3.1. The Involved Precipitation Reactions Analysis
The complexation of Fe
3+ with phosphate is complicated in solution [
19,
20]. In this work, complexation ions such as Fe(PO
4)
23−, Fe(OH)PO
4−, FeH
3(PO
4)
33−, Fe(PO
4)
23−, Fe
2PO
43+, etc., were not taken into account owing to small equilibrium constants (<10
−8). The possible reactions in solution and corresponding equilibrium constants are listed in
Table 3 [
21,
22,
23]. On the basis of mass conservation and reactions in
Table 3, the mass balance of Fe and P can be expressed, as given in formulas (1) and (2). The total concentration of Fe and P was measured by redox titration of potassium dichromate and the gravimetric method. According to pH and the equilibrium constants of possible reactions, the corresponding concentration of complexation ions can be represented by free Fe
3+ ion and H
3PO
4 molecules.
where [Fe]
T and [P]
T is the total concentration of Fe and P in solution; [
x] is the concentration of
x;
x is the species of complexation.
And the concentration of free Fe
3+ ion and H
3PO
4 molecules, under pH ranging from 0.2 to 1.3, can be calculated by the total concentration and mass balance of Fe and P. The equations for the concentration of Fe
3+ and H
3PO
4 were presented in Formulas (3) and (4). In addition, the concentration of various complexation species with phosphate groups under different pH environments is listed in
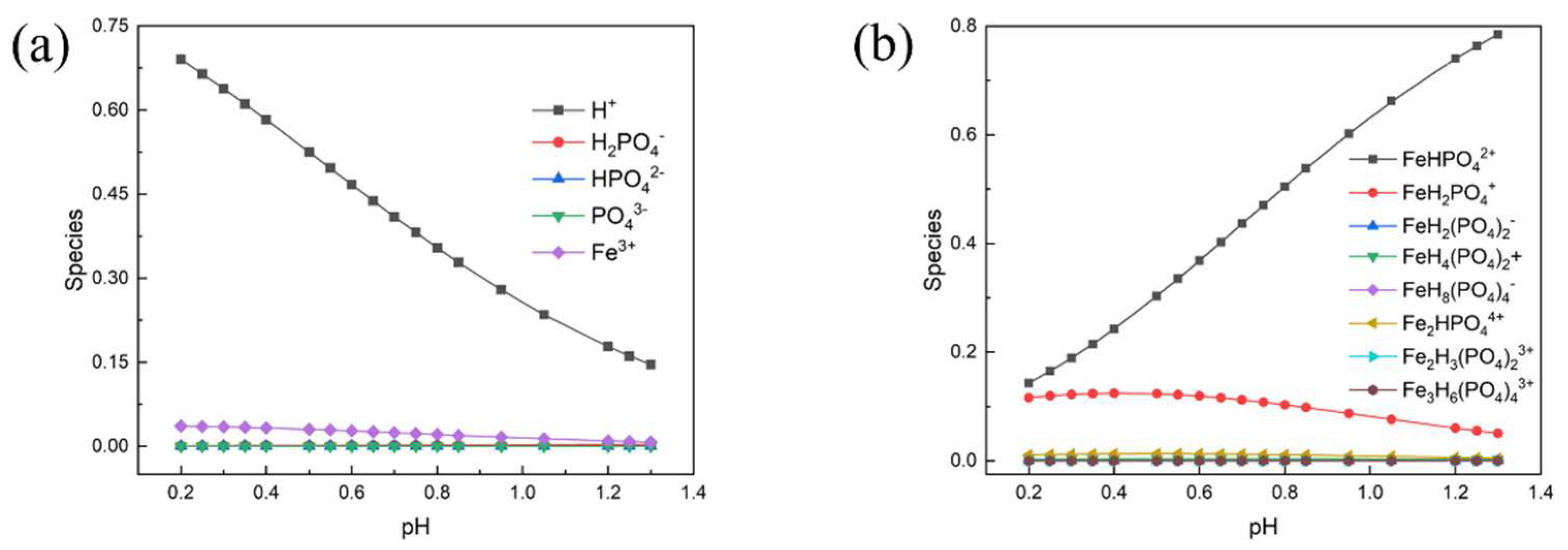
Tables S1–S4 (Supporting Information). The distribution of the various complexation species is presented in
Figure 2.
where
x is the concentration of Fe
3+ and
y is the concentration of H
3PO
4;
a,
b,
A,
B,
C,
D,
E,
F,
G,
H are the constants in different pH environment based on the equilibrium constants of possible reactions respectively.
