Unravelling the Chemistry of the [Cu(4,7-Dichloroquinoline)2Br2]2 Dimeric Complex through Structural Analysis: A Borderline Ligand Field Case



Abstract
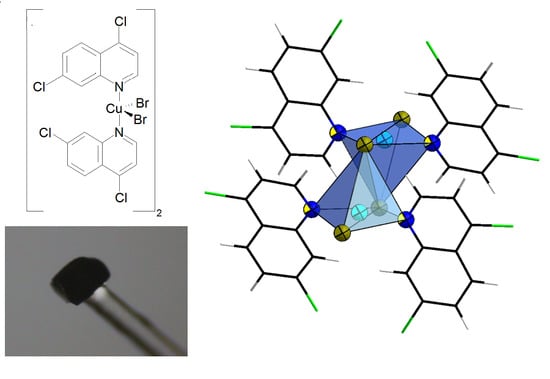
1. Introduction
2. Methods
2.1. Synthesis
2.2. Single-Crystal X-ray Diffraction and Model Refinement
2.3. Magnetic Measurements
2.4. Quantum Simulations (Gas Phase)
2.5. Experimental Charge Density Analysis
3. Results
Crystal Packing
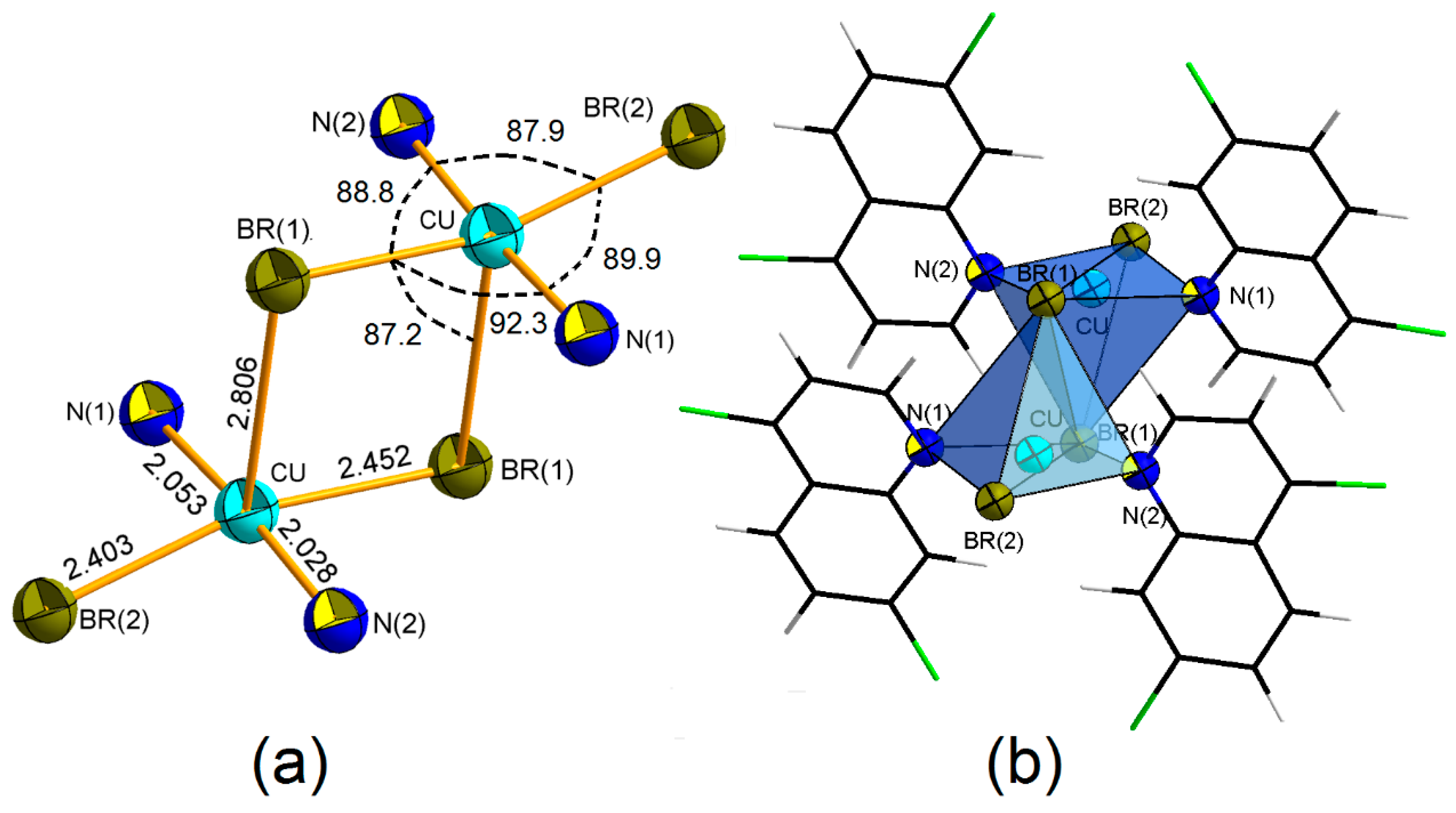
4. Coordination Geometry
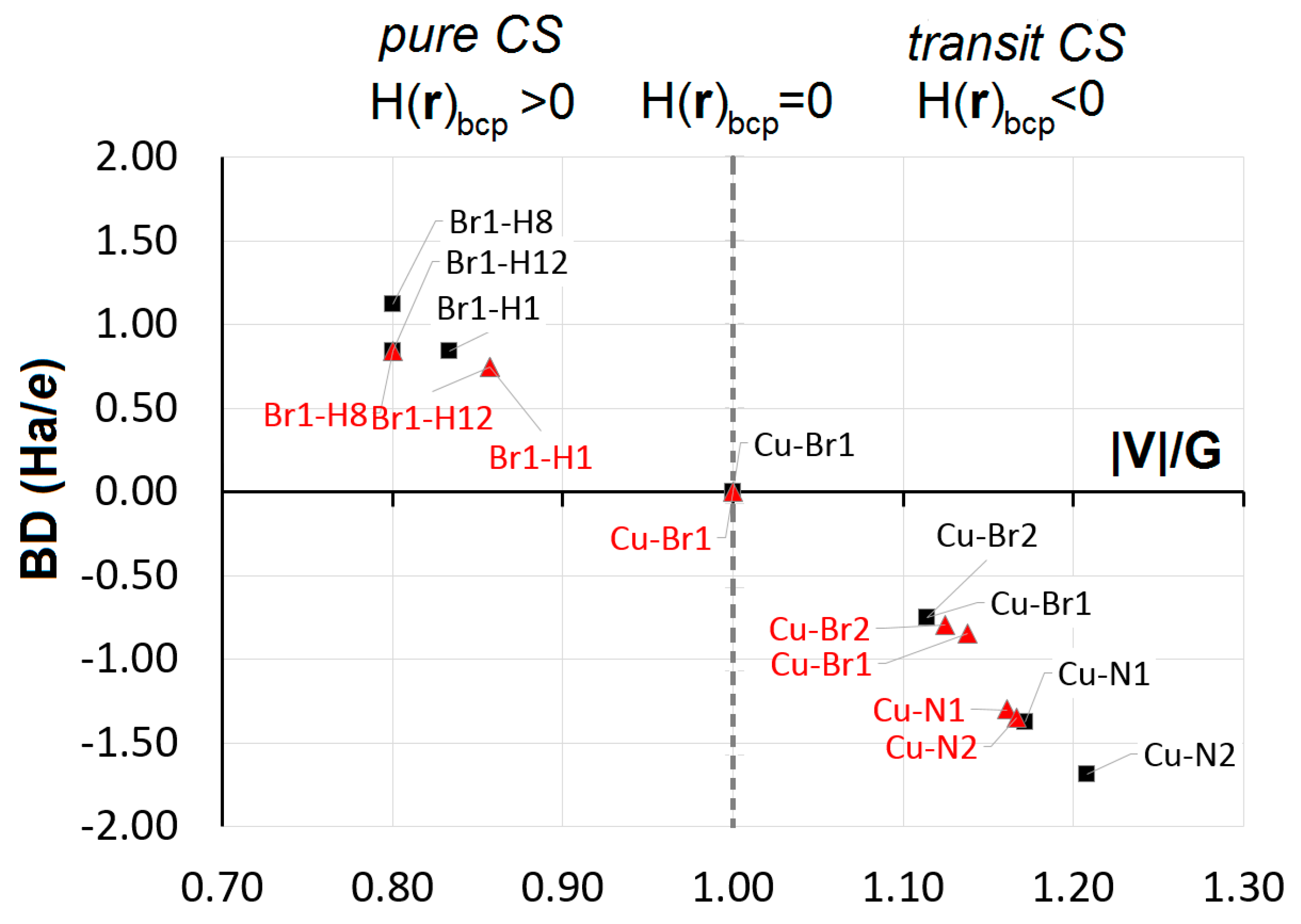
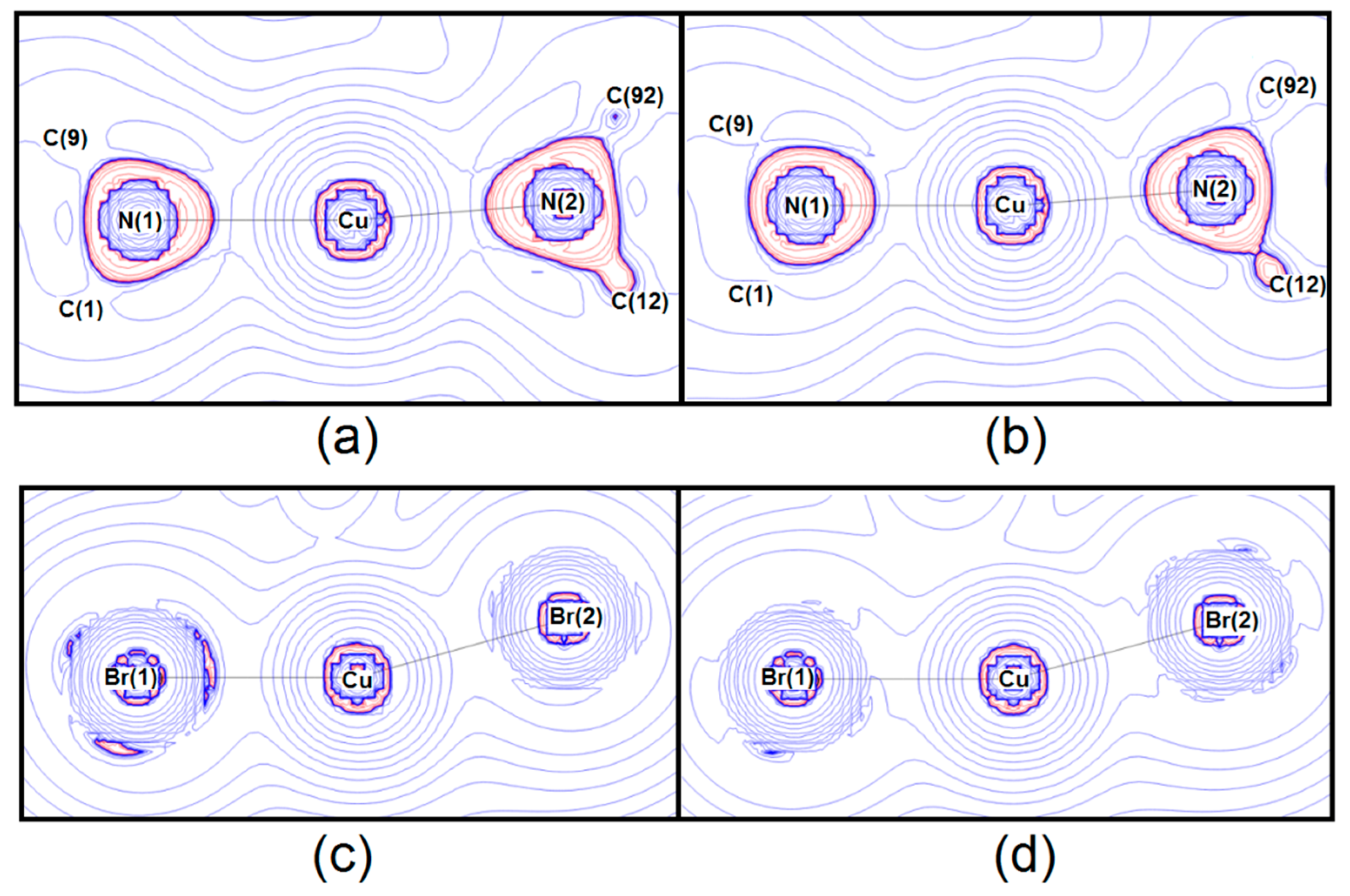
5. Chemical Bonding
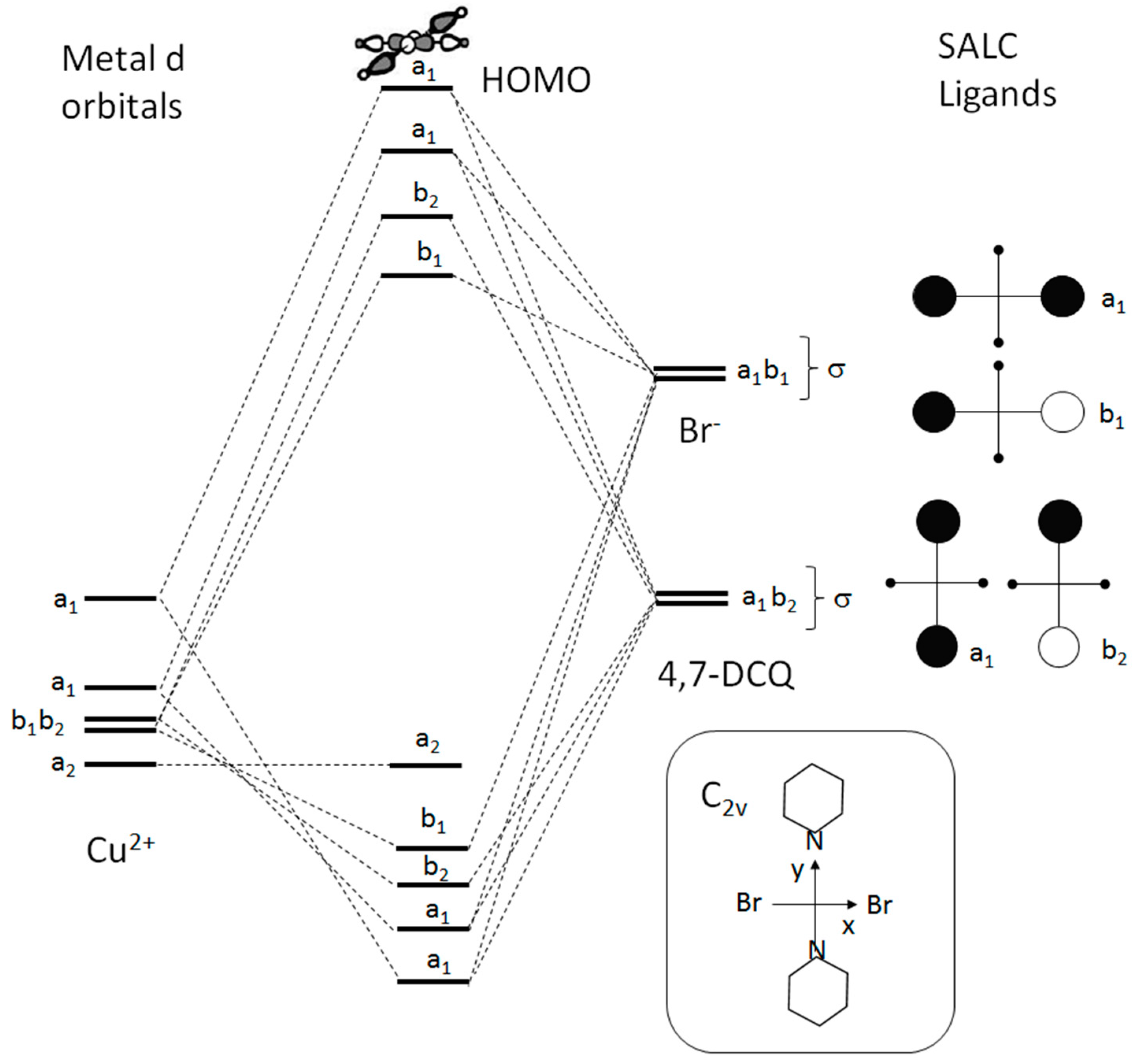
6. Electronic States
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reinen, D. Cu2+, a Chameleon in Coordination Chemistry. Comments Inorg. Chem. 1983, 2, 227–246. [Google Scholar] [CrossRef]
- Persson, I.; Persson, P.; Sandström, M.; Ullström, A.-S. Structure of Jahn–Teller distorted solvated copper(ii) ions in solution, and in solids with apparently regular octahedral coordination geometry. J. Chem. Soc. Dalton Trans. 2002, 7, 1256–1265. [Google Scholar] [CrossRef]
- Chaboy, J.; Muñoz-Páez, A.; Merkling, P.J.; Sánchez Marcos, E. The hydration of Cu2+: Can the Jahn-Teller effect be detected in liquid solution? J. Chem. Phys. 2006, 124, 064509. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, F. Unveiling the Local Structure of Cu2+ Ions from d-Orbital Splitting. Application to K 2 ZnF 4: Cu2+ and KZnF 3: Cu2+. Inorg. Chem. 2017, 56, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.P. Elusiveness of CuIII Complexation; Preference for Trifluoromethyl Oxidation in the Formation of[CuI(CF3)4]− Salts. Angew. Chem. Int. Ed. Engl. 1995, 34, 80–81. [Google Scholar] [CrossRef]
- Aullón, G.; Alvarez, S. Oxidation states, atomic charges and orbital populations in transition metal complexes. Theor. Chem. Acc. 2009, 123, 67–73. [Google Scholar] [CrossRef]
- Walroth, R.C.; Lukens, J.T.; MacMillan, S.N.; Finkelstein, K.D.; Lancaster, K.M. Spectroscopic Evidence for a 3d 10 Ground State Electronic Configuration and Ligand Field Inversion in [Cu(CF3)4]1–. J. Am. Chem. Soc. 2016, 138, 1922–1931. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alvarez, S.; Mealli, C.; Falceto, A.; Cahill, T.J.; Zeng, T.; Manca, G. From Widely Accepted Concepts in Coordination Chemistry to Inverted Ligand Fields. Chem. Rev. 2016, 116, 8173–8192. [Google Scholar] [CrossRef]
