
A Fast, Low-Cost and Simple Method for Predicting Atomic/Inter-Atomic Properties by Combining a Low Dimensional Deep Learning Model with a Fragment Based Graph Convolutional Network

Abstract
:1. Introduction
2. Computational Details
2.1. Structure of the F-GCN
2.2. Utilisation of QM Descriptors by a Low Dimensional DNN
3. Results and Discussion
3.1. Performance of the QM Augmented F-GCN in NMR Chemical Shift Predictions
3.2. Performance of the QM Augmented F-GCN in BDE Predictions
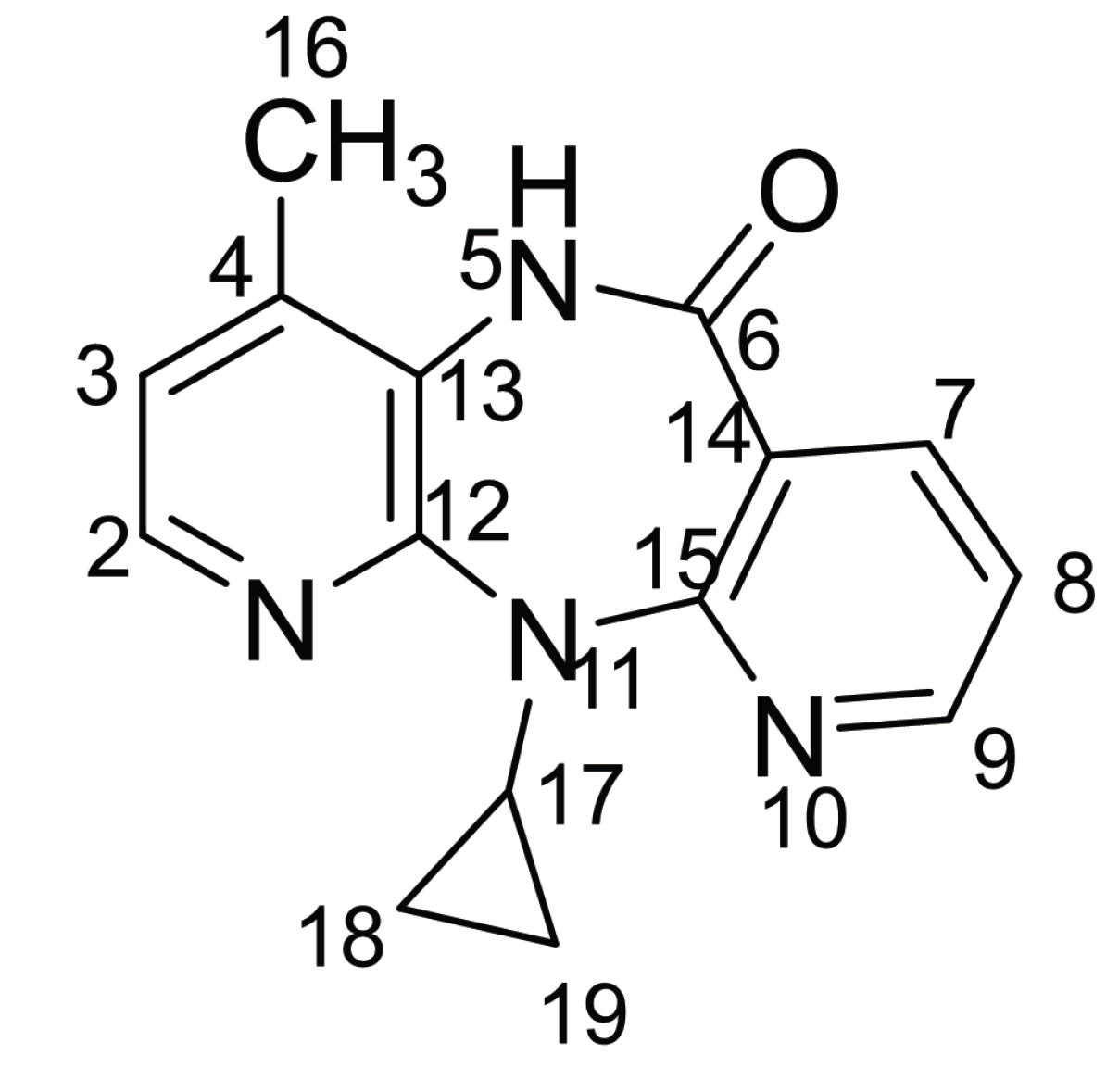
3.3. Nevirapine Structure Elucidation by the QM Augmented F-GCN Architecture
3.4. Calculations of Phenol O-H BDEs by the QM Augmented F-GCN Architecture
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gani, T.Z.H.; Kulik, H.J. Understanding and Breaking Scaling Relations in Single-Site Catalysis: Methane to Methanol Conversion by FeIV=O. ACS Catal. 2018, 8, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Marque, S.R.A.; Matyjaszewski, K.; Coote, M.L. Linear-Free Energy Relationships for Modeling Structure–Reactivity Trends in Controlled Radical Polymerization. Macromolecules 2011, 44, 7568–7583. [Google Scholar] [CrossRef]
- Bian, C.; Wang, S.; Liu, Y.; Jing, X. Thermal stability of phenolic resin: New insights based on bond dissociation energy and reactivity of functional groups. RSC Adv. 2016, 6, 55007–55016. [Google Scholar] [CrossRef]
- Kim, S.; Chmely, S.C.; Nimlos, M.R.; Bomble, Y.J.; Foust, T.D.; Paton, R.S.; Beckham, G.T. Computational Study of Bond Dissociation Enthalpies for a Large Range of Native and Modified Lignins. J. Phys. Chem. Lett. 2011, 2, 2846–2852. [Google Scholar] [CrossRef]
- Drew, K.L.; Reynisson, J. The impact of carbon–hydrogen bond dissociation energies on the prediction of the cytochrome P450 mediated major metabolic site of drug-like compounds. Eur. J. Med. Chem. 2012, 56, 48–55. [Google Scholar] [CrossRef]
- Blanksby, S.J.; Ellison, G.B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef]
- Hartwig, J.F. Catalyst-Controlled Site-Selective Bond Activation. Acc. Chem. Res. 2017, 50, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Yao, K.; Herr, J.E.; Brown, S.N.; Parkhill, J. Intrinsic Bond Energies from a Bonds-in-Molecules Neural Network. J. Phys. Chem. Lett. 2017, 8, 2689–2694. [Google Scholar] [CrossRef]
- Gao, P.; Wang, X.; Yu, H. Towards an Accurate Prediction of Nitrogen Chemical Shifts by Density Functional Theory and Gauge-Including Atomic Orbital. Adv. Theory Simul. 2019, 2, 1800148. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Wang, X.; Huang, Z.; Yu, H. 11B NMR Chemical Shift Predictions via Density Functional Theory and Gauge-Including Atomic Orbital Approach: Applications to Structural Elucidations of Boron-Containing Molecules. ACS Omega 2019, 4, 12385–12392. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational Prediction of 1H and 13C Chemical Shifts: A Useful Tool for Natural Product, Mechanistic, and Synthetic Organic Chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, J.; Chen, H. A systematic benchmarking of 31P and 19F NMR chemical shift predictions using different DFT/GIAO methods and applying linear regression to improve the prediction accuracy. Int. J. Quantum Chem. 2020, 121, e26482. [Google Scholar] [CrossRef]
- Behler, J. Perspective: Machine learning potentials for atomistic simulations. J. Chem. Phys. 2016, 145, 170901. [Google Scholar] [CrossRef] [Green Version]
- Behler, J. First Principles Neural Network Potentials for Reactive Simulations of Large Molecular and Condensed Systems. Angew. Chem. Int. Ed. 2017, 56, 12828–12840. [Google Scholar] [CrossRef]
- Wang, J.; Olsson, S.; Wehmeyer, C.; Pérez, A.; Charron, N.E.; de Fabritiis, G.; Noé, F.; Clementi, C. Machine Learning of Coarse-Grained Molecular Dynamics Force Fields. ACS Cent. Sci. 2019, 5, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Botu, V.; Batra, R.; Chapman, J.; Ramprasad, R. Machine Learning Force Fields: Construction, Validation, and Outlook. J. Phys. Chem. C 2017, 121, 511–522. [Google Scholar] [CrossRef]
- Meldgaard, S.A.; Kolsbjerg, E.L.; Hammer, B. Machine learning enhanced global optimization by clustering local environments to enable bundled atomic energies. J. Chem. Phys. 2018, 149, 134104. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, R.; Xie, Y.; Jiang, D.E. Global minimization of gold clusters by combining neural network potentials and the basin-hopping method. Nanoscale 2015, 7, 14817–14821. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, K.H.; Jørgensen, M.S.; Bruix, A.; Hammer, B. Accelerating atomic structure search with cluster regularization. J. Chem. Phys. 2018, 148, 241734. [Google Scholar] [CrossRef]
- Wexler, R.B.; Martirez, J.M.P.; Rappe, A.M. Chemical Pressure-Driven Enhancement of the Hydrogen Evolving Activity of Ni2P from Nonmetal Surface Doping Interpreted via Machine Learning. J. Am. Chem. Soc. 2018, 140, 4678–4683. [Google Scholar] [CrossRef] [PubMed]
- Mansouri Tehrani, A.; Oliynyk, A.O.; Parry, M.; Rizvi, Z.; Couper, S.; Lin, F.; Miyagi, L.; Sparks, T.D.; Brgoch, J. Machine Learning Directed Search for Ultraincompressible, Superhard Materials. J. Am. Chem. Soc. 2018, 140, 9844–9853. [Google Scholar] [CrossRef] [PubMed]
- Panapitiya, G.; Avendaño-Franco, G.; Ren, P.; Wen, X.; Li, Y.; Lewis, J.P. Machine-Learning Prediction of CO Adsorption in Thiolated, Ag-Alloyed Au Nanoclusters. J. Am. Chem. Soc. 2018, 140, 17508–17514. [Google Scholar] [CrossRef] [PubMed]