It can be found that Fe and P are predominantly presented as FeHPO
4+ and FeH
2PO
42+ in the pH range. The pH of the mixture at room temperature in this work is around 0.85 and the proportion of main complexation ions are calculated to be 53.82% FeHPO
4+, and 9.79% FeH
2PO
42+. Therefore, the main precipitation reactions, even during the process of raising the temperature, can be written as the following reactions in
Table 4 (the pH decreased with the increase of temperature).
3.3. Kinetic of Phase Transformation
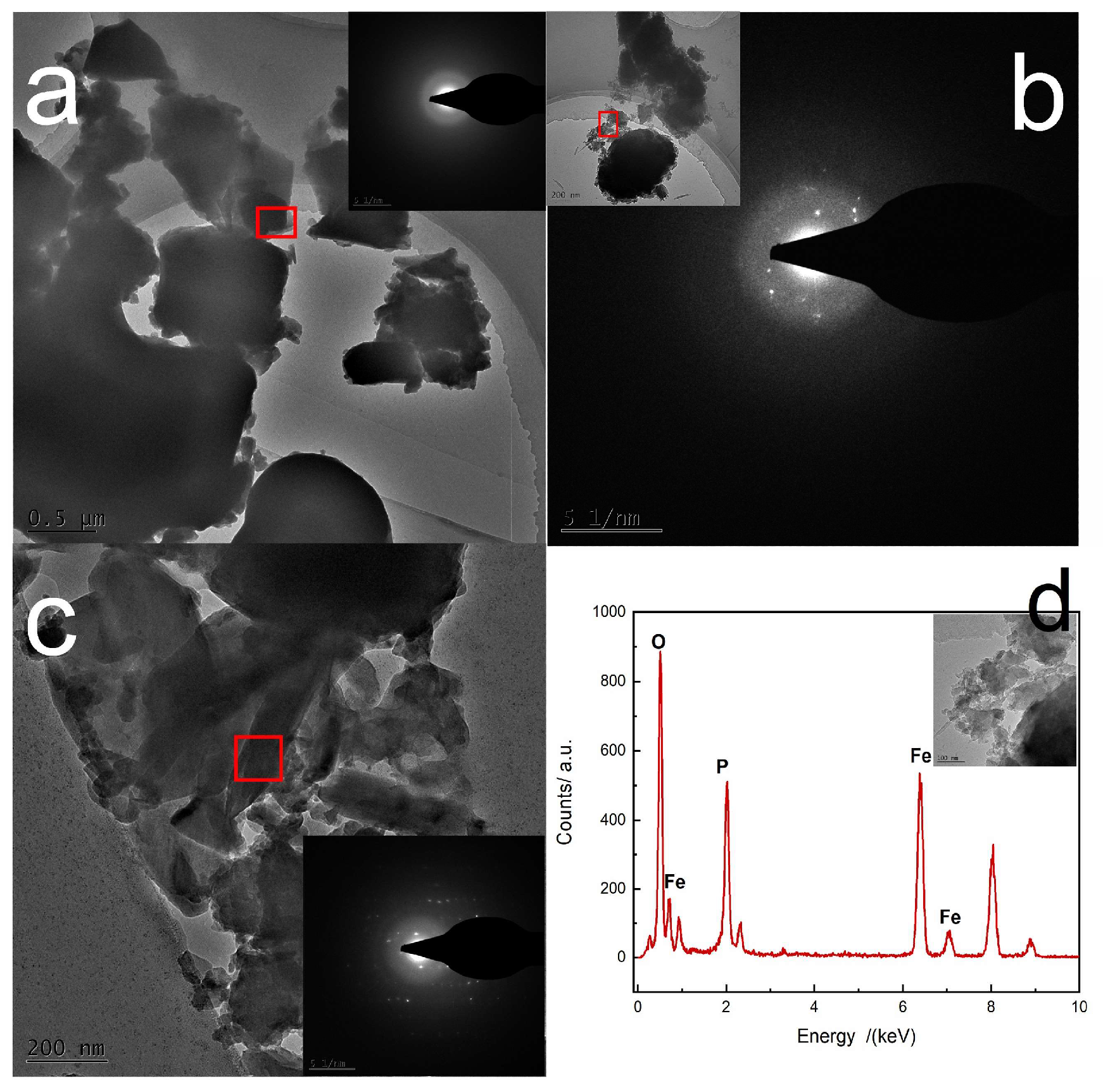
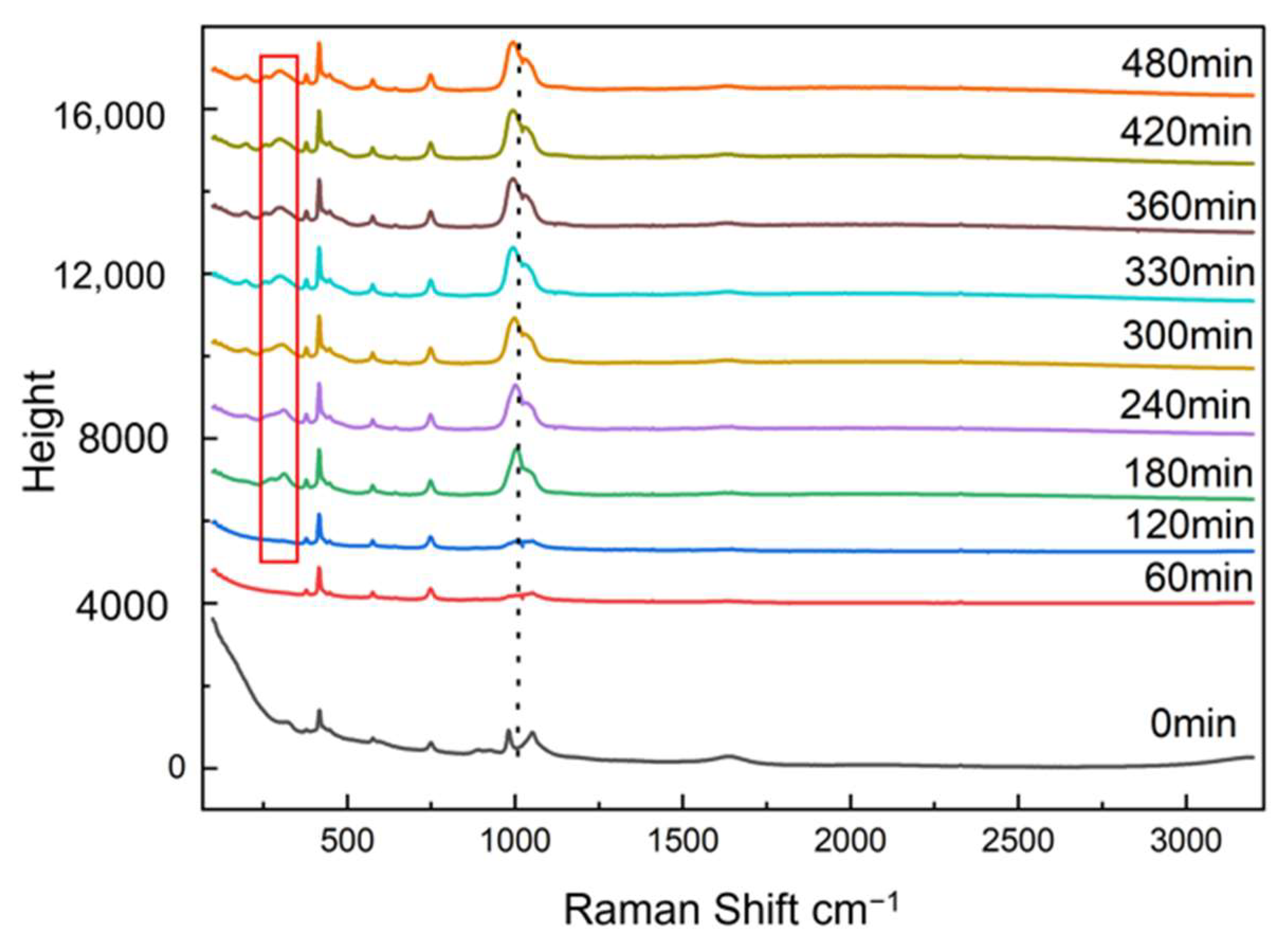
Raman was used to monitor the transformation of amorphous FePO
4·2H
2O, as shown in
Figure 10. Transformation of amorphous FePO
4·2H
2O was followed by the appearance of monoclinic FePO
4·2H
2O characteristic peaks and the changes in height of the characteristic peaks: a new peak at 303 cm
−1 appeared in the Raman spectra and its intensity increased gradually as the monoclinic FePO
4·2H
2O content increased during transformation.
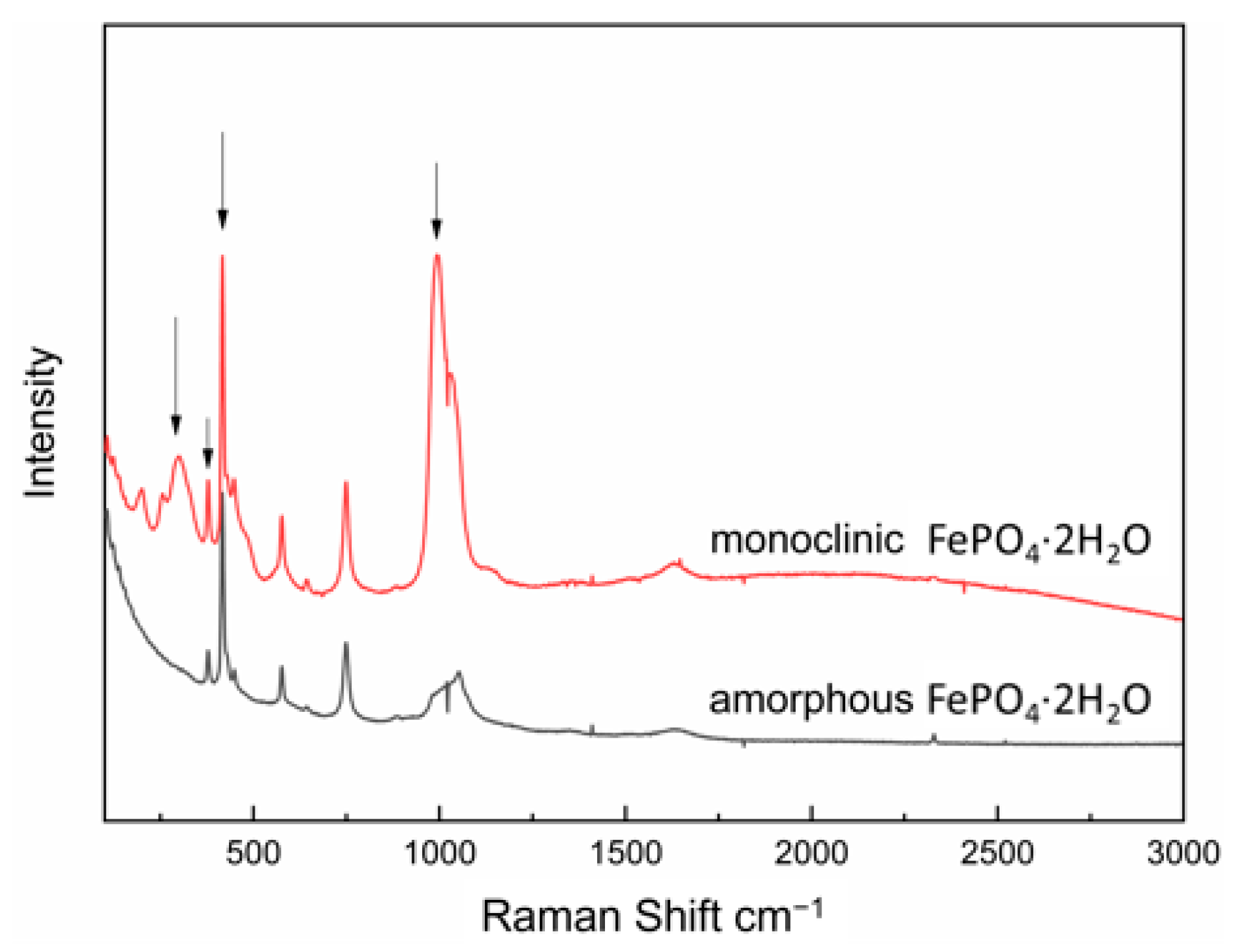
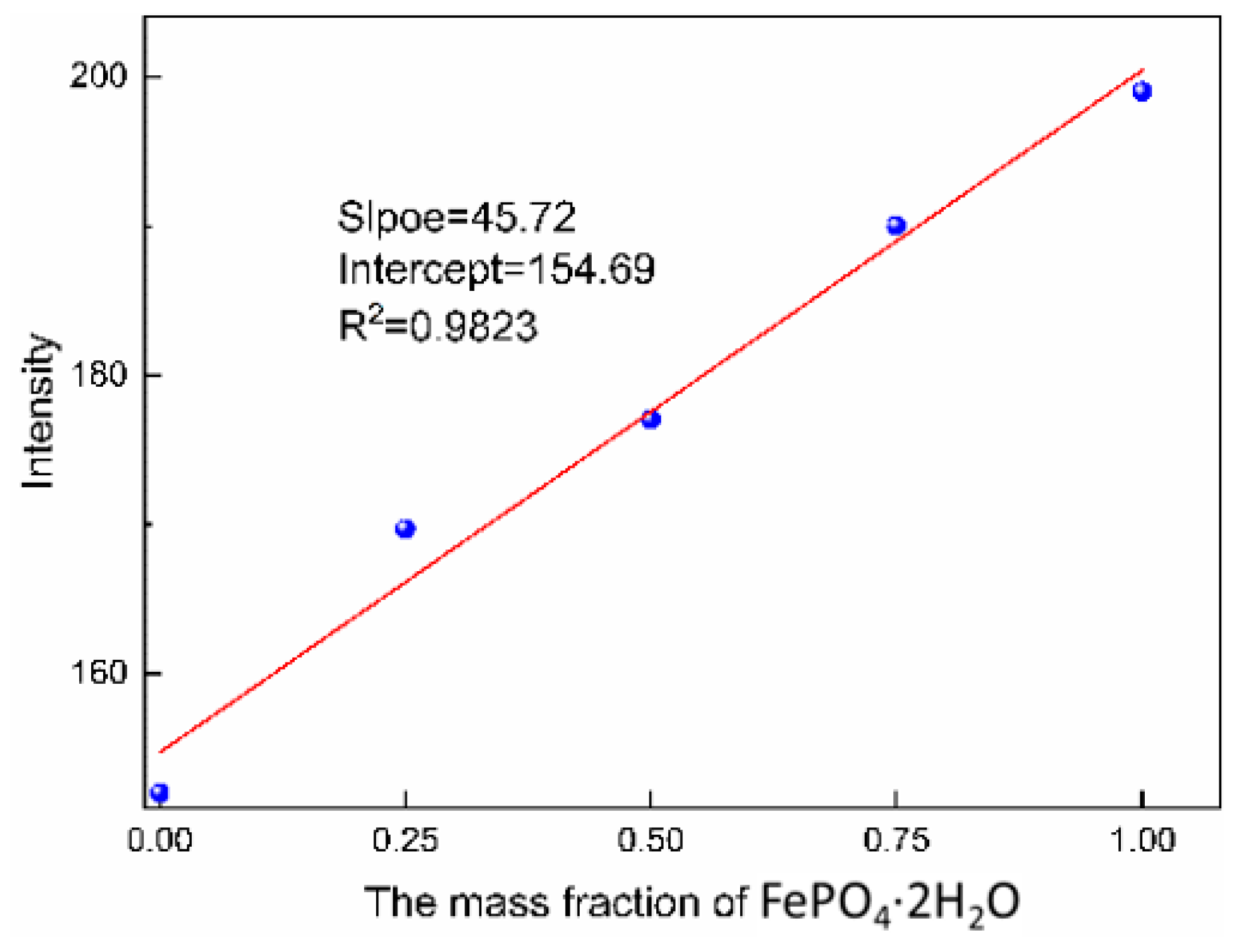
The corresponding characteristic peak intensity of the standard solution was measured by Raman spectroscopy (shown in
Figure 11) and the standard curve could be obtained according to the relationship between intensity of characteristic peak and the mass fraction of monoclinic FePO
4·2H
2O, as shown in
Figure 12. The transformation process was quantitatively evaluated by the relationship, which can be seen in
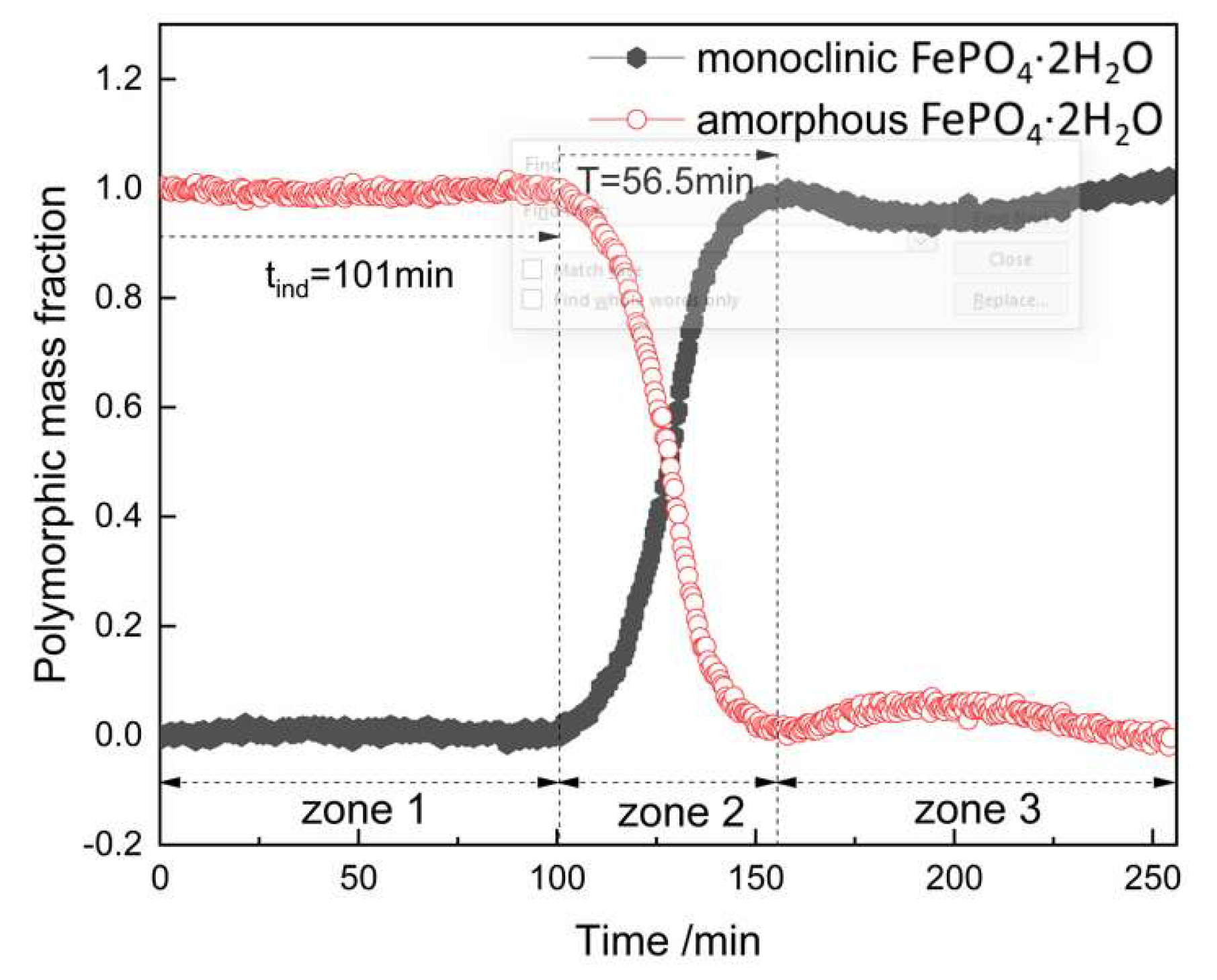
Figure 13. It shows the changes of polymorphic composition during transformation. The mass fraction of the amorphous FePO
4·2H
2O decreased gradually after the induction time of 101 min, which could be attributed to the nucleation of monoclinic FePO
4·2H
2O and the transformation of the amorphous FePO
4·2H
2O. The curves are stable in zone 3 for the completion of transformation.
The Johnson–Mehl–Avrami (JMA) dynamics model was used for describing the phase transformation, as shown in equation 4. It was considered that the solution is well-mixed and the growth rate of crystals is independent of time. It assumed that the nucleation sites were located in the well-mixed reactant bases on solid-state reactions [
37,
38]. The calculated parameters were shown in the
Table 6 and the fitting relevance value of R
2 was over 0.97 indicating that the fitting result was great consistent with experimental results.
where
x is the percentage content of monoclinic FePO
4·2H
2O at reaction time
t;
tind is the induction period of FePO
4·2H
2O transformation; K is the rate constant of transformation; n (called as Avrami exponent) is a constant related to the behaviors of nucleation and growth of FePO
4·2H
2O; if the value of n is over 1, the nucleation is the key factor for phase transformation or the migration of the chemicals to the nucleation point is dominant.
