Cyclic Automated Model Building (CAB) Applied to Nucleic Acids


 , ,
, , 
Abstract
1. Introduction
2. CAB Algorithm for Locating Ligand Heavy Atoms
3. The Recursive Algorithm
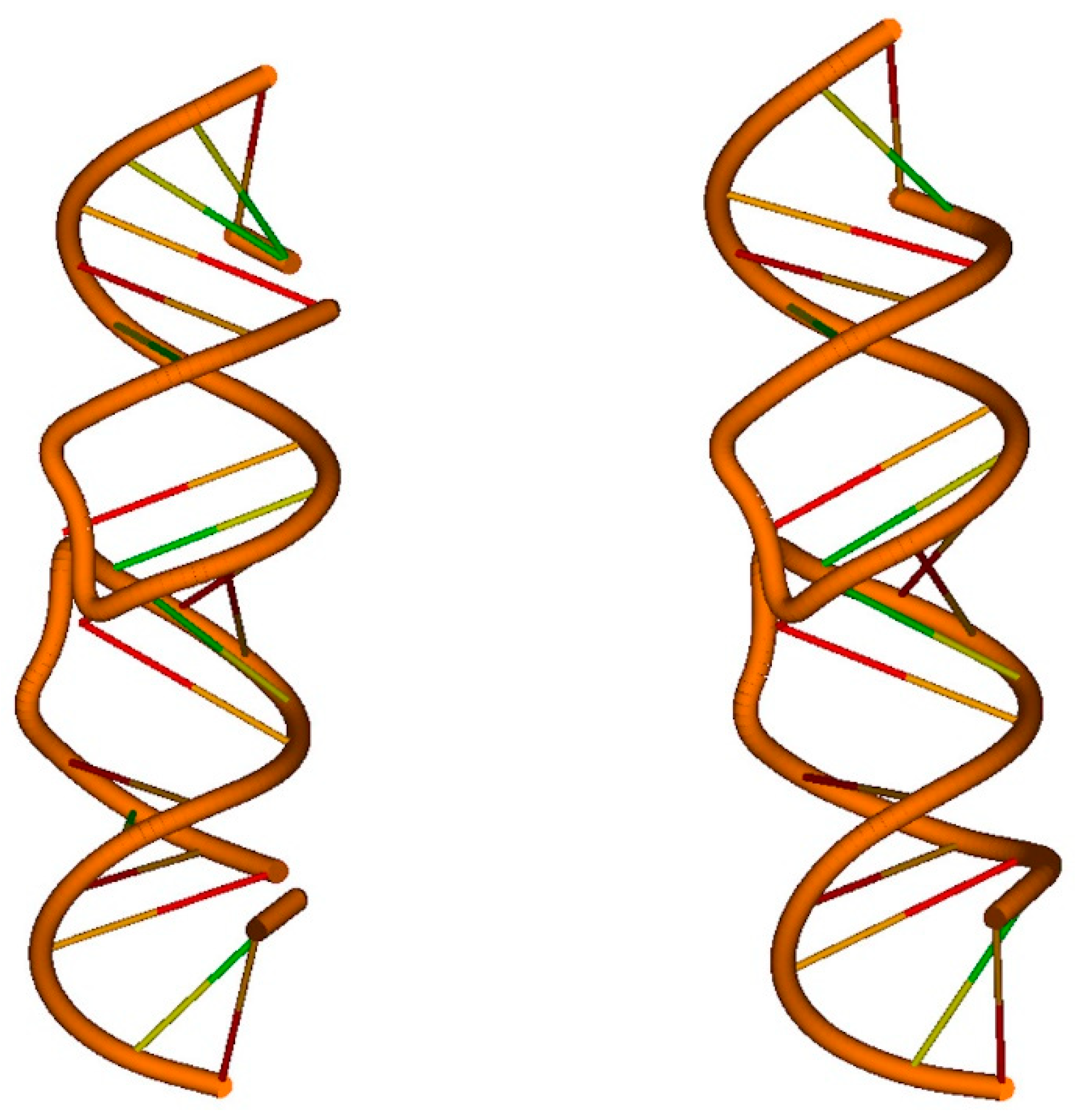
4. Applications
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMB | automated model building. |
| MR | molecular replacement. |
| R | crystallographic residual between observed and calculated structure factor amplitudes (for all of the experimental data). |
| Rf | cross validation R-value for the free data set [63]. |
| MA | ratio “number of residues with P atoms within 0.6Å distance from the published positions/number of residues in the asymmetric unit”, according to the published sequence. It is an indication of the accuracy of the model. |
| MAM ratio | “number of non-hydrogen atoms within 0.6Å distance from published positions/number of non-hydrogen atoms in the asymmetric unit”. |
References
- Cowtan, K.D. The Buccaneer software for automated model building. Tracing protein chains. Acta Cryst. 2006, D62, 1002–1011. [Google Scholar] [CrossRef]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.-W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst. 2008, D64, 61–69. [Google Scholar] [CrossRef]
- Wang, X.; Kapral, G.; Murray, L.; Richardson, D.; Richardson, J.; Snoeying, J. RNABC: Forward kinematics to reduce all-atom steric clashes in RNA backbone. J. Math. Biol. 2008, 56, 253–278. [Google Scholar] [CrossRef][Green Version]
- Yamashita, K.; Zhou, Y.; Tanaka, I.; Yao, M. New model-fitting and model-completion programs for automated iterative nucleic acid refinement. Acta Cryst. 2013, D69, 1171–1179. [Google Scholar] [CrossRef]
- Keating, K.S.; Pyle, A.M. RCrane: Semi-automated RNA model building. Acta Cryst. 2012, D68, 985–995. [Google Scholar] [CrossRef]
- Keating, K.S.; Pyle, A.M. Semi-automated model building for RNA crystallography using a directed rotameric approach. Proc. Natl. Acad. Sci. USA 2010, 107, 8177–8182. [Google Scholar] [CrossRef]
- Murray, L.J.; Arendall, W.B.; Richardson, D.C.; Richardson, J.S. RNA backbone is rotameric. Proc. Natl. Acad. Sci. USA 2003, 100, 13904–13909. [Google Scholar] [CrossRef]
- Hattne, J.; Lamzin, V.S. Pattern-recognition-based detection of planar objects in three-dimensional electron-density maps. Acta Cryst. 2008, D64, 834–842. [Google Scholar] [CrossRef]
- Cowtan, K. Automated nucleic acid chain tracing in real time. IUCR J. 2014, 1, 387–392. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Lebedev, A.; Wilson, K.S.; Dodson, E.J. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Cryst. 1999, D55, 247–255. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst. 2012, D68, 352–367. [Google Scholar] [CrossRef]
