
Polymorphic Crystallization Design to Prevent the Degradation of the β-Lactam Structure of a Carbapenem



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Crystallization
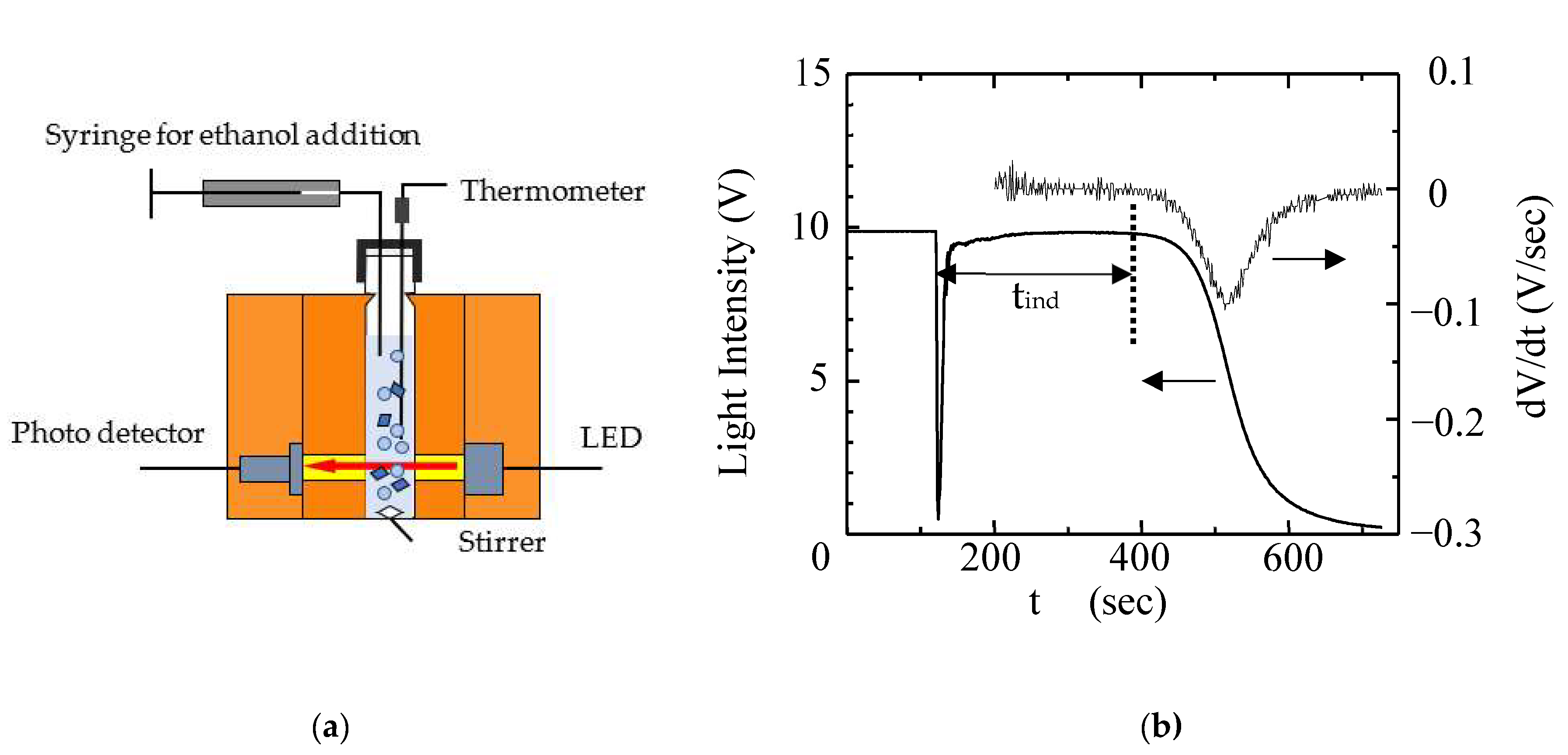
2.3. Determination of the Induction Period (tind)
2.4. Measurement of the Stability of CS-023 Dissolved in Water and Crystalline Products
2.5. Thermal Analysis
2.6. Phase Transition on Instrumental Analysis
2.7. Phase Transition by Absorption and Emission of Water and Ethanol
3. Results and Discussion
3.1. Cooling Crystallization Based on an Aqueous Solution
