
Controlling Protein Crystallization by Free Energy Guided Design of Interactions at Crystal Contacts

 , , , , , , and
, , , , , , and
Abstract
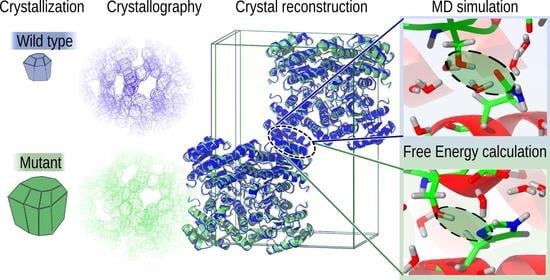
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental and Computational Details
2.1. Protein Mutagenesis, Production, Purification and Analysis
2.2. Crystallization
2.3. X-ray Data Collection and Structure Solution
2.4. System Construction for Atomistic Molecular Dynamics (MD) Simulations
2.5. MD System Equilibration
2.6. MD Free Energy Calculations
2.7. Method Consistency Evaluation and System Reduction Procedure
3. Results
3.1. Experimentally Observed Crystallizability Order
3.2. Preservation of Crystal Packing and Crystal Building Blocks
3.3. Theoretically Calculated Crystallizability Order
3.4. Transforming a “Wet Contact” into a “Dry Contact”
3.5. Optimizing a Hydrogen Bonding Interaction
3.6. Destroying Interactions
3.7. Introducing an Ionic Interaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Graphical Preparation
References
- Doye, J.P.; Louis, A.A.; Vendruscolo, M. Inhibition of protein crystallization by evolutionary negative design. Phys. Biol. 2004, 1, P9–P13. [Google Scholar] [CrossRef]
- Siezen, R.J.; Fisch, M.R.; Slingsby, C.; Benedek, G.B. Opacification of gamma-crystallin solutions from calf lens in relation to cold cataract formation. Proc. Natl. Acad. Sci. USA 1985, 82, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Pande, A.; Pande, J.; Asherie, N.; Lomakin, A.; Ogun, O.; King, J.; Benedek, G.B. Crystal cataracts: Human genetic cataract caused by protein crystallization. Proc. Natl. Acad. Sci. USA 2001, 98, 6116–6120. [Google Scholar] [CrossRef] [PubMed]
- Lessin, L.S.; Jensen, W.N.; Ponder, E. Molecular mechanism of hemolytic anemia in homozygous hemoglobin C disease: Electron microscopic study by the freeze-etching technique. J. Exp. Med. 1969, 130, 443–466. [Google Scholar] [CrossRef] [PubMed]
- Charcot, J.M.; Robin, C. Observation of leukocytosis. TCRMem. Soc. Biol. 1853, 4, 450–454. [Google Scholar]
- von Leyden, E.V. Zur Kenntnis des Asthma bronchiale. Arch. Pathol. Anat. Physiol. Klin. Med. Berl. 1872, 54, 324–344; 346–352. [Google Scholar] [CrossRef]
- Schnepf, E.; Crickmore, N.; Van Rie, J.; Lereclus, D.; Baum, J.; Feitelson, J.; Zeigler, D.; Dean, D. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol. Mol. Biol. Rev. 1998, 62, 775–806. [Google Scholar] [CrossRef]
- Perutz, M.F.; Rossmann, M.G.; Cullis, A.F.; Muirhead, H.; Will, G.; North, A. Structure of hæmoglobin: A three-dimensional Fourier synthesis at 5.5-Å. resolution, obtained by X-ray analysis. Nature 1960, 185, 416–422. [Google Scholar] [CrossRef]
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, R.; Wyckoff, H.; Phillips, D.C. A three-dimensional model of the myoglobin molecule obtained by X-ray analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef]
- Osborne, T.B. Crystallized vegetable proteids. Am. J. Physiol. 1892, 14, 662–689. [Google Scholar]
- Ritthausen, H.; Kellner, O. Über die Eiweisskörper verschiedener Oelsamen. Z. Anal. Chem. 1882, 21, 461–462. [Google Scholar] [CrossRef]
- Ritthausen, H. Krystallinische Eiweisskörper aus verschiedenen Oelsamen. J. Prakt. Chem. 1881, 23, 481–486. [Google Scholar] [CrossRef]