- Burla, M.C.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G. CAB: A cyclic automatic model building procedure. Acta Cryst. 2018, D74, 1096–1104. [Google Scholar] [CrossRef]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mazzone, A.; Siliqi, D. Molecular replacement: The probabilistic approach of the program REMO09 and its applications. Acta Cryst. 2009, A65, 512–527. [Google Scholar] [CrossRef]
- Burla, M.C.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G. Synergy among phase-refinement techniques in macromolecular crystallography. Acta Cryst. 2017, D73, 877–888. [Google Scholar] [CrossRef]
- Cowtan, K. Fast Fourier feature recognition. Acta Cryst. 2001, D57, 1435–1444. [Google Scholar] [CrossRef]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Siliqi, D. Phasing at resolution higher than the experimental resolution. Acta Cryst. 2005, D61, 556–565. [Google Scholar] [CrossRef]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Siliqi, D. Ab initio phasing at resolution higher than experimental resolution. Acta Cryst. 2005, D61, 1080–1087. [Google Scholar] [CrossRef]
- Giacovazzo, C.; Siliqi, D. Improving Direct Methods phases by Heavy-Atom information and Solvent Flattening. Acta Cryst. 1997, D53, 789–798. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Giacovazzo, C.; Polidori, G. The difference electron density: A probabilistic reformulation. Acta Cryst. 2010, A66, 347–361. [Google Scholar] [CrossRef]
- Burla, M.C.; Giacovazzo, C.; Polidori, G. From a random to the correct structure: The VLD algorithm. J. Appl. Cryst. 2010, 43, 825–836. [Google Scholar] [CrossRef]
- Giacovazzo, C. Solution of the phase problem at non-atomic resolution by the Phantom Derivative method. Acta Cryst. 2015, D71, 483–512. [Google Scholar] [CrossRef] [PubMed]
- Giacovazzo, C. From direct-space discrepancy functions to crystallographic least squares. Acta Cryst. 2015, D71, 36–45. [Google Scholar] [CrossRef]
- Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C. Phase improvement via the Phantom Derivative technique: Ancils that are related to the target structure. Acta Cryst. 2016, A72, 551–557. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Cryst. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Burla, M.C.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G. How far are we from automatic crystal structure solution via molecular-replacement techniques? Acta Cryst. 2020, D76, 9–18. [Google Scholar] [CrossRef]