3.1.1. Crystal Polymorphs Obtained using Cooling Crystallization
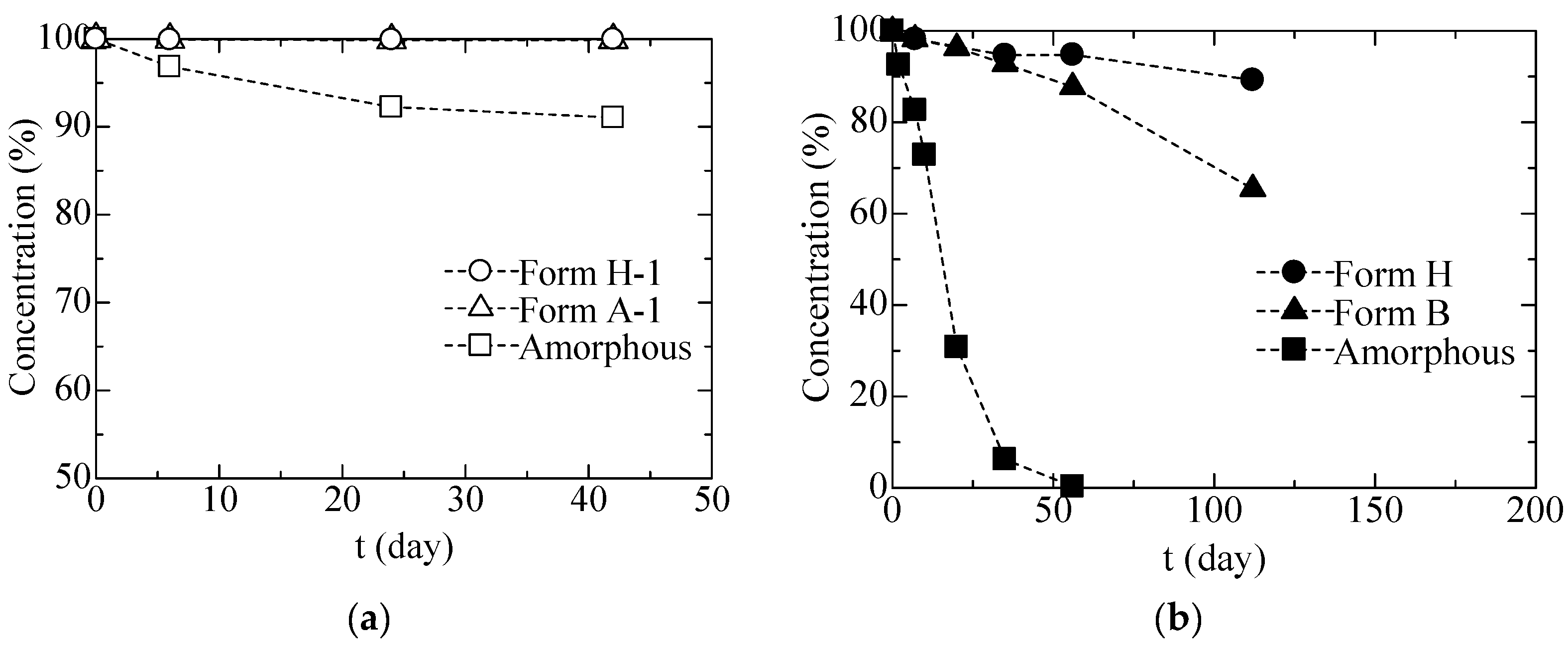
3.1.2. Stabilities of CS-023 Molecules in Aqueous Solution and the Polymorph Form H Crystal
3.2. Poor Solvent Crystallization
3.2.1. Appearance of New Polymorph Form A
3.2.2. Solid Phase Transition of Form A to a New Hydrate Crystal Form B
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, Q3C. Impurities: Guideline for Residual Solvents. Available online: https://www.pmda.go.jp/int-activities/int-harmony/ich/0043.html (accessed on 18 July 2021).