- Macetti, G.; Rizzato, S.; Beghi, F.; Silvestrini, L.; Presti, L. Lo On the molecular basis of the activity of the antimalarial drug chloroquine: EXAFS-assisted DFT evidence of a direct Fe–N bond with free heme in solution. Phys. Scr. 2016, 91, 023001. [Google Scholar] [CrossRef]
- Macetti, G.; Loconte, L.; Rizzato, S.; Gatti, C.; Lo Presti, L. Intermolecular Recognition of the Antimalarial Drug Chloroquine: A Quantum Theory of Atoms in Molecules–Density Functional Theory Investigation of the Hydrated Dihydrogen Phosphate Salt from the 103 K X-ray Structure. Cryst. Growth Des. 2016, 16, 6043–6054. [Google Scholar] [CrossRef]
- Sacchi, P.; Loconte, L.; Macetti, G.; Rizzato, S.; Lo Presti, L. Correlations of Crystal Structure and Solubility in Organic Salts: The Case of the Antiplasmodial Drug Piperaquine. Cryst. Growth Des. 2019, 19, 1399–1410. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch. Biochem. Biophys. 2016, 611, 100–112. [Google Scholar] [CrossRef]
- Di Vaira, M.; Bazzicalupi, C.; Orioli, P.; Messori, L.; Bruni, B.; Zatta, P. Clioquinol, a Drug for Alzheimer’s Disease Specifically Interfering with Brain Metal Metabolism: Structural Characterization of Its Zinc(II) and Copper(II) Complexes. Inorg. Chem. 2004, 43, 3795–3797. [Google Scholar] [CrossRef] [PubMed]
- Zanon, V.S.; Lima, J.A.; Cuya, T.; Lima, F.R.S.; da Fonseca, A.C.C.; Gomez, J.G.; Ribeiro, R.R.; França, T.C.C.; Vargas, M.D. In-vitro evaluation studies of 7-chloro-4-aminoquinoline Schiff bases and their copper complexes as cholinesterase inhibitors. J. Inorg. Biochem. 2019, 191, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.S. Cholinesterase inhibitors for Alzheimer’s disease. In Cochrane Database of Systematic Reviews; Birks, J.S., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2006. [Google Scholar]
- Touret, F.; de Lamballerie, X. Of chloroquine and COVID-19. Antivir. Res. 2020, 177, 104762. [Google Scholar] [CrossRef]