- Campbell, N.H.; Parkinson, G.N.; Reszka, A.P.; Neidle, S. Structural basis of DNA quadruplex recognition by an acridine drug. J. Am. Chem. Soc. 2008, 130, 6722–6724. [Google Scholar] [CrossRef]
- Millonig, H.; Pous, J.; Gouyette, C.; Subirana, J.A.; Campos, J.L. The interaction of manganese ions with DNA. J. Inorg. Biochem. 2009, 103, 876–880. [Google Scholar] [CrossRef]
- Juan, E.C.M.; Shimizu, S.; Ma, X.; Kurose, T.; Haraguchi, T.; Zhang, F.; Tsunoda, M.; Ohkubo, A.; Sekine, M.; Shibata, T.; et al. Insights into the DNA stabilizing contributions of a bicyclic cytosine analogue: Crystal structures of DNA duplexes containing 7,8-dihydropyrido [2,3-d]pyrimidin-2-one. Nucleic Acids Res. 2010, 38, 6737–6745. [Google Scholar] [CrossRef]
- Carter, M.; Ho, P.S. Assaying the energies of biological halogen bonds. Cryst. Growth Des. 2011, 11, 5087–5095. [Google Scholar] [CrossRef]
- Carter, M.; Voth, A.R.; Scholfield, M.R.; Rummel, B.; Sowers, L.C.; Ho, P.S. Enthalpy-entropy compensation in biomolecular halogen bonds measured in DNA junctions. Biochemistry 2013, 52, 4891–4903. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.P. The Effects of Disubstitution on the Binding of Ruthenium Complexes to DNA. Ph.D. Thesis, University of Reading, Reading, UK, 2014. [Google Scholar]
- Rohner, M.; Medina-Molner, A.; Spingler, B. N,N,O and N,O,N meridional cis coordination of two guanines to copper(II) by d(CGCGCG)2. Inorg. Chem. 2016, 55, 6130–6140. [Google Scholar] [CrossRef] [PubMed]
- Russo Krauss, I.; Ramaswamy, S.; Neidle, S.; Haider, S.; Parkinson, G.N. Structural insights into the quadruplex-duplex 3′ interface formed from a telomeric repeat: A potential molecular target. J. Am. Chem. Soc. 2016, 138, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Ahmad Sobri, A.F.; Brady, R.L. Non-natural DNA pair Z (6-amino-5-nitro-2[1H] pyridone heterocycle)-guanosine. 2016; Unpublished work. [Google Scholar]
- Sbirkova, H.I.; Schivachev, B.L. Crystal structure of a DNA sequence d(CGTGAATTCACG) at 130K. Bulg. Chem. Commun. 2016, 48, 589–593. [Google Scholar]
- Reichenbach, L.F.; Sobri, A.A.; Zaccai, N.R.; Agnew, C.R.J.; Burton, N.; Eperon, L.P.; de Ornellas, S.; Eperon, I.C.; Brady, R.L.; Burley, G.A. Structural basis of the mispairing of an artificially expanded genetic information system. Chemistry 2016, 1, 946–958. [Google Scholar] [CrossRef][Green Version]
- Hardwick, J.S.; Ptchelkine, D.; El-Sagheer, A.H.; Tear, I.; Singleton, D.; Phillips, S.E.V.; Lane, A.N.; Brown, T. 5-Formylcytosine does not change the global structure of DNA. Nat. Struct. Mol. Biol. 2017, 24, 544–552. [Google Scholar] [CrossRef]
