Abdelmoneim Sulieman. 2022. "The Exchange-Correlation Effects on the Electronic Bands of Hybrid Armchair Single-Walled Carbon Boron Nitride Nanostructure" Crystals 12, no. 3: 394. https://doi.org/10.3390/cryst12030394
APA StyleItas, Y. S., Suleiman, A. B., Ndikilar, C. E., Lawal, A., Razali, R., Khandaker, M. U., Ahmad, P., Tamam, N., & Sulieman, A. (2022). The Exchange-Correlation Effects on the Electronic Bands of Hybrid Armchair Single-Walled Carbon Boron Nitride Nanostructure. Crystals, 12(3), 394. https://doi.org/10.3390/cryst12030394