- Rupp, M.; Ramakrishnan, R.; von Lilienfeld, O.A. Machine Learning for Quantum Mechanical Properties of Atoms in Molecules. J. Phys. Chem. Lett. 2015, 6, 3309–3313. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Wilbraham, L.; Slater, B.J.; Zwijnenburg, M.A.; Sprick, R.S.; Cooper, A.I. Accelerated Discovery of Organic Polymer Photocatalysts for Hydrogen Evolution from Water through the Integration of Experiment and Theory. J. Am. Chem. Soc. 2019, 141, 9063–9071. [Google Scholar] [CrossRef] [Green Version]
- Ahneman, D.T.; Estrada, J.G.; Lin, S.; Dreher, S.D.; Doyle, A.G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 2018, 360, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Mater, A.C.; Coote, M.L. Deep Learning in Chemistry. J. Chem. Inf. Model. 2019, 59, 2545–2559. [Google Scholar] [CrossRef]
- Faber, F.A.; Hutchison, L.; Huang, B.; Gilmer, J.; Schoenholz, S.S.; Dahl, G.E.; Vinyals, O.; Kearnes, S.; Riley, P.F.; von Lilienfeld, O.A. Prediction Errors of Molecular Machine Learning Models Lower than Hybrid DFT Error. J. Chem. Theory Comput. 2017, 13, 5255–5264. [Google Scholar] [CrossRef]
- Kendall, A.; Gal, Y. What Uncertainties Do We Need in Bayesian Deep Learning for Computer Vision? In Advances in Neural Information Processing Systems; Curran Associates, Inc.: Red Hook, NY, USA, 2017; Volume 30. [Google Scholar]
- De Sousa Ribeiro, F.; Calivá, F.; Swainson, M.; Gudmundsson, K.; Leontidis, G.; Kollias, S. Deep Bayesian Self-Training. Neural Comput. Appl. 2020, 32, 4275–4291. [Google Scholar] [CrossRef] [Green Version]
- Schütt, K.T.; Sauceda, H.E.; Kindermans, P.J.; Tkatchenko, A.; Müller, K.R. SchNet – A deep learning architecture for molecules and materials. J. Chem. Phys. 2018, 148, 241722. [Google Scholar] [CrossRef]
- Lu, C.; Liu, Q.; Wang, C.; Huang, Z.; Lin, P.; He, L. Molecular Property Prediction: A Multilevel Quantum Interactions Modeling Perspective. arXiv 2019, arXiv:1906.11081. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Zhang, J.; Peng, Q.; Zhang, J.; Glezakou, V.A. General Protocol for the Accurate Prediction of Molecular 13C/1H NMR Chemical Shifts via Machine Learning Augmented DFT. J. Chem. Inf. Model. 2020, 60, 3746–3754. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, J.; Sun, Y.; Yu, J. Accurate predictions of aqueous solubility of drug molecules via the multilevel graph convolutional network (MGCN) and SchNet architectures. Phys. Chem. Chem. Phys. 2020, 22, 23766–23772. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, J.; Sun, Y.; Yu, J. Toward Accurate Predictions of Atomic Properties via Quantum Mechanics Descriptors Augmented Graph Convolutional Neural Network: Application of This Novel Approach in NMR Chemical Shifts Predictions. J. Phys. Chem. Lett. 2020, 0, 9812–9818. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, J.; Qiu, H.; Zhao, S. A general QSPR protocol for the prediction of atomic/inter-atomic properties: A fragment based graph convolutional neural network (F-GCN). Phys. Chem. Chem. Phys. 2021, 23, 13242–13249. [Google Scholar] [CrossRef]