- Sbirkova, H.I.; Schivachev, B.L. The effect of berenil and cacodylate on the crystal structure of d(CGTGAATTCACG). 2017; Unpublished work. [Google Scholar]
- Sbirkova-Dimitrova, H.I.; Shivachev, B.L. Crystal structure of the DNA sequence d(CGTGAATTCACG)2 with DAPI. Acta Cryst. 2017, 73, 500–504. [Google Scholar] [CrossRef]
- Cruse, W.; Saludjian, P.; Neuman, A.; Prange, T. Destabilizing effect of a fluorouracil extra base in a hybrid RNA duplex compared with bromo and chloro analogues. Acta Cryst. 2001, D57, 1609–1613. [Google Scholar] [CrossRef]
- Gherghe, C.M.; Krahn, J.M.; Weeks, K.M. Crystal structures, reactivity and inferred acylation transition states for 2′-amine substituted RNA. J. Am. Chem. Soc. 2005, 127, 13622–13628. [Google Scholar] [CrossRef]
- Haeberli, P.; Berger, I.; Pallan, P.S.; Egli, M. Syntheses of 4′-thioribonucleosides and thermodynamic stability and crystal structure of RNA oligomers with incorporated 4′-thiocytosine. Nucleic Acids Res. 2005, 33, 3965–3975. [Google Scholar] [CrossRef][Green Version]
- Ennifar, E.; Paillart, J.C.; Bodlenner, A.; Walter, P.; Weibel, J.-M.; Aubertin, A.-M.; Pale, P.; Dumas, P.; Marquet, R. Targeting the dimerization initiation site of HIV-1 RNA with aminoglycosides: From crystal to cell. Nucleic Acids Res. 2006, 34, 2328–2339. [Google Scholar] [CrossRef]
- Zhao, Q.; Han, Q.; Kissinger, C.R.; Hermann, T.; Thompson, P.A. Structure of hepatitis C virus IRES subdomain IIa. Acta Cryst. 2008, D64, 436–443. [Google Scholar] [CrossRef]
- Thore, S.; Frick, C.; Ban, N. Structural basis of thiamine pyrophosphate analogues binding to the eukaryotic riboswitch. J. Am. Chem. Soc. 2008, 130, 8116–8117. [Google Scholar] [CrossRef]
- Pitt, J.N.; Ferre-D’Amare, A.R. Structure-guided engineering of the regioselectivity of RNA ligase ribozymes. J. Am. Chem. Soc. 2009, 131, 3532–3540. [Google Scholar] [CrossRef]
- Ren, A.; Rajashankar, K.R.; Patel, D.J. Fluoride ion encapsulation by Mg2+ ions and phosphates in a fluoride riboswitch. Nature 2012, 486, 85–89. [Google Scholar] [CrossRef]
- Vorobiev, S.M.; Ma, L.-C.; Montelione, G.T. Crystal structure of the 16-mer double stranded RNA. 2016; Unpublished work. [Google Scholar]
- Monestier, A.; Aleksandrov, A.; Coureux, P.D.; Panvert, M.; Mechulam, Y.; Schmitt, E. The structure of an E. coli tRNAf(Met) A1-U72 variant shows an unusual conformation of the A1-U72 base pair. RNA 2017, 23, 673–682. [Google Scholar] [CrossRef]
- Huang, L.; Wang, J.; Wilson, T.J.; Lilley, D.M.J. Structure of the guanidine III riboswitch. Cell Chem. Biol. 2017, 24, 1407–1415. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Z. DNA 8mer containing two 2SeT modifications. 2016; Unpublished work. [Google Scholar]