- Hickey, M.B.; Peterson, M.L.; Manas, E.S.; Alvarez, J.; Haeffner, F.; Almarsson, Ö. Hydrates and Solid-State Reactivity, A Survey of β-Lactam Antibiotics. J. Pharm. Sci. 2007, 96, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Takebayashi, Y.; Sunagawa, M.; Isobe, Y.; Hamazume, Y.; Uemura, A.; Noguchi, T. The stability of a novel carbapenem antibiotic, meropenem (SM-7338), in a solid-state formulation for injection. Chem. Pharm. Bull. 1993, 41, 1998–2002. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Needham, F.; Crowder, C.E.; Reid, J.W.; Fawcett, T.G.; Faber, J. X-ray powder diffraction analysis of imipenem monohydrate. Powder Diffr. 2012, 27, 20–24. [Google Scholar] [CrossRef]
- Valentina, C.; Norberto, M.; Giovanni, P. Crystal Chemistry of the Antibiotic Doripenem. J. Pharm. Sci. 2014, 103, 3641–3647. [Google Scholar]
- Nangia, A.; Biradha, K.; Desiraju, G.R. Correlation of biological activity in β-lactam antibiotics with Woodward and Cohen structural parameters: A Cambridge database study. J. Chem. Soc. Perkin Trans. 2 1996, 5, 943–953. [Google Scholar] [CrossRef]
- Vangala, V.R.; Chow, P.S.; Tan, R.B.H. The solvates and salt of antibiotic agent, nitrofurantoin: Structural, thermochemical and desolvation studies. CrystEngComm 2013, 15, 878–889. [Google Scholar] [CrossRef]
- Kawamoto, I.; Shimoji, Y.; Kanno, O.; Kojima, K.; Ishikawa, K.; Matsuyama, E.; Ashida, Y.; Shibayama, T.; Fukuoka, T.; Ohya, S. Synthesis and structure-activity relationships of novel parenteral carbapenems, CS-023 (R-115685) and related compounds containing an amidine moiety. J. Antibiot. 2003, 56, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, T.; Sugiyama, D.; Kamiyama, E.; Tokui, T.; Hirota, T.; Ikeda, T. Characterization of CS-023 (RO4908463), a Novel Parenteral Carbapenem Antibiotic, and Meropenem as Substrates. Drug Metab. Pharmacokinet. 2007, 22, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Yoshifumi, K.; Masakuni, M. Surface Phenomena and Mechanism of Polymorphic Transition in Theophylline. J. Chem. Eng. Jpn. 2010, 43, 253–260. [Google Scholar]
- Loftsson, T. Drug Stability for Pharmaceutical Scientists, 1st ed.; Academic Press: Oxford, UK, 2014; pp. 63–102. [Google Scholar]
- Mullin, J.W. Crystallization, 4th ed.; Butterworth-Heinemann: Oxford, UK, 2001; pp. 181–215. [Google Scholar]
- Saito, A.; Igarashi, K.; Azuma, M.; Ooshima, H. Aggregation of p-Acetanisidide Molecules in the Under- and Super-saturated Solution and Its Effect on Crystallization. J. Chem. Eng. Jpn. 2002, 35, 1133–1139. [Google Scholar] [CrossRef]
- Maruyama, S.; Ooshima, H.; Kato, J. Crystal structures and solvent-mediated transformation of Taltirelin polymorphs. Chem. Eng. J. 1999, 75, 193–200. [Google Scholar] [CrossRef]
- Hirano, M.; Igarashi, K.; Machiya, K.; Tamura, R.; Tue, H.; Ooshima, H. Relationship between Crystal Polymorphism and Solution Structure of an Imidazopyridine Derivative as a Drug Substance for Osteoporosis. J. Chem. Eng. Jpn. 2009, 42, 204–211. [Google Scholar] [CrossRef]
- Xing, Z.; Igarashi, K.; Morioka, A.; Ooshima, H. Repeated Cooling Crystallization for Production of Microcrystals with a Narrow Size Distribution. J. Chem. Eng. Jpn. 2012, 45, 811–815. [Google Scholar] [CrossRef]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuura, S.; Igarashi, K.; Azuma, M.; Ooshima, H. Polymorphic Crystallization Design to Prevent the Degradation of the β-Lactam Structure of a Carbapenem. Crystals 2021, 11, 931. https://doi.org/10.3390/cryst11080931
Matsuura S, Igarashi K, Azuma M, Ooshima H. Polymorphic Crystallization Design to Prevent the Degradation of the β-Lactam Structure of a Carbapenem. Crystals. 2021; 11(8):931. https://doi.org/10.3390/cryst11080931
Chicago/Turabian StyleMatsuura, Shinji, Koichi Igarashi, Masayuki Azuma, and Hiroshi Ooshima. 2021. "Polymorphic Crystallization Design to Prevent the Degradation of the β-Lactam Structure of a Carbapenem" Crystals 11, no. 8: 931. https://doi.org/10.3390/cryst11080931
APA StyleMatsuura, S., Igarashi, K., Azuma, M., & Ooshima, H. (2021). Polymorphic Crystallization Design to Prevent the Degradation of the β-Lactam Structure of a Carbapenem. Crystals, 11(8), 931. https://doi.org/10.3390/cryst11080931