According to the spectra presented in
Figure 13 and the Equation (5), the relationship of time and crystallinity and the parameters of JMA dynamics model were obtained, which were shown in
Figure 14 and
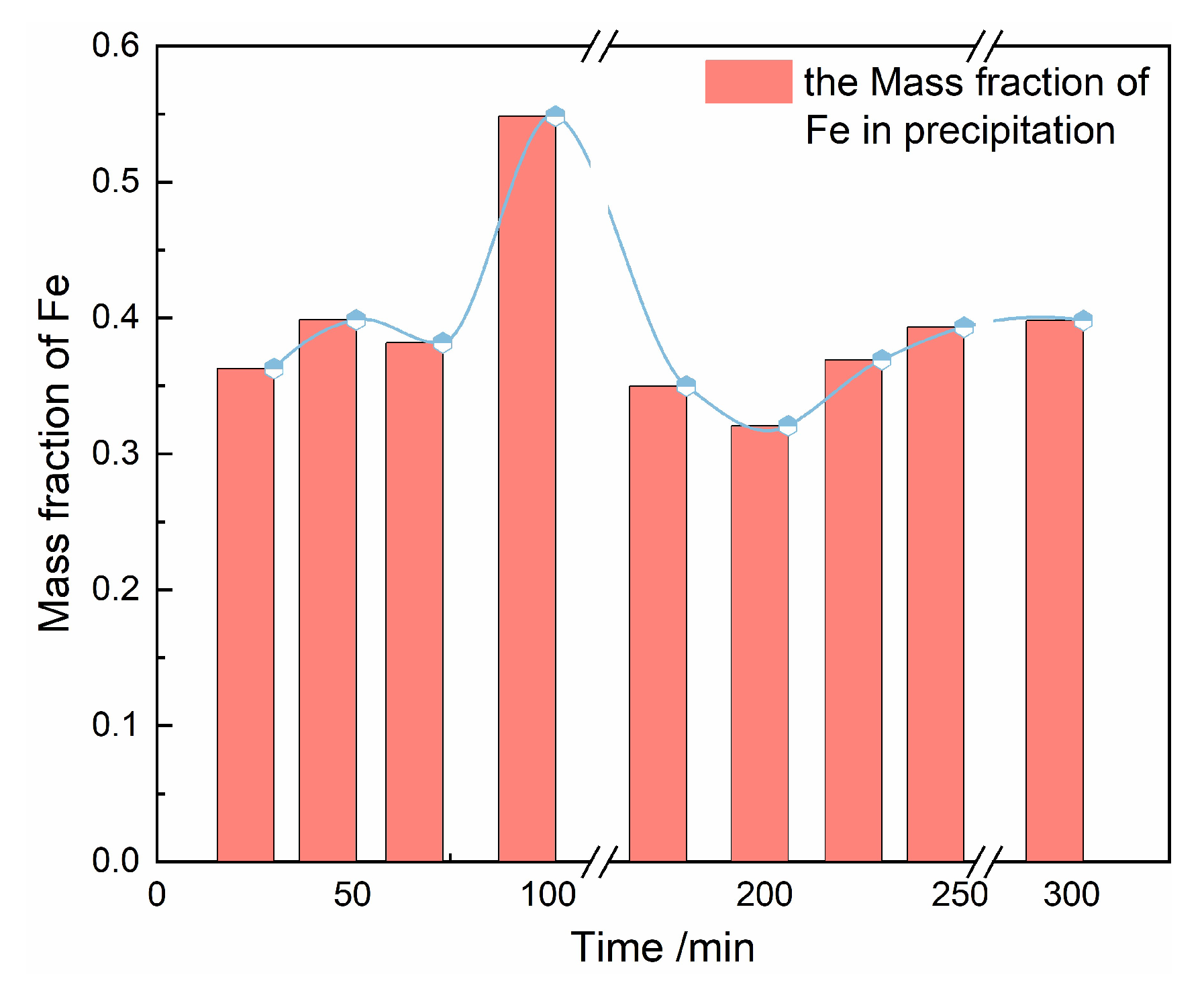
Table 6. The time required for transformation was 56.5 min and the induction period of this process was 101 min. Moreover, the concentration of Fe in the solution and the intensity of the corresponding characteristic peak at 303 cm
−1 changed almost simultaneously: the concentration of Fe in solution decreased accompanying by the increase of the corresponding peak intensity with the time evolution (
Supporting Information, Figure S2). It was indicated that the dissolution rate of amorphous FePO
4·2H
2O was slowest during phase transformation. Therefore, the dissolution rate of amorphous FePO
4·2H
2O is considered to be the control step of the transformation rate.
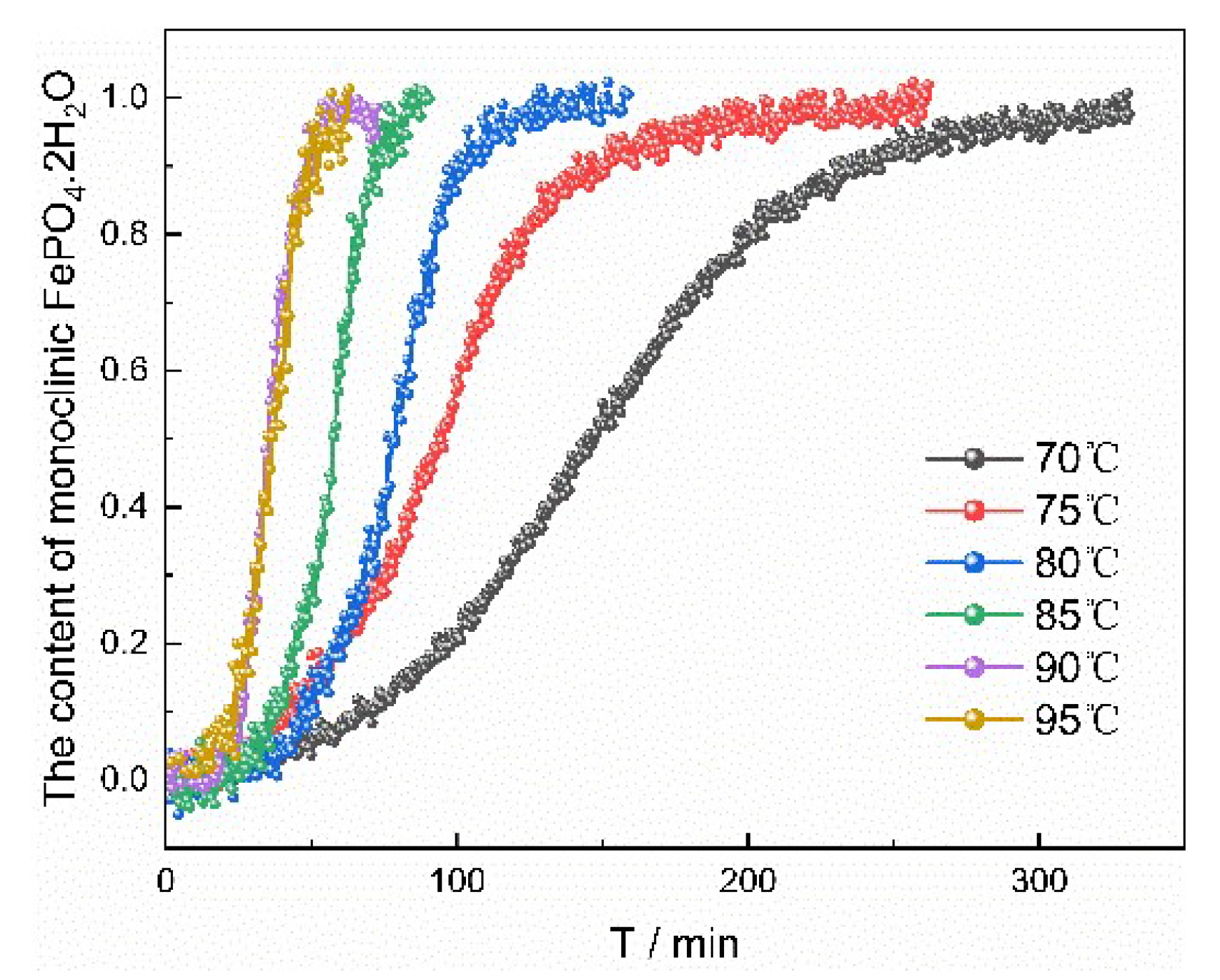
The experiments at different temperatures were carried out in order to evaluate the kinetics of the transformation process. The fitting results at different temperatures evaluated by the JMA model between temperature and the rate constant of transformation are shown in
Table 7, and the changes trends of characteristic peak under different temperatures were presented in
Figure S2 in Supporting Information. The activation energy of the transformation process was obtained by the Arrhenius equation, which was shown as Equation (6):
where
K is the rate constant of transformation; R is the molar gas constant;
T is the thermodynamic temperature; E
a is the apparent activation energy and A is the pre-exponential factor which is also called the frequency factor.
The parameters of reaction calculated by the JMA model are listed in
Table 7. With the increase of reaction temperature, the induction period and the time required for transformation decreased, and rate constant of transformation increased. Increasing temperature can enhance molecular movement and reduce the interfacial energy of solid–liquid interface. Therefore, the nucleation rate of monoclinic FePO
4·2H
2O was accelerated and correspondingly the induction period and the time required for transformation was shortened. The value of the apparent activation energy E
a in the transformation process can be derived by plotting the logarithm of
K against 1/
T and analyzing the slope and intercept (
Figure S3 in Supporting Information). The value of the activation energy was calculated to be 9.619 kJ/mol.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}