- Zhang, W.; Tam, C.P.; Zhou, L.; Oh, S.S.; Wang, J.; Szostak, J.W. Structural Rationale for the Enhanced Catalysis of Nonenzymatic RNA Primer Extension by a Downstream Oligonucleotide. J. Am. Chem. Soc. 2018, 140, 2829–2840. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. 2010, D66, 486–501. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.I.I.I.; Darden, T.; Gohlke, H.; Luo, R.; Merz KMJr Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Chou, F.C.; Sripakdeevong, P.; Dibrov, S.M.; Hermann, T.; Das, R. Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat. Methods 2013, 10, 74–76. [Google Scholar] [CrossRef]
- Das, R.; Karanicolas, J.; Baker, D. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat. Methods 2010, 7, 291–294. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Chou, F.-C.; Das, R. Modeling Complex RNA Tertiary Folds with Rosetta. Methods Enzymol. 2015, 553, 35–64. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.W.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar] [CrossRef]
- Pavelcik, F.; Schneider, B. Building of RNA and DNA double helices into electron density. Acta Cryst. 2008, D64, 620–626. [Google Scholar] [CrossRef]
- Pavelcik, F. Application of constrained real-space refinement of flexible molecular fragments to automatic model building of RNA structures. J. Appl. Cryst. 2012, 45, 309–315. [Google Scholar] [CrossRef]
- Lu, X.L.; Olson, W.K. 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef]
- Brünger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| PDB | SG | RES | nN | Model | Ligand(s) |
|---|---|---|---|---|---|
| 3ce5 [27] | I 4 | 2.50 | 2·12 | 1k8p | 2·K + BRA |
| 3eil [28] | P 32 | 2.60 | 6·12 | 3·463d | 7·Mn |
| 3n4o [29] | P 21 21 21 | 2.90 | 2·12 | 1dnh | 2·B7C + HT |
| 3tok [30] | C 2 | 1.74 | 10 + 10 | 2org | Na |
| 4gsg [31] | C 2 | 2.00 | 2·(10 + 10) | 2·2org | UCL |
| 4ms5 [32] | P 43 21 2 | 2.23 | 1·10 | 3qrn | Ba + RKF |
| 4xqz [33] | P 21 | 2.15 | 8·6 | 2·5ihd | 6·Cu + 4·Ca + 7·Cl + MES + MOH |
| 5dwx [34] | P 4 21 2 | 2.71 | 24 + 8 | 1kf1 | K |
| 5i4s [35] | R 3 | 2.46 | 2·12 | 476d | 8·Ca + 2·1W5 |
| 5ihd [33] | P 21 | 1.57 | 4·6 | 2·2dcg | 4·Cu + 2·Ca + 2·2OP + SIN |
| 5ju4 [36] | P 21 21 21 | 2.00 | 2·12 | 1d29 | Mg + Cl |
| 5lj4 [37] | R 3 | 2.17 | 2·12 | 463d | 4·Ca + 2·1W5 + 2·1WA |
| 5mvt [38] | P 31 2 1 | 1.89 | 2·12 | 5mvl | 3·Co |
| 5nt5 [39] | P 21 21 21 | 2.30 | 2·12 | 1d29 | Na + CAC |
| 5t4w [40] | P 21 21 21 | 2.30 | 2·12 | 5jua | DAP |
| 1iha [41] | C 2 | 1.60 | 2·9 | 165d | 2·Cl + 2·BRