Tetrathiafulvalene: A Gate to the Mechanochemical Mechanisms of Electron Transfer Reactions


Abstract
1. Introduction
2. Materials and Methods
2.1. Mechanochemical Syntheses
2.2. Laboratory X-ray Powder Diffraction (XRPD)
2.3. Isothermal Kinetic Profiles for the Neat and LAG Mechanochemical Synthesis of TTF-CA
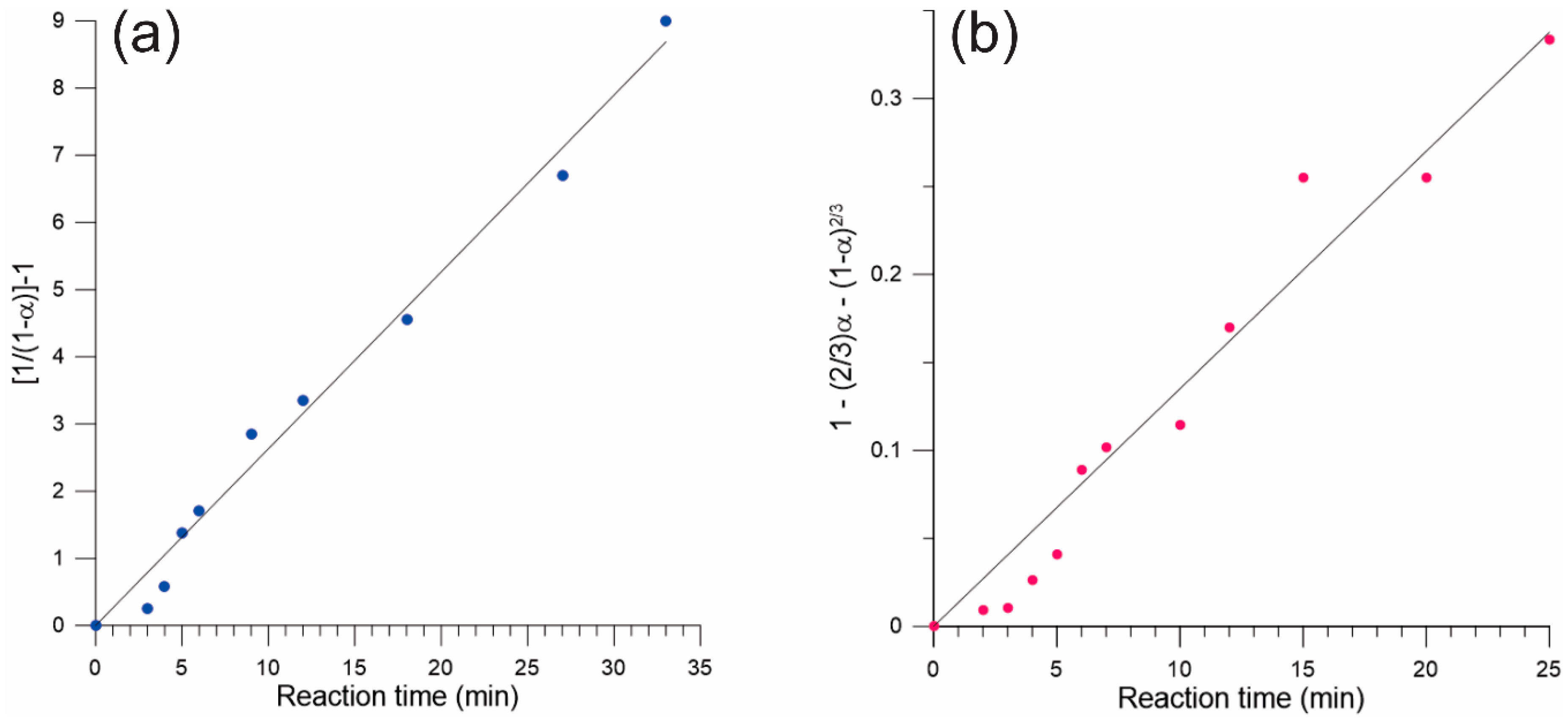
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Todres, Z.V. Organic Mechanochemistry and Its Practical Applications; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Margetic, D.; Strukil, V. Mechanochemical Organic Synthesis; Elsevier Inc.: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Trask, A.V.; Jones, W. Crystal engineering of organic cocrystals by the solid-state grinding approach. Top. Curr. Chem. 2005, 254, 41–70. [Google Scholar] [CrossRef]
- Friščić, T. New opportunities for materials synthesis using mechanochemistry. J. Mater. Chem. 2010, 20, 7599–7605. [Google Scholar] [CrossRef]
- Friščić, T.; Jones, W. Recent advances in understanding the mechanism of cocrystal formation via grinding. Cryst. Growth Des. 2009, 9, 1621–1637. [Google Scholar] [CrossRef]
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W.; et al. Mechanochemistry: Opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. [Google Scholar] [CrossRef]
- Fischer, F.; Wenzel, K.-J.; Rademann, K.; Emmerling, F. Quantitative determination of activation energies in mechanochemical reactions. Phys. Chem. Chem. Phys. 2016, 18, 23320–23325. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Grepioni, F. Mechanochemical Preparation of Co-Crystals. Chem. Soc. Rev. 2013, 42, 7638–7648. [Google Scholar] [CrossRef]
- Weyna, D.R.; Shattock, T.; Vishweshwar, P.; Zaworotko, M.J. Synthesis and structural characterization of cocrystals and pharmaceutical cocrystals: Mechanochemistry vs. slow evaporation from solution. Cryst. Growth Des. 2009, 9, 1106–1123. [Google Scholar] [CrossRef]
- Hasa, D.; Jones, W. Screening for new pharmaceutical solid forms using mechanochemistry: A practical guide. Adv. Drug Deliv. Rev. 2017, 117, 147–161. [Google Scholar] [CrossRef]
- Ling, A.R.; Baker, J.L. Derivatives of Quinhydrone. J. Am. Chem. Soc. 1893, 63, 1314–1327. [Google Scholar] [CrossRef]
- Kuroda, R.; Imai, Y.; Tajima, N. Generation of a co-crystal phase with novel coloristic properties via solid state grinding procedures. Chem. Commun. 2002, 2848–2849. [Google Scholar] [CrossRef]
- Imai, Y.; Tajima, N.; Sato, T.; Kuroda, R. Molecular recognition in solid-state crystallization: Colored chiral adduct formations of 1,1′-Bi-2-napthol derivatives and benzoquinone with a third component. Chirality 2002, 14, 604–609. [Google Scholar] [CrossRef]
- Chemerisov, S.D.; Grinberg, O.Y.; Tipikin, D.S.; Lebedev, Y.S.; Kurreck, H.; Möbius, K. Mechanically induced radical pair formation in porphyrin-quinone and related donor-acceptor mixtures. unusual stability and zero-field splittings. Chem. Phys. Lett. 1994, 218, 353–361. [Google Scholar] [CrossRef]
