
First-Principles Study on Lattice Dynamics and Thermal Conductivity of Thermoelectric Intermetallics Fe3Al2Si3


Abstract
1. Introduction
2. Computational Method
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goldsmid, H. Bismuth Telluride and Its Alloys as Materials for Thermoelectric Generation. Materials 2014, 7, 2577–2592. [Google Scholar] [CrossRef]
- Shiota, Y.; Muta, H.; Yamamoto, K.; Ohishi, Y.; Kurosaki, K.; Yamanaka, S. A new semiconductor Al2Fe3Si3 with complex crystal structure. Intermetallics 2017, 89, 51–56. [Google Scholar] [CrossRef]
- Takagiwa, Y.; Isoda, Y.; Goto, M.; Shinohara, Y. Electronic structure and thermoelectric properties of narrow-band-gap intermetallic compound Al2Fe3Si3. J. Therm. Anal. Calorim. 2018, 131, 281–287. [Google Scholar] [CrossRef]
- Hou, Z.; Takagiwa, Y.; Shinohara, Y.; Xu, Y.; Tsuda, K. Machine-Learning-Assisted Development and Theoretical Consideration for the Al2Fe3Si3 Thermoelectric Material. ACS Appl. Mater. Interfaces 2019, 11, 11545–11554. [Google Scholar] [CrossRef] [PubMed]
- Shiota, Y.; Yamamoto, K.; Ohishi, Y.; Kurosaki, K.; Muta, H. Thermoelectric Properties of Co- and Mn-Doped Al2Fe3Si3. J. Electron. Mater. 2019, 48, 475–482. [Google Scholar] [CrossRef]
- Takagiwa, Y.; Shinohara, Y. A practical appraisal of thermoelectric materials for use in an autonomous power supply. Scr. Mater. 2019, 172, 98–104. [Google Scholar] [CrossRef]
- Takagiwa, Y.; Ikeda, T.; Kojima, H. Earth-Abundant Fe–Al–Si Thermoelectric (FAST) Materials: From Fundamental Materials Research to Module Development. ACS Appl. Mater. Interfaces 2020, 12, 48804–48810. [Google Scholar] [CrossRef]
- Waldecker, G.; Meinhold, H.; Birkholz, U. Thermal conductivity of semiconducting and metallic FeSi2. Phys. Status Solidi 1973, 15, 143–149. [Google Scholar] [CrossRef]
- Kim, S.W.; Cho, M.K.; Mishima, Y.; Choi, D.C. High temperature thermoelectric properties of p- and n-type β-FeSi2 with some dopants. Intermetallics 2003, 11, 399–405. [Google Scholar] [CrossRef]
- Lue, C.S.; Kuo, Y.K. Thermoelectric properties of the semimetallic Heusler compounds Fe2−xV1+xM (M = Al, Ga). Phys. Rev. B 2002, 66, 085121. [Google Scholar] [CrossRef]
- Nishino, Y.; Deguchi, S.; Mizutani, U. Thermal and transport properties of the Heusler-type Fe2VAl1−xGex (0 ≤ x ≤ 0.20) alloys: Effect of doping on lattice thermal conductivity, electrical resistivity, and Seebeck coefficient. Phys. Rev. B 2006, 74, 115115. [Google Scholar] [CrossRef]
- Tobita, K.; Sato, N.; Katsura, Y.; Kitahara, K.; Nishio-Hamane, D.; Gotou, H.; Kimura, K. High-pressure synthesis of tetragonal iron aluminide FeAl2. Scr. Mater. 2017, 141, 107–110. [Google Scholar] [CrossRef]
- Tobita, K.; Kitahara, K.; Katsura, Y.; Sato, N.; Nishio-Hamane, D.; Gotou, H.; Kimura, K. Phase stability and thermoelectric properties of semiconductor-like tetragonal FeAl2. Sci. Technol. Adv. Mater. 2019, 20, 937–948. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- VESTA. Available online: https://jp-minerals.org/vesta/en/ (accessed on 4 April 2021).
- Marker, M.C.J.; Skolyszewska-Kühberger, B.; Effenberger, H.S.; Schmetterer, C.; Richter, K.W. Phase equilibria and structural investigations in the system Al–Fe–Si. Intermetallics 2011, 19, 1919–1929. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- QUANTUM ESPRESSO. Available online: https://www.quantum-espresso.org/ (accessed on 4 April 2021).