- Cohen, T.; Schambach, R.A. The Copper-Quinoline Decarboxylation. J. Am. Chem. Soc. 1970, 92, 3189–3190. [Google Scholar] [CrossRef]
- Cahiez, G.; Moyeux, A.; Gager, O.; Poizat, M. Copper-Catalyzed Decarboxylation of Aromatic Carboxylic Acids: En Route to Milder Reaction Conditions. Adv. Synth. Catal. 2013, 355, 790–796. [Google Scholar] [CrossRef]
- Kawata, T.; Uekusa, H.; Ohba, S.; Furukawa, T.; Tokii, T.; Muto, Y.; Kato, M. Magneto-structural correlation in dimeric copper(II) benzoates. Acta Crystallogr. Sect. B 1992, 48, 253–261. [Google Scholar] [CrossRef]
- Hiller, W. The Crystal Structure of [(Quinoline)2CuI]2, CH3CN. Z. Naturforsch. 1984, 39b, 861–863. [Google Scholar] [CrossRef][Green Version]
- Engelhardt, L.M.; Healy, P.C.; Kildea, J.D.; White, A.H. Lewis-base adducts of group 11 metal(i) compounds. LIII: Synthesis and structural characterization of binuclear μ, μ’ -dichloro-, dibrorno- and diiodo-bis[(pyridine)(triphenylphosphine)copper(i)] complexes, and their pyridine-4-carbonitrile analogues. Aust. J. Chem. 1989, 42, 913–922. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory, International Series of Monographs on Chemistry 22; Clarendon Press: Oxford, UK, 1990; ISBN 9780198558651. [Google Scholar]
- Madsen, A.Ø. SHADE web server for estimation of hydrogen anisotropic displacement parameters. J. Appl. Crystallogr. 2006, 39, 757–758. [Google Scholar] [CrossRef]
- Bruker AXS Inc. APEX II SAINT and SADABS User Manuals; Bruker AXS Inc.: Madison, WI, USA, 2014. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Bain, G.A.; Berry, J.F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V. Gaussian 09, Revis. D.01; Gaussian Inc.: Wallingford, UK, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Gionda, A.; Macetti, G.; Loconte, L.; Rizzato, S.; Orlando, A.M.; Gatti, C.; Lo Presti, L. A variable-temperature X-ray diffraction and theoretical study of conformational polymorphism in a complex organic molecule (DTC). RSC Adv. 2018, 8, 38445–38454. [Google Scholar] [CrossRef]
- Peintinger, M.F.; Oliveira, D.V.; Bredow, T. Consistent Gaussian basis sets of triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2013, 34, 451–459. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X—H bond lengths from neutron diffraction data. Acta Crystallogr. Sect. B Struct. Sci. 2010, 66, 380–386. [Google Scholar] [CrossRef]
- Roversi, P.; Destro, R. Approximate anisotropic displacement parameters for H atoms in molecular crystals. Chem. Phys. Lett. 2004, 386, 472–478. [Google Scholar] [CrossRef]
- Hansen, N.K.; Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Crystallogr. Sect. A 1978, 34, 909–921. [Google Scholar] [CrossRef]
- Koritsanszky, T.; Mallinson, P.R.; Macchi, P.; Volkov, A.; Gatti, C.; Richter, T.; Farrugia, L.J. XD2006-A Computer Program Package for Multipole Refinement, Topological Analysis of Charge Densities and Evaluation of Intermolecular Energies from Experimental and Theoretical Structure Factors. In Users’ Manual; University at Buffalo: Buffalo, NY, USA, 2006. [Google Scholar]
- Su, Z.; Coppens, P. Nonlinear Least-Squares Fitting of Numerical Relativistic Atomic Wave Functions by a Linear Combination of Slater-Type Functions for Atoms with Z = 1–36. Acta Crystallogr. Sect. A Found. Crystallogr. 1998, 54, 646–652. [Google Scholar] [CrossRef]
- Lo Presti, L.; Sist, M.; Loconte, L.; Pinto, A.; Tamborini, L.; Gatti, C. Rationalizing the lacking of inversion symmetry in a noncentrosymmetric polar racemate: An experimental and theoretical study. Cryst. Growth Des. 2014, 14, 5822–5833. [Google Scholar] [CrossRef]
- Destro, R.; Ruffo, R.; Roversi, P.; Soave, R.; Loconte, L.; Lo Presti, L. Anharmonic motions versus dynamic disorder at the Mg ion from the charge densities in pyrope (Mg3Al2Si3O12) crystals at 30 K: Six of one, half a dozen of the other. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, L. On the significance of weak hydrogen bonds in crystal packing: A large databank comparison of polymorphic structures. CrystEngComm 2018, 20, 5976–5989. [Google Scholar] [CrossRef]
- Destro, R.; Sartirana, E.; Loconte, L.; Soave, R.; Colombo, P.; Destro, C.; Lo Presti, L. Competing C=O⋯C=O, C-H⋯O, Cl⋯O, and Cl⋯Cl interactions governing the structural phase transition of 2,6-dichloro-p- benzoquinone at T c = 122.6 K. Cryst. Growth Des. 2013, 13, 4571–4582. [Google Scholar] [CrossRef]
- Colombo, V.; Lo Presti, L.; Gavezzotti, A. Two-component organic crystals without hydrogen bonding: Structure and intermolecular interactions in bimolecular stacking. CrystEngComm 2017, 19, 2413–2423. [Google Scholar] [CrossRef]
- Zhao, J.P.; Xie, Y.; Li, J.R.; Bu, X.H. Structural and magnetic modulations of copper(II) azido complexes: Unexpected in situ reactions of mono-N-donor pyridine-based co-ligands. Dalton Trans. 2016, 45, 1514–1524. [Google Scholar] [CrossRef]
- Villa-Pérez, C.; Cadavid-Vargas, J.F.; Di Virgilio, A.L.; Echeverría, G.A.; Camí, G.E.; Soria, D.B. Crystal structure, Hirshfeld surface analysis, spectroscopic and biological studies on sulfamethazine and sulfaquinoxaline ternary complexes with 2,2′-biquinoline. New J. Chem. 2018, 42, 891–901. [Google Scholar] [CrossRef]
- Viossat, B.; Gaucher, J.F.; Mazurier, A.; Selkti, M.; Tomas, A. Crystals tructure of bis(μ-chloro)bis[chloro-(o-phenanthroline-N,N’)-coppeг(II)],Cu2(C12H8N2)2(Cl2)2. Z. Krist. New Cryst. Struct. 1998, 213, 21–23. [Google Scholar] [CrossRef]
- Rösener, T.; Hoffmann, A.; Herres-Pawlis, S. Next Generation of Guanidine Quinoline Copper Complexes for Highly Controlled ATRP: Influence of Backbone Substitution on Redox Chemistry and Solubility. Eur. J. Inorg. Chem. 2018, 2018, 3164–3175. [Google Scholar] [CrossRef]
- Clark, G.R.; Waters, J.M.; Waters, T.N.; Williams, G.J. Polymorphism in Schiff base complexes of copper(II): The crystal and molecular structure of a second brown form of bis(N-methyl-2-hydroxy-1-naphthaldiminato)copper(II). J. Inorg. Nucl. Chem. 1977, 39, 1971–1975. [Google Scholar] [CrossRef]
- Creaven, B.S.; Devereux, M.; Karcz, D.; Kellett, A.; McCann, M.; Noble, A.; Walsh, M. Copper(II) complexes of coumarin-derived Schiff bases and their anti-Candida activity. J. Inorg. Biochem. 2009, 103, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Sydoruk, T.V.; Buvaylo, E.A.; Kokozay, V.N.; Vassilyeva, O.Y.; Skelton, B.W. Bis{μ-2-methoxy-6-[(methylimino)methyl]phenolato}bis({2-methoxy-6-[(methylimino)methyl]phenolato}copper(II)). Acta Crystallogr. Sect. E Struct. Reports Online 2013, 69, m551–m552. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.L.; Lin, X.S. Synthesis and crystal structures of two Schiff base copper(II) complexes derived from 4-chloro-2-[(2-morpholin-4-ylethylimino)methyl]phenol. Russ. J. Coord. Chem. 2010, 36, 472–476. [Google Scholar] [CrossRef]