- Tipikin, D.S.; Lebedev, Y.S.; Rieker, A. Mechanochemical generation of stable radical species. Oxidation of pyrocatechols. Chem. Phys. Lett. 1997, 272, 399–404. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F. Reactions between or within Molecular Crystals. Angew. Chem.-Int. Ed. 2004, 43, 4002–4011. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; Clarendon Press: Oxford, UK, 2002. [Google Scholar]
- Lapidus, S.H.; Naik, A.; Wixtrom, A.; Massa, N.E.; Ta Phuoc, V.; Del Campo, L.; Lebègue, S.; Ángyán, J.G.; Abdel-Fattah, T.; Pagola, S. The black polymorph of TTF-CA: TTF polymorphism and solvent effects in mechanochemical and vapor digestion syntheses, FT-IR, crystal packing, and electronic structure. Cryst. Growth Des. 2014, 14, 91–100. [Google Scholar] [CrossRef]
- Mohamud, S.; Ta Phuoc, V.; Massa, N.E.; Pagola, S. TTF-DDQ: Two “Green” synthetic routes, crystal structure and band gap from FT-IR spectroscopy. Synth. Met. 2016, 214, 71–75. [Google Scholar] [CrossRef]
- Jones, J.; Ta Phuoc, V.; Del Campo, L.; Massa, N.E.; Brown, C.M.; Pagola, S. Accessing new charge transfer complexes by mechanochemistry: A tetrathiafulvalene chloranilic acid polymorph containing segregated tetrathiafulvalene stacks. Cryst. Growth Des. 2019, 19, 4970–4980. [Google Scholar] [CrossRef]
- Pagola, S.; Polymeros, A.; Kourkoumelis, N. WinPSSP: A revamp of the computer program PSSP and its performance solving the crystal structures of small organic compounds and solids of biological and pharmaceutical interest. J. Appl. Crystallogr. 2017, 50, 293–303. [Google Scholar] [CrossRef]
- Pagola, S.; Stephens, P.W. PSSP, a computer program for the crystal structure solution of molecular materials from X-ray powder diffraction data. J. Appl. Crystallogr. 2010, 43, 370–376. [Google Scholar] [CrossRef]
- Altheimer, B.D.; Pagola, S.; Zeller, M.; Mehta, M.A. Mechanochemical conversions between crystalline polymorphs of a complex organic solid. Cryst. Growth Des. 2013, 13, 3447–3453. [Google Scholar] [CrossRef]
- Bendikov, M.; Wudl, F.; Perepichka, D.F. Tetrathiafulvalenes, oligoacenenes, and their buckminsterfullerene derivatives: The brick and mortar of organic electronics. Chem. Rev. 2004, 104, 4891–4945. [Google Scholar] [CrossRef]
- Schultz, A.J.; Stucky, G.D.; Blessing, R.H.; Coppens, P. The temperature dependence of the crystal and molecular structure of. D 2, 2′-Bi-1,3-dithiole [TTF] 7,7,8,8-tetracyano-p-quinodimethane [TCNQ]. J. Am. Chem. Soc. 1976, 98, 3194–3201. [Google Scholar] [CrossRef]
- Zhang, L.; Fakhouri, S.M.; Liu, F.; Timmons, J.C.; Ran, N.A.; Briseno, A.L. Chalcogenoarene semiconductors: New ideas from old materials. J. Mater. Chem. 2011, 21, 1329–1337. [Google Scholar] [CrossRef]
- Ellern, A.; Bernstein, J.; Becker, J.Y.; Zamir, S.; Shahal, L.; Cohen, L. A new polymorphic modification of tetrathiafulvalene. Crystal structure, lattice energy and intermolecular interactions. Chem. Matter. 1994, 6, 1378–1385. [Google Scholar] [CrossRef]