- St. John, P.C.; Guan, Y.; Kim, Y.; Kim, S.; Paton, R.S. Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost. Nat. Commun. 2020, 11, 2328. [Google Scholar] [CrossRef]
- Kwon, Y.; Lee, D.; Choi, Y.S.; Kang, M.; Kang, S. Neural Message Passing for NMR Chemical Shift Prediction. J. Chem. Inf. Model. 2020, 60, 2024–2030. [Google Scholar] [CrossRef]
- Gerrard, W.; Bratholm, L.A.; Packer, M.J.; Mulholland, A.J.; Glowacki, D.R.; Butts, C.P. IMPRESSION—Prediction of NMR parameters for 3-dimensional chemical structures using machine learning with near quantum chemical accuracy. Chem. Sci. 2020, 11, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zheng, D.; Ye, Z.; Gan, Q.; Li, M.; Song, X.; Zhou, J.; Ma, C.; Yu, L.; Gai, Y.; et al. Deep Graph Library: A Graph-Centric, Highly-Performant Package for Graph Neural Networks. arXiv 2019, arXiv:cs.LG/1909.01315. [Google Scholar]
- Chen, G.; Chen, P.; Hsieh, C.Y.; Lee, C.K.; Liao, B.; Liao, R.; Liu, W.; Qiu, J.; Sun, Q.; Tang, J.; et al. Alchemy: A Quantum Chemistry Dataset for Benchmarking AI Models. arXiv 2019, arXiv:1906.09427. [Google Scholar]
- Pople, J.A.; Bernstein, H.J.; Schneider, W.G. High-Resolution Nuclear Magnetic Resonance; McGraw-Hill: New York, NY, USA, 1959. [Google Scholar]
- Becker, E. High Resolution NMR: Theory and Chemical Applications; Elsevier Science: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Slichter, C. Principles of Magnetic Resonance; Springer Series in Solid-State Sciences; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Lodewyk, M.W.; Soldi, C.; Jones, P.B.; Olmstead, M.M.; Rita, J.; Shaw, J.T.; Tantillo, D.J. The Correct Structure of Aquatolide—Experimental Validation of a Theoretically-Predicted Structural Revision. J. Am. Chem. Soc. 2012, 134, 18550–18553. [Google Scholar] [CrossRef] [PubMed]
- Xin, D.; Sader, C.A.; Chaudhary, O.; Jones, P.J.; Wagner, K.; Tautermann, C.S.; Yang, Z.; Busacca, C.A.; Saraceno, R.A.; Fandrick, K.R.; et al. Development of a 13C NMR Chemical Shift Prediction Procedure Using B3LYP/cc-pVDZ and Empirically Derived Systematic Error Correction Terms: A Computational Small Molecule Structure Elucidation Method. J. Org. Chem. 2017, 82, 5135–5145. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision E.01.; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Internet Bond-Energy Databank (pKa and BDE)—iBonD Home Page. 2020. Available online: http://ibond.nankai.edu.cn/ (accessed on 22 October 2022).