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Kennard, O.; Watson, D.G.; Taylor, R. Supplement. Tables of bond lengths determined by X-ray and neutron diffraction. Part 2. Organometallic compounds and co-ordination complexes of the d-and f-block metals. J. Chem. Soc. Dalton Trans. 1989, 12, S1–S83. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. J. Chem. Soc. Dalton Trans. 2008, 21, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, L.; Destro, R. Experimental and theoretical charge density distribution of the colossal magnetoresistive transition metal sulfide Fe Cr2 S4. J. Chem. Phys. 2008, 128, 044710. [Google Scholar] [CrossRef] [PubMed]
- Blanchi, R.; Gervasio, G.; Marabello, D. Experimental electron density in the triclinic phase of Co2(CO)6(μ-CO)(μ-C4O2H2) at 120 K. Acta Crystallogr. Sect. B Struct. Sci. 2001, 57, 638–645. [Google Scholar] [CrossRef]
- Abramov, Y.A. On the Possibility of Kinetic Energy Density Evaluation from the Experimental Electron-Density Distribution. Acta Crystallogr. Sect. A Found. Crystallogr. 1997, 53, 264–272. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X-H⋯F-Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Bianchi, R.; Gervasio, G.; Marabello, D. The experimental charge density in transition metal compounds. C. R. Chim. 2005, 8, 1392–1399. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Hallman, M.; Kolczyńska, K.; Stepniewski, T.M. On the Harmonic Oscillator Model of Electron Delocalization (HOMED) index and its application to heteroatomic π-electron systems. Symmetry 2010, 2, 1485–1509. [Google Scholar] [CrossRef]
- Gatti, C.; Orlando, A.M.; Monza, E.; Lo Presti, L. Exploring Chemistry Through the Source Function for the Electron and the Electron Spin Densities. In Applications of Topological Methods in Molecular Chemistry; Chauvin, R., Lepetit, C., Silvi, B., Alikhani, E., Eds.; Springer: Cham, Switzerland, 2016; pp. 101–129. [Google Scholar]
- Tariq, M. Electrochemistry of Br-/Br2 Redox Couple in Acetonitrile, Methanol and Mix Media of Acetonitrile-Methanol: An Insight into Redox Behavior of Bromide on Platinum (Pt) and Gold (Au) Electrode. Z. Phys. Chem. 2020, 234, 295–312. [Google Scholar] [CrossRef]