- Batsanov, A.S. Tetrathiafulvalene revisited. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2006, C62, o501–o504. [Google Scholar] [CrossRef]
- Herranz, M.Á.; Sánchez, L.; Martín, N. Tetrathiafulvalene: A paradigmatic electron donor molecule. Phosphorus. Sulfur. Silicon Relat. Elem. 2005, 180, 1133–1148. [Google Scholar] [CrossRef]
- Kharisov, B.I.; Méndez-Rojas, M.A.; Garnovskii, A.D.; Ivakhnenko, E.P.; Ortiz-Méndez, U. Review: Coordination chemistry of o-quinone complexes. J. Coord. Chem. 2002, 55, 745–770. [Google Scholar] [CrossRef]
- Dressel, M. Ordering phenomena in quasi-one-dimensional organic conductors. Naturwissenschaften 2007, 94, 527–541. [Google Scholar] [CrossRef]
- Ouahab, L.; Yagubskii, E. (Eds.) Organic Conductors, Superconductors and Magnets: From Synthesis to Molecular Electronics; NATO Science Series; Kluwer Academic Publishers: Dordrecht, The Neatherlands, 2004. [Google Scholar]
- Masino, M.; Castagnetti, N.; Girlando, A.; Chimiche, S.; Ambientale, S.; Parma, I.; Area, P.; Parma, U.; Parma, I. Phenomenology of the neutral-ionic valence instability in mixed stack charge-transfer crystals. Crystals 2017, 7, 108. [Google Scholar] [CrossRef]
- Le Cointe, M.; Lemée-Cailleau, M.H.; Cailleau, H.; Toudic, B.; Toupet, L.; Heger, G.; Moussa, F.; Schweiss, P.; Kraft, K.H.; Karl, N. Symmetry breaking and structural changes at the neutral-to-ionic transition in tetrathiafulvalene-p-chloranil. Phys. Rev. B 1995, 51, 3374–3386. [Google Scholar] [CrossRef]
- Girlando, A.; Marzola, F.; Pecile, C. Vibrational spectroscopy of mixed stack organic semiconductors: Neutral and ionic phases of tetrathiafulvalene-chloranil (TTF-CA) charge transfer complex. J. Chem. Phys. 1983, 79, 1075–1085. [Google Scholar] [CrossRef]
- Li, H.; Wen, H.; Stowell, J.G.; Morris, K.R.; Byrn, S.R. Crystal quality and physical reactivity in the case of flufenamic acid (FFA). J. Pharm. Sci. 2010, 99, 3839–3848. [Google Scholar] [CrossRef]
- Kaupp, G. Solid-state molecular syntheses: Complete reactions without auxiliaries based on the new solid-state mechanism. CrystEngComm 2003, 5, 117–133. [Google Scholar] [CrossRef]
- Paul, I.C.; Curtin, D.Y. Thermally induced organic reactions in the solid state. Acc. Chem. Res. 1973, 6, 217–225. [Google Scholar] [CrossRef]
- Chen, X.; Li, T.; Morris, K.R.; Byrn, S.R. Crystal Packing and chemical reactivity of two polymorphs of flufenamic acid with ammonia. Mol. Cryst. Liq. Cryst. 2002, 381, 121–131. [Google Scholar] [CrossRef]
- Khawan, A.; Flanagan, D.R. Basics and applications of solid-state kinetics: A pharmaceutical perspective. J. Pharm. Sci. 2006, 95, 472–498. [Google Scholar] [CrossRef]
- Chang, R. ESR study of organic electron transfer reactions. J. Chem. Educ. 1970, 47, 563–568. [Google Scholar] [CrossRef]
- Anslyn, E.V.; Dougherty, D.A. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, USA, 2006. [Google Scholar]
- Buncel, E.; Stairs, R.A. Solvent Effects in Chemistry, 2nd ed.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Buncel, E.; Wilson, H. Solvent effects on rates and equilibria: A practical approach. J. Chem. Educ. 1980, 57, 629. [Google Scholar] [CrossRef]
- Bowmaker, G.A. Solvent-assisted mechanochemistry. Chem. Commun. 2013, 49, 334–348. [Google Scholar] [CrossRef]
- Chen, X.; Stowell, J.G.; Morris, K.R.; Byrn, S.R. Quantitative study of solid-state acid-base reactions between polymorphs of flufenamic acid and magnesium oxide using X-ray powder diffraction. J. Pharm. Biomed. Anal. 2010, 51, 866–874. [Google Scholar] [CrossRef]
- Varga, K.; Volarić, J.; Vančik, H. Crystal disordering and organic solid-state reactions. CrystEngComm 2015, 17, 1434–1438. [Google Scholar] [CrossRef]
- Bamford, C.H.; Tipper, C.F.H. (Eds.) Comprehensive Chemical Kinetics. Reactions in the Solid State; Elsevier Scientific Publishing Company: Amsterdam, The Netherlands, 1980. [Google Scholar]
- Khawam, A.; Flanagan, D.R. Solid-state kinetic models: Basics and mathematical fundamentals. J. Phys. Chem. B 2006, 110, 17315–17328. [Google Scholar] [CrossRef]
- Byrn, S.R.; Pfeiffer, R.R.; Stowell, J.G. Solid-State Chemistry of Drugs, 2nd ed.; SSCI, Inc.: West Lafayette, IN, USA, 1999. [Google Scholar]
- Friščić, T.; Halasz, I.; Beldon, P.J.; Belenguer, A.M.; Adams, F.; Kimber, S.A.; Honkimaki, V.; Dinnebier, R.E. Real-time and in situ monitoring of mechanochemical milling reactions. Nat. Chem. 2013, 5, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Halasz, I.; Kimber, S.A.J.; Beldon, P.J.; Belenguer, A.M.; Adams, F.; Honkimäki, V.; Nightingale, R.C.; Dinnebier, R.E.; Friščić, T. In situ and real-time monitoring of mechanochemical milling reactions using synchrotron X-ray diffraction. Nat. Protoc. 2013, 8, 1718–1729. [Google Scholar] [CrossRef]
- Užarević, K.; Štrukil, V.; Mottillo, C.; Julien, P.A.; Puškarić, A.; Friščić, T.; Halasz, I. Exploring the effect of temperature on a mechanochemical reaction by in situ synchrotron powder X-ray diffraction. Cryst. Growth Des. 2016, 16, 2342–2347. [Google Scholar] [CrossRef]
- Halasz, I.; Puskaric, A.; Kimber, S.A.J.; Beldon, P.J.; Belenguer, A.M.; Adams, F.; Honkimaki, V.; Dinnebier, R.E.; Patel, B.; Jones, W.; et al. Real-time in situ powder X-ray diffraction monitoring of mechanochemical synthesis of pharmaceutical cocrystals. Angew. Chem. Int. Ed. 2013, 52, 11538–11541. [Google Scholar] [CrossRef]
- Užarević, K.; Halasz, I.; Friščić, T. Real-time and in situ monitoring of mechanochemical reactions: A new playground for all chemists. J. Phys. Chem. Lett. 2015, 6, 4129–4140. [Google Scholar] [CrossRef]
- Gracin, D.; Štrukil, V.; Friščić, T.; Halasz, I.; Užarević, K. Laboratory real-time and in situ monitoring of mechanochemical milling reactions by Raman spectroscopy. Angew. Chem.-Int. Ed. 2014, 53, 6193–6197. [Google Scholar] [CrossRef]