- Denisov, E. A new semiempirical method of estimation of activity and bond dissociation energies of antioxidants. Polym. Degrad. Stab. 1995, 49, 71–75. [Google Scholar] [CrossRef]
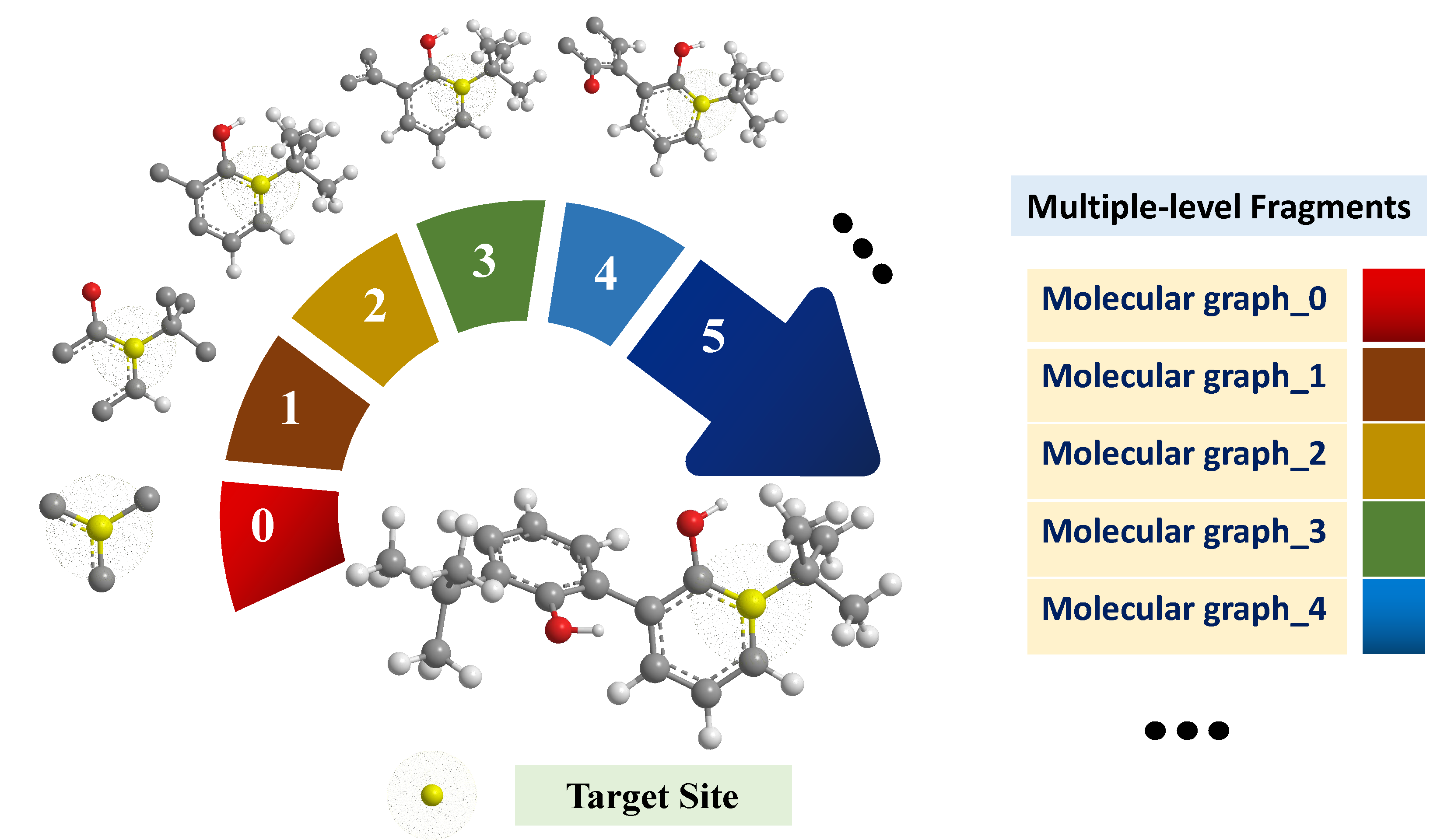







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Exptl. | Pred. | Error |
|---|---|---|---|
| 2 | 140.36 | 143.33 | 2.97 |
| 3 | 120.35 | 122.41 | 2.06 |
| 4 | 139.52 | 137.42 | 2.10 |
| 6 | 169.06 | 165.45 | 3.61 |
| 7 | 144.47 | 138.38 | 6.09 |
| 8 | 118.99 | 119.93 | 0.94 |
| 9 | 152.15 | 155.36 | 3.21 |
| 12 | 154.17 | 152.68 | 1.49 |
| 13 | 124.97 | 126.29 | 1.32 |
| 14 | 122.13 | 120.39 | 1.74 |
| 15 | 160.73 | 159.95 | 0.78 |
| 16 | 17.86 | 20.39 | 2.53 |
| 17 | 29.65 | 32.49 | 2.84 |
| 18 | 8.88 | 9.07 | 0.19 |
| 19 | 9.15 | 9.45 | 0.30 |
| Position | Exptl. | Pred. | Error |
|---|---|---|---|
| 2 | 8.08 | 7.80 | 0.28 |
| 3 | 7.07 | 6.71 | 0.36 |
| 7 | 8.02 | 7.93 | 0.09 |
| 8 | 7.20 | 6.77 | 0.43 |
| 9 | 8.51 | 8.25 | 0.26 |
| 16 | 2.34 | 2.25 | 0.09 |
| 17 | 3.62 | 3.60 | 0.02 |
| 18 | 0.35 | 0.51 | 0.16 |