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, N.J.; Walters, L.N.; Hellman, O.; Gall, D.; Khare, S.V. Dynamical stabilization in delafossite nitrides for solar energy conversion. J. Mater. Chem. A 2018, 6, 20852–20860. [Google Scholar] [CrossRef]
- Tadano, T.; Tsuneyuki, S. Self-consistent phonon calculations of lattice dynamical properties in cubic SrTiO3 with first-principles anharmonic force constants. Phys. Rev. B 2015, 92, 054301. [Google Scholar] [CrossRef]
- Tadano, T.; Gohda, Y.; Tsuneyuki, S. Anharmonic force constants extracted from first-principles molecular dynamics: Applications to heat transfer simulations. J. Phys. Condens. Matter 2014, 26, 225402. [Google Scholar] [CrossRef]
- ALAMODE. Available online: https://alamode.readthedocs.io/en/latest/index.html (accessed on 4 April 2021).
- Hou, Z.; Takagiwa, Y.; Shinohara, Y.; Xu, Y.; Tsuda, K. First-principles study of electronic structures and elasticity of Al2Fe3Si3. J. Phys. Condens. Matter 2021. accepted manuscript. [Google Scholar] [CrossRef] [PubMed]
- Yanson, T.I.; Manyako, M.B.; Bodak, O.I.; German, N.V.; Zarechnyuk, O.S.; Cerný, R.; Pacheco, J.V.; Yvon, K. Triclinic Fe3Al2Si3 and Orthorhombic Fe3Al2Si4 with New Structure Types. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1996, 52, 2964–2967. [Google Scholar] [CrossRef]
- Li, W.; Mingo, N. Ultralow lattice thermal conductivity of the fully filled skutterudite YbFe4Sb12 due to the flat avoided-crossing filler modes. Phys. Rev. B-Condens. Matter Mater. Phys. 2015, 91, 144304. [Google Scholar] [CrossRef]
- Lee, C.H.; Hase, I.; Sugawara, H.; Yoshizawa, H.; Sato, H. Low-Lying Optical Phonon Modes in the Filled Skutterudite CeRu4Sb12. J. Phys. Soc. Jpn. 2006, 75, 123602. [Google Scholar] [CrossRef]
- Tse, J.S.; Shpakov, V.P.; Murashov, V.V.; Belosludov, V.R. The low frequency vibrations in clathrate hydrates. J. Chem. Phys. 1997, 107, 9271–9274. [Google Scholar] [CrossRef]
- Christensen, M.; Abrahamsen, A.B.; Christensen, N.B.; Juranyi, F.; Andersen, N.H.; Lefmann, K.; Andreasson, J.; Bahl, C.R.H.; Iversen, B.B. Avoided crossing of rattler modes in thermoelectric materials. Nat. Mater. 2008, 7, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Tadano, T.; Tsuneyuki, S. Quartic Anharmonicity of Rattlers and Its Effect on Lattice Thermal Conductivity of Clathrates from First Principles. Phys. Rev. Lett. 2018, 120, 105901. [Google Scholar] [CrossRef] [PubMed]
- Esfarjani, K.; Chen, G.; Stokes, H.T. Heat transport in silicon from first-principles calculations. Phys. Rev. B 2011, 84, 085204. [Google Scholar] [CrossRef]
- Shiomi, J.; Esfarjani, K.; Chen, G. Thermal conductivity of half-Heusler compounds from first-principles calculations. Phys. Rev. B 2011, 84, 104302. [Google Scholar] [CrossRef]
- Snyder, G.J.; Toberer, E.S. Complex thermoelectric materials. Nat. Mater. 2008, 7, 105–114. [Google Scholar] [CrossRef] [PubMed]







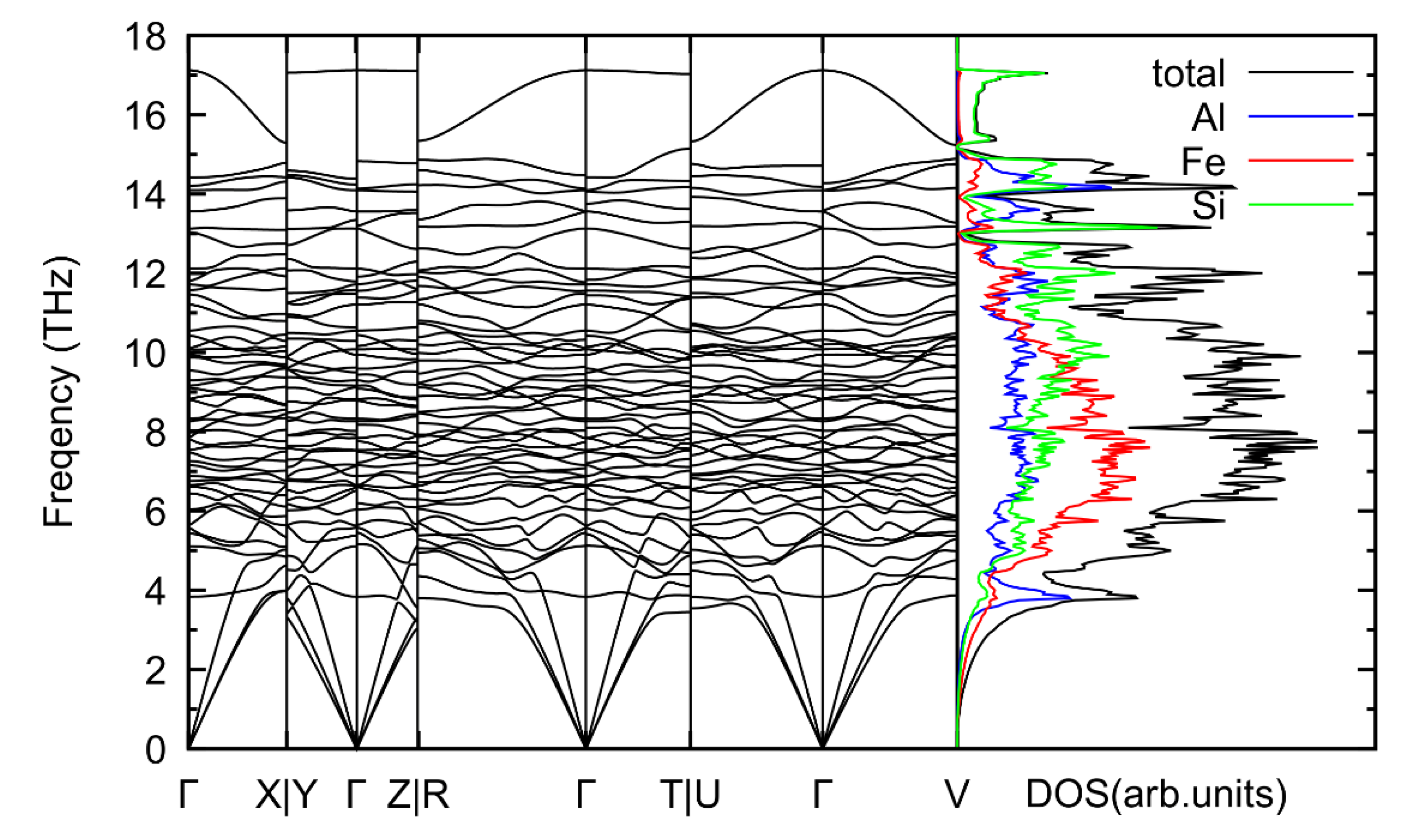{kind=link}
{kind=link}
{kind=link}
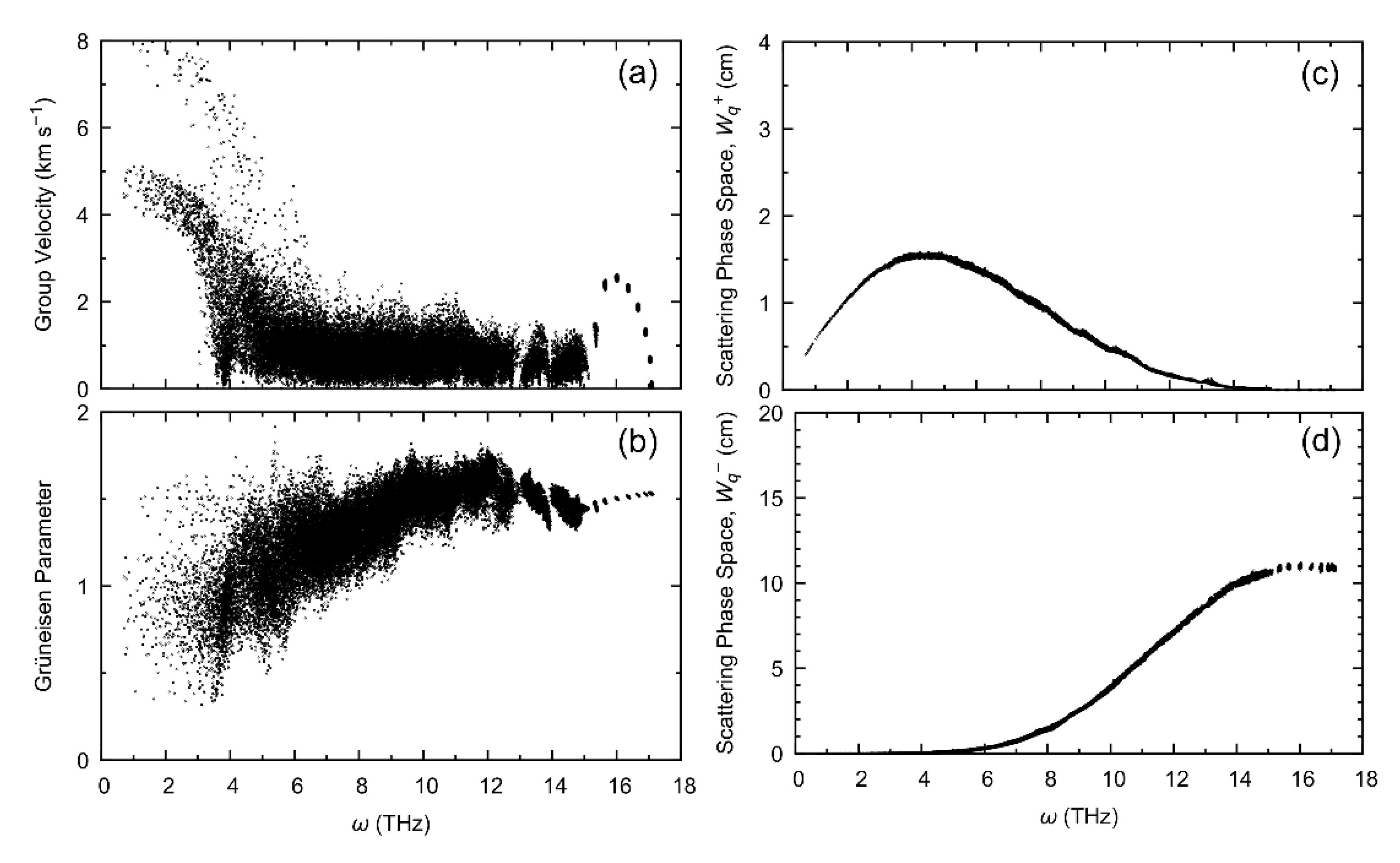{kind=link}
{kind=link}
{kind=link}
| Parameter | Calculated | Experimental | |||
|---|---|---|---|---|---|
| This Study (QE, PBE) | Reference [26] (VASP, PBE) | Reference [4] | Reference [16] | Reference [27] | |
| a | 4.6011 | 4.6032 | 4.5995 | 4.684 | 4.6512 |
| b | 6.3244 | 6.3256 | 6.3352 | 6.325 | 6.3261 |
| c | 7.4578 | 7.4594 | 7.521 | 7.498 | 7.499 |
| α | 101.91 | 101.90 | 101.827 | 100.99 | 101.375 |
| β | 106.78 | 106.79 | 106.427 | 105.6 | 105.923 |
| γ | 100.59 | 100.59 | 100.729 | 101.62 | 101.237 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, N.; Takagiwa, Y. First-Principles Study on Lattice Dynamics and Thermal Conductivity of Thermoelectric Intermetallics Fe3Al2Si3. Crystals 2021, 11, 388. https://doi.org/10.3390/cryst11040388
Sato N, Takagiwa Y. First-Principles Study on Lattice Dynamics and Thermal Conductivity of Thermoelectric Intermetallics Fe3Al2Si3. Crystals. 2021; 11(4):388. https://doi.org/10.3390/cryst11040388
Chicago/Turabian StyleSato, Naoki, and Yoshiki Takagiwa. 2021. "First-Principles Study on Lattice Dynamics and Thermal Conductivity of Thermoelectric Intermetallics Fe3Al2Si3" Crystals 11, no. 4: 388. https://doi.org/10.3390/cryst11040388
APA StyleSato, N., & Takagiwa, Y. (2021). First-Principles Study on Lattice Dynamics and Thermal Conductivity of Thermoelectric Intermetallics Fe3Al2Si3. Crystals, 11(4), 388. https://doi.org/10.3390/cryst11040388