- Zevin, L.S.; Kimmel, G. Quantitative X-ray Diffractometry; Mureinik, I., Ed.; Springer: New York, NY, USA, 1995. [Google Scholar]
- Ma, X.; Yuan, W.; Bell, S.E.J.; James, S.L. Better understanding of mechanochemical reactions: Raman monitoring reveals surprisingly simple “pseudo-fluid” model for a ball milling reaction. Chem. Commun. 2014, 50, 1585–1587. [Google Scholar] [CrossRef]
- Julien, P.A.; Malvestiti, I.; Friščić, T. The effect of milling frequency on a mechanochemical organic reaction monitored by in situ Raman spectroscopy. Beilstein J. Org. Chem. 2017, 13, 2160–2168. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | σ | α ± Δα | ||||
|---|---|---|---|---|---|---|
| 0 | - | - | - | - | - | 0 ± 0 |
| 3 | 0.033 | 0.060 | 0.040 | 0.044 | 0.01 | 0.20 ± 0.02 |
| 4 | 0.066 | 0.103 | 0.080 | 0.083 | 0.02 | 0.37 ± 0.04 |
| 5 | 0.121 | 0.137 | 0.131 | 0.130 | 0.02 | 0.58 ± 0.03 |
| 6 | 0.109 | 0.158 | 0.160 | 0.143 | 0.04 | 0.63 ± 0.05 |
| 9 | 0.168 | 0.168 | 0.160 | 0.166 | 0.01 | 0.74 ± 0.02 |
| 12 | 0.154 | 0.190 | 0.179 | 0.174 | 0.02 | 0.77 ± 0.03 |
| 18 | 0.164 | 0.179 | 0.211 | 0.185 | 0.03 | 0.82 ± 0.04 |
| 27 | 0.192 | 0.210 | 0.188 | 0.197 | 0.02 | 0.87 ± 0.03 |
| 33 | 0.183 | 0.203 | 0.219 | 0.202 | 0.02 | 0.90 ± 0.04 |
| Time (min) | σ | α ± Δα | ||||
|---|---|---|---|---|---|---|
| 0 | - | - | - | - | - | 0 ± 0 |
| 2 | 0.077 | 0.056 | 0.049 | 0.061 | 0.02 | 0.27 ± 0.04 |
| 3 | 0.087 | 0.052 | 0.052 | 0.064 | 0.02 | 0.28 ± 0.03 |
| 4 | 0.100 | 0.101 | 0.091 | 0.097 | 0.02 | 0.43 ± 0.03 |
| 5 | 0.126 | 0.115 | 0.112 | 0.118 | 0.02 | 0.52 ± 0.02 |
| 6 | 0.164 | 0.163 | 0.153 | 0.160 | 0.02 | 0.71 ± 0.14 |
| 7 | 0.157 | 0.194 | 0.151 | 0.168 | 0.02 | 0.75 ± 0.04 |
| 10 | 0.179 | 0.188 | 0.159 | 0.175 | 0.02 | 0.78 ± 0.03 |
| 12 | 0.209 | 0.195 | 0.189 | 0.198 | 0.02 | 0.88 ± 0.03 |
| 15 | 0.211 | 0.227 | 0.216 | 0.218 | 0.02 | 0.97 ± 0.03 |
| 20 | 0.198 | 0.242 | 0.215 | 0.218 | 0.04 | 0.97 ± 0.05 |
| 25 | 0.197 | 0.253 | 0.225 | 0.225 | 0.03 | 1.00 ± 0.04 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, R.; Gokus, M.K.; Pagola, S. Tetrathiafulvalene: A Gate to the Mechanochemical Mechanisms of Electron Transfer Reactions. Crystals 2020, 10, 482. https://doi.org/10.3390/cryst10060482
Chen R, Gokus MK, Pagola S. Tetrathiafulvalene: A Gate to the Mechanochemical Mechanisms of Electron Transfer Reactions. Crystals. 2020; 10(6):482. https://doi.org/10.3390/cryst10060482
Chicago/Turabian StyleChen, Richard, Mehmet Kerem Gokus, and Silvina Pagola. 2020. "Tetrathiafulvalene: A Gate to the Mechanochemical Mechanisms of Electron Transfer Reactions" Crystals 10, no. 6: 482. https://doi.org/10.3390/cryst10060482
APA StyleChen, R., Gokus, M. K., & Pagola, S. (2020). Tetrathiafulvalene: A Gate to the Mechanochemical Mechanisms of Electron Transfer Reactions. Crystals, 10(6), 482. https://doi.org/10.3390/cryst10060482