- Hekmat, D. Large-scale crystallization of proteins for purification and formulation. Bioprocess Biosyst. Eng. 2015, 38, 1209–1231. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.K.; Govardhan, C.P.; Jung, C.W.; Margolin, A.L. Protein crystals for the delivery of biopharmaceuticals. Expert Opin. Biol. Ther. 2004, 4, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.W. On the Equilibrium of Heterogeneous Systems, Part II. Trans. Conn. Acad. Sci. 1878, 3, 343–524. [Google Scholar]
- Volmer, M.; Weber, A. Nucleus Formation in Supersaturated Systems. Z. Phys. Chem. 1926, 119, 277–301. [Google Scholar]
- Becker, R.; Döring, W. Kinetische behandlung der keimbildung in übersättigten dämpfen. Ann. Phys. 1935, 416, 719–752. [Google Scholar] [CrossRef]
- ten Wolde, P.R.; Frenkel, D. Enhancement of protein crystal nucleation by critical density fluctuations. Science 1997, 277, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Malkin, A.; Kuznetsov, Y.G.; Land, T.; DeYoreo, J.; McPherson, A. Mechanisms of growth for protein and virus crystals. Nat. Struct. Biol. 1995, 2, 956–959. [Google Scholar] [CrossRef]
- McPherson, A.; Malkin, A.; Kuznetsov, Y.G.; Plomp, M. Atomic force microscopy applications in macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 1053–1060. [Google Scholar] [CrossRef]
- McPherson, A.; Kuznetsov, Y.G.; Malkin, A.; Plomp, M. Macromolecular crystal growth as revealed by atomic force microscopy. J. Struct. Biol. 2003, 142, 32–46. [Google Scholar] [CrossRef]
- Vekilov, P.G.; Chernov, A.A. The physics of protein crystallization. In Solid State Physics; Elsevier: Amsterdam, The Netherlands, 2003; Volume 57, pp. 1–147. [Google Scholar]
- Vekilov, P.; Rosenberger, F. Dependence of lysozyme growth kinetics on step sources and impurities. J. Cryst. Growth 1996, 158, 540–551. [Google Scholar] [CrossRef]
- Burton, W.K.; Cabrera, N.; Frank, F. The growth of crystals and the equilibrium structure of their surfaces. Philos. Trans. R. Soc. London. Ser. A Math. Phys. Sci. 1951, 243, 299–358. [Google Scholar]
- Durbin, S.; Feher, G. Crystal growth studies of lysozyme as a model for protein crystallization. J. Cryst. Growth 1986, 76, 583–592. [Google Scholar] [CrossRef]
- Schmit, J.D.; Dill, K. Growth rates of protein crystals. J. Am. Chem. Soc. 2012, 134, 3934–3937. [Google Scholar] [CrossRef] [PubMed]
- Luft, J.R.; Snell, E.H.; DeTitta, G.T. Lessons from high-throughput protein crystallization screening: 10 years of practical experience. Expert Opin. Drug Discov. 2011, 6, 465–480. [Google Scholar] [CrossRef]
- Goldschmidt, L.; Cooper, D.R.; Derewenda, Z.S.; Eisenberg, D. Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci. 2007, 16, 1569–1576. [Google Scholar] [CrossRef]
- Derewenda, Z.S. Rational protein crystallization by mutational surface engineering. Structure 2004, 12, 529–535. [Google Scholar] [CrossRef]
- Derewenda, Z.S. Application of protein engineering to enhance crystallizability and improve crystal properties. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 604–615. [Google Scholar] [CrossRef]
- Cooper, D.R.; Boczek, T.; Grelewska, K.; Pinkowska, M.; Sikorska, M.; Zawadzki, M.; Derewenda, Z. Protein crystallization by surface entropy reduction: Optimization of the SER strategy. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 636–645. [Google Scholar] [CrossRef]