| 19 | 0.88 | 0.87 | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, P.; Liu, Z.; Zhang, J.; Wang, J.-A.; Henkelman, G. A Fast, Low-Cost and Simple Method for Predicting Atomic/Inter-Atomic Properties by Combining a Low Dimensional Deep Learning Model with a Fragment Based Graph Convolutional Network. Crystals 2022, 12, 1740. https://doi.org/10.3390/cryst12121740
Gao P, Liu Z, Zhang J, Wang J-A, Henkelman G. A Fast, Low-Cost and Simple Method for Predicting Atomic/Inter-Atomic Properties by Combining a Low Dimensional Deep Learning Model with a Fragment Based Graph Convolutional Network. Crystals. 2022; 12(12):1740. https://doi.org/10.3390/cryst12121740
Chicago/Turabian StyleGao, Peng, Zonghang Liu, Jie Zhang, Jia-Ao Wang, and Graeme Henkelman. 2022. "A Fast, Low-Cost and Simple Method for Predicting Atomic/Inter-Atomic Properties by Combining a Low Dimensional Deep Learning Model with a Fragment Based Graph Convolutional Network" Crystals 12, no. 12: 1740. https://doi.org/10.3390/cryst12121740
APA StyleGao, P., Liu, Z., Zhang, J., Wang, J.-A., & Henkelman, G. (2022). A Fast, Low-Cost and Simple Method for Predicting Atomic/Inter-Atomic Properties by Combining a Low Dimensional Deep Learning Model with a Fragment Based Graph Convolutional Network. Crystals, 12(12), 1740. https://doi.org/10.3390/cryst12121740