- Desmarquest, J.P.; Trinh-Dinh, C.; Bloch, O. Determination electrochimique des potentiels normaux des couples oxydo-reducteurs Cu2+/Cu+ et Cu+/Cu0 dans le methanol. J. Electroanal. Chem. 1970, 27, 101–108. [Google Scholar] [CrossRef]
- Goodname, D.M.L.; Goodname, M.; Canham, G.W.R. Reversible Oxidation of Copper(I) Iodide in the Presence of Imidazole. Nature 1969, 222, 866. [Google Scholar]
- Banthia, S.; Samanta, A. In Situ Reduction of Copper(II) Forming an Unusually Air Stable Linear Complex of Copper(I) with a Fluorescent Tag. Inorg. Chem. 2004, 43, 6890–6892. [Google Scholar] [CrossRef] [PubMed]
- Ziesak, A.; Wesp, T.; Hübner, O.; Kaifer, E.; Wadepohl, H.; Himmel, H.-J. Counter-ligand control of the electronic structure in dinuclear copper-tetrakisguanidine complexes. Dalton Trans. 2015, 44, 19111–19125. [Google Scholar] [CrossRef]
- Ukpong, E.J.; Akpanudo, N.W.; Prasad, J. Redox and spectral behaviour of copper (II)-chloro and bromo complexes in some nonaqueous solvents. Afr. J. Pure Appl. Chem. 2010, 4, 38–43. [Google Scholar]
- Evans, D.A.; Burgey, C.S.; Kozlowski, M.C.; Tregay, S.W. C2-Symmetric Copper(II) Complexes as Chiral Lewis Acids. Scope and Mechanism of the Catalytic Enantioselective Aldol Additions of Enolsilanes to Pyruvate Esters. J. Am. Chem. Soc. 1999, 121, 686–699. [Google Scholar] [CrossRef]







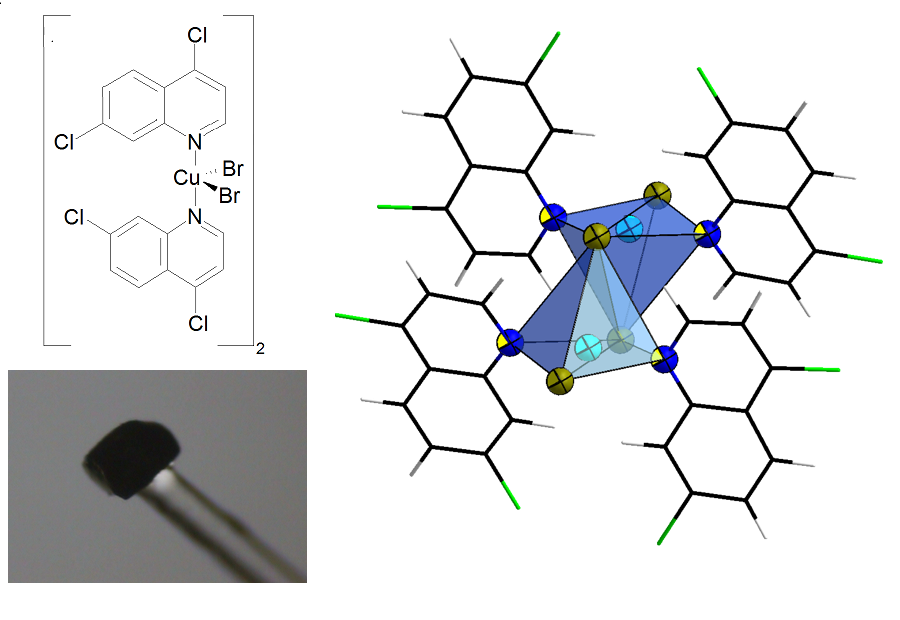{kind=link}
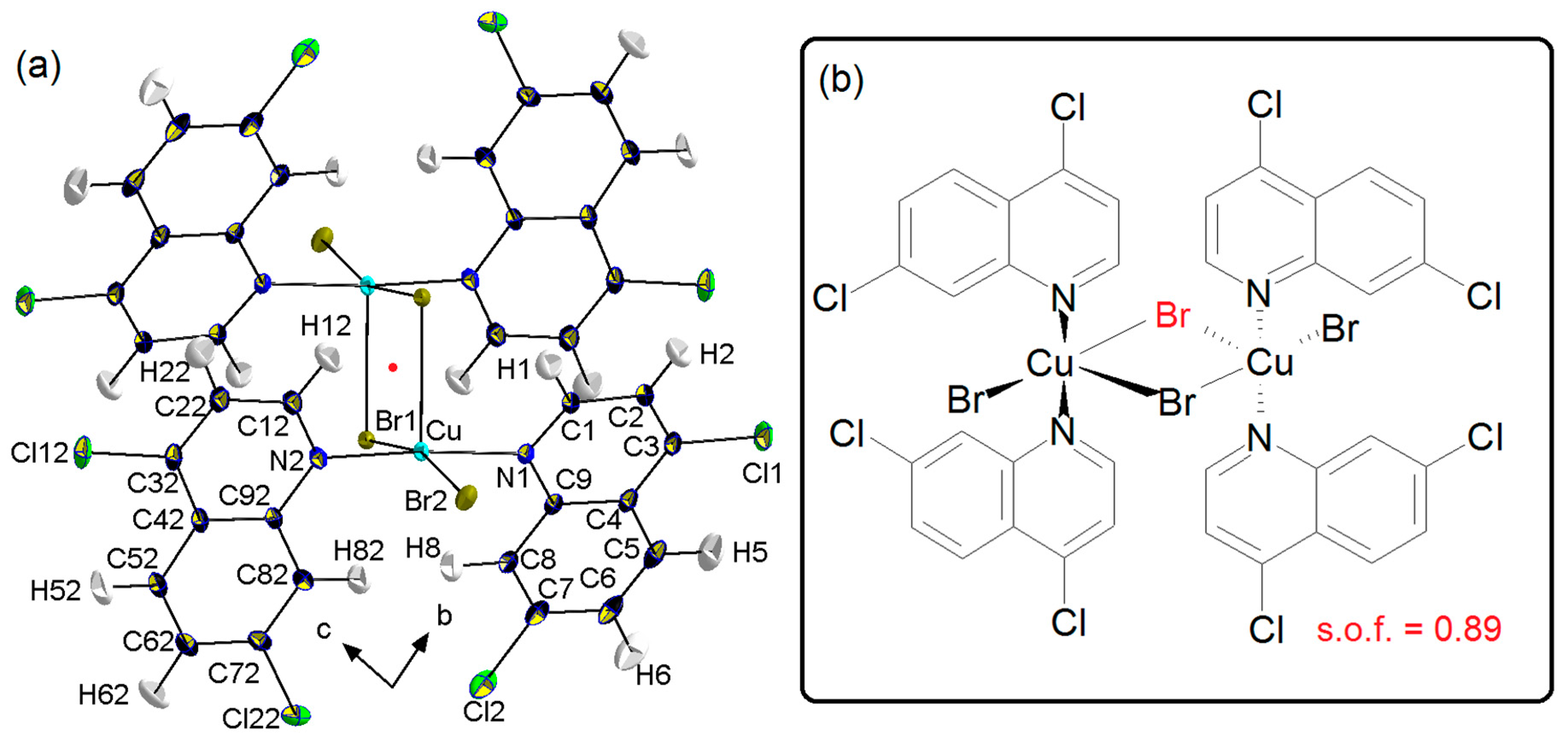{kind=link}
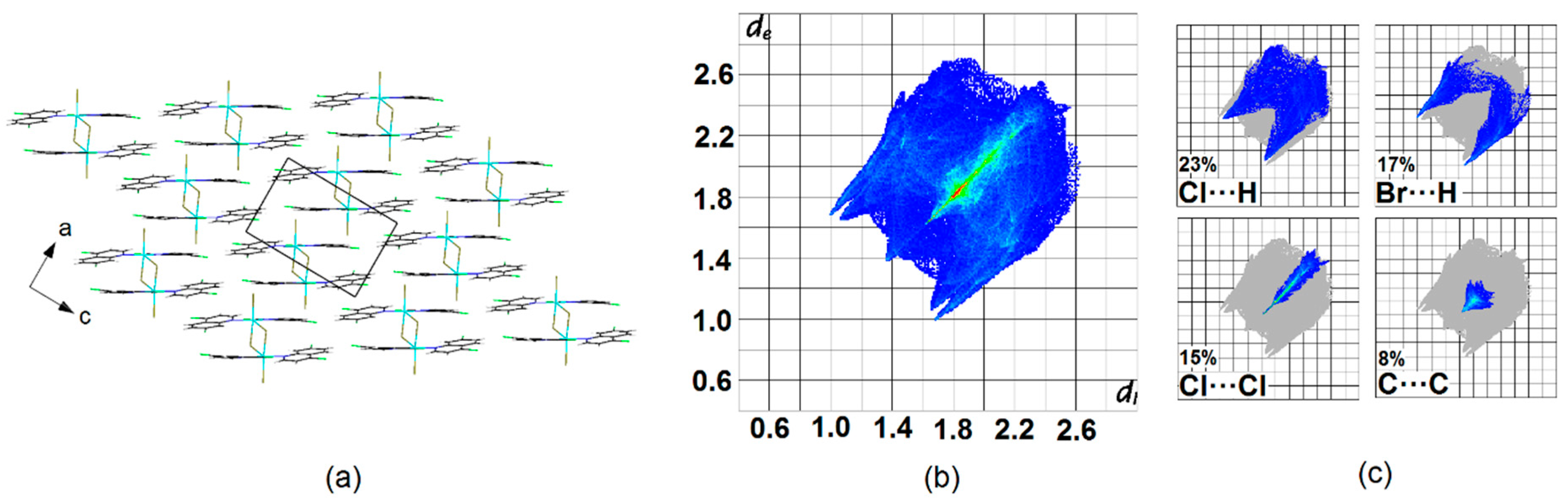{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | ||
|---|---|---|
| a (Å) | 7.8673(5) | |
| b (Å) | 11.0031(7) | |
| c (Å) | 11.7131(6) | |
| α (deg) | 82.325(2) | |
| β (deg) | 89.994(2) | |
| γ (deg) | 81.179(2) | |
| V (Å3) | 992.76(10) | |
| Density (g·cm−3) | 2.041 | |
| Crystal size (mm) | 0.350 × 0.225 × 0.200 | |
| Data collection | ||
| Measured/Unique refls. | 81,711/16,169 | |
| Observed refls. (I > 2σ(I)) | 13,645 | |
| (sinθ/λ)max/Å−1 | 1.00 | |
| Completeness | 0.973 | |
| Rint | 0.0264 | |
| Refinement details | IAM model b | Multipole Refinement c |