- Derewenda, Z.S.; Vekilov, P.G. Entropy and surface engineering in protein crystallization. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 116–124. [Google Scholar] [CrossRef]
- Hermann, J.; Nowotny, P.; Schrader, T.E.; Biggel, P.; Hekmat, D.; Weuster-Botz, D. Neutron and X-ray crystal structures of Lactobacillus brevis alcohol dehydrogenase reveal new insights into hydrogen-bonding pathways. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Yau, S.T.; Vekilov, P.G. Quasi-planar nucleus structure in apoferritin crystallization. Nature 2000, 406, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmüller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef]
- Goetz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Gooetz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Brünger, A.T.; Kuriyan, J.; Karplus, M. Crystallographic R factor refinement by molecular dynamics. Science 1987, 235, 458–460. [Google Scholar] [CrossRef]
- Cerutti, D.S.; Freddolino, P.L.; Duke, R.E., Jr.; Case, D.A. Simulations of a protein crystal with a high resolution X-ray structure: Evaluation of force fields and water models. J. Phys. Chem. B 2010, 114, 12811–12824. [Google Scholar] [CrossRef]
- Janowski, P.A.; Liu, C.; Deckman, J.; Case, D.A. Molecular dynamics simulation of triclinic lysozyme in a crystal lattice. Protein Sci. 2016, 25, 87–102. [Google Scholar] [CrossRef]
- Stocker, U.; Spiegel, K.; van Gunsteren, W. On the similarity of properties in solution or in the crystalline state: A molecular dynamics study of hen lysozyme. J. Biomol. NMR 2000, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hummel, W. New alcohol dehydrogenases for the synthesis of chiral compounds. In New Enzymes for Organic Synthesis; Springer: Cham, Switzerland, 1997; pp. 145–184. [Google Scholar]
- Niefind, K.; Müller, J.; Riebel, B.; Hummel, W.; Schomburg, D. The crystal structure of R-specific alcohol dehydrogenase from Lactobacillus brevis suggests the structural basis of its metal dependency. J. Mol. Biol. 2003, 327, 317–328. [Google Scholar] [CrossRef]
- Schlieben, N.H.; Niefind, K.; Müller, J.; Riebel, B.; Hummel, W.; Schomburg, D. Atomic resolution structures of R-specific alcohol dehydrogenase from Lactobacillus brevis provide the structural bases of its substrate and cosubstrate specificity. J. Mol. Biol. 2005, 349, 801–813. [Google Scholar] [CrossRef]
- Nowotny, P.; Hermann, J.; Li, J.; Krautenbacher, A.; Kloepfer, K.; Hekmat, D.; Weuster-Botz, D. Rational Crystal Contact Engineering of Lactobacillus brevis Alcohol Dehydrogenase To Promote Technical Protein Crystallization. Cryst. Growth Des. 2019, 19, 2380–2387. [Google Scholar] [CrossRef]
- Grob, P.; Huber, M.; Walla, B.; Hermann, J.; Janowski, R.; Niessing, D.; Hekmat, D.; Weuster-Botz, D. Crystal Contact Engineering Enables Efficient Capture and Purification of an Oxidoreductase by Technical Crystallization. Biotechnol. J. 2020, 15, 2000010. [Google Scholar] [CrossRef]
- Zheng, L.; Baumann, U.; Reymond, J.L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004, 32, e115. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Collaborative, C.P. The CCP4 suite: Programs for protein crystallography. Acta Crystallographica. Sect. D Biol. Crystallogr. 1994, 50, 760. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef]
- Gapsys, V.; Michielssens, S.; Seeliger, D.; de Groot, B.L. pmx: Automated protein structure and topology generation for alchemical perturbations. J. Comput. Chem. 2015, 36, 348–354. [Google Scholar] [CrossRef]
- Gapsys, V.; de Groot, B.L. pmx Webserver: A User Friendly Interface for Alchemistry. J. Chem. Inf. Model. 2017, 57, 109–114. [Google Scholar] [CrossRef]
- Bennett, C.H. Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- Chen, W.; Deng, Y.; Russell, E.; Wu, Y.; Abel, R.; Wang, L. Accurate calculation of relative binding free energies between ligands with different net charges. J. Chem. Theory Comput. 2018, 14, 6346–6358. [Google Scholar] [CrossRef]
- Janin, J. Wet and dry interfaces: The role of solvent in protein–protein and protein–DNA recognition. Structure 1999, 7, R277–R279. [Google Scholar] [CrossRef]
- Heyda, J.; Mason, P.E.; Jungwirth, P. Attractive interactions between side chains of histidine-histidine and histidine-arginine-based cationic dipeptides in water. J. Phys. Chem. B 2010, 114, 8744–8749. [Google Scholar] [CrossRef]
- Kashchiev, D.; Vekilov, P.G.; Kolomeisky, A.B. Kinetics of two-step nucleation of crystals. J. Chem. Phys. 2005, 122, 244706. [Google Scholar] [CrossRef]
- Vekilov, P.G. The two-step mechanism of nucleation of crystals in solution. Nanoscale 2010, 2, 2346–2357. [Google Scholar] [CrossRef]
- Bordner, A.J.; Abagyan, R. Statistical analysis and prediction of protein–protein interfaces. Proteins Struct. Funct. Bioinform. 2005, 60, 353–366. [Google Scholar] [CrossRef]
- Conte, L.L.; Chothia, C.; Janin, J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999, 285, 2177–2198. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, P.; Janin, J. Dissecting protein–protein recognition sites. Proteins: Struct. Funct. Bioinform. 2002, 47, 334–343. [Google Scholar] [CrossRef]
- Bahadur, R.P.; Chakrabarti, P.; Rodier, F.; Janin, J. A dissection of specific and non-specific protein–protein interfaces. J. Mol. Biol. 2004, 336, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Ofran, Y.; Rost, B. Analysing six types of protein–protein interfaces. J. Mol. Biol. 2003, 325, 377–387. [Google Scholar] [CrossRef]
- McNicholas, S.; Potterton, E.; Wilson, K.; Noble, M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hermann, J.; Bischoff, D.; Grob, P.; Janowski, R.; Hekmat, D.; Niessing, D.; Zacharias, M.; Weuster-Botz, D. Controlling Protein Crystallization by Free Energy Guided Design of Interactions at Crystal Contacts. Crystals 2021, 11, 588. https://doi.org/10.3390/cryst11060588
Hermann J, Bischoff D, Grob P, Janowski R, Hekmat D, Niessing D, Zacharias M, Weuster-Botz D. Controlling Protein Crystallization by Free Energy Guided Design of Interactions at Crystal Contacts. Crystals. 2021; 11(6):588. https://doi.org/10.3390/cryst11060588
Chicago/Turabian StyleHermann, Johannes, Daniel Bischoff, Phillip Grob, Robert Janowski, Dariusch Hekmat, Dierk Niessing, Martin Zacharias, and Dirk Weuster-Botz. 2021. "Controlling Protein Crystallization by Free Energy Guided Design of Interactions at Crystal Contacts" Crystals 11, no. 6: 588. https://doi.org/10.3390/cryst11060588
APA StyleHermann, J., Bischoff, D., Grob, P., Janowski, R., Hekmat, D., Niessing, D., Zacharias, M., & Weuster-Botz, D. (2021). Controlling Protein Crystallization by Free Energy Guided Design of Interactions at Crystal Contacts. Crystals, 11(6), 588. https://doi.org/10.3390/cryst11060588