| R(F)/wR(F2)/goodness–of–fit d | 0.0339/0.0670/1.061 | 0.0239/0.0333/1.011 |
| Δρmax/Δρmin (e·Å−3) | +1.356/−0.913 | +0.497/−0.418 |
| Spherical (ζ = κα) and deformation (ζ’ = κ’α’) atomic exponents e | // | Cu: κ = 1.004(1), κ’ = 1.01(1) Br: κ = 1.052(2), κ’ = 1.12(2) Cl: κ = 1.020(1), κ’ = 0.99(2) N: κ = 1.004(1), κ’ = 0.90(2) C: κ = 1.013(1), κ’ = 0.952(4) H: κ = 1.230(6), κ’ = 1.13(1) |
| System | X-ray a | Isolated Complex, DFT b | Isolated Ligand, DFT b |
|---|---|---|---|
| Quinoline 1 c (total) | 0.88 | 0.87 | 0.88 |
| Quinoline 2 c (total) | 0.88 | 0.88 | 0.88 |
| Quinoline 1, N-bearing ring d | 0.87 | 0.87 | 0.88 |
| Quinoline 1, C6 ring e | 0.95 | 0.94 | 0.94 |
| Quinoline 2, N-bearing ring d | 0.88 | 0.89 | 0.88 |
| Quinoline 2, C6 ring e | 0.96 | 0.95 | 0.94 |
| Fragment | X-ray Geometry b | DFT-Optimized b |
|---|---|---|
| μ2–Br− (Br(1)) | −0.781 | −0.780 |
| Br−, equatorial (Br(2)) | −0.695 | −0.681 |
| Cu | 1.298 | 1.309 |
| 4,7-DCQ 1 | 0.084 | 0.075 |
| 4,7-DCQ 2 | 0.091 | 0.075 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finocchio, G.; Rizzato, S.; Macetti, G.; Tusha, G.; Lo Presti, L. Unravelling the Chemistry of the [Cu(4,7-Dichloroquinoline)2Br2]2 Dimeric Complex through Structural Analysis: A Borderline Ligand Field Case. Crystals 2020, 10, 477. https://doi.org/10.3390/cryst10060477
Finocchio G, Rizzato S, Macetti G, Tusha G, Lo Presti L. Unravelling the Chemistry of the [Cu(4,7-Dichloroquinoline)2Br2]2 Dimeric Complex through Structural Analysis: A Borderline Ligand Field Case. Crystals. 2020; 10(6):477. https://doi.org/10.3390/cryst10060477
Chicago/Turabian StyleFinocchio, Giada, Silvia Rizzato, Giovanni Macetti, Gers Tusha, and Leonardo Lo Presti. 2020. "Unravelling the Chemistry of the [Cu(4,7-Dichloroquinoline)2Br2]2 Dimeric Complex through Structural Analysis: A Borderline Ligand Field Case" Crystals 10, no. 6: 477. https://doi.org/10.3390/cryst10060477
APA StyleFinocchio, G., Rizzato, S., Macetti, G., Tusha, G., & Lo Presti, L. (2020). Unravelling the Chemistry of the [Cu(4,7-Dichloroquinoline)2Br2]2 Dimeric Complex through Structural Analysis: A Borderline Ligand Field Case. Crystals, 10(6), 477. https://doi.org/10.3390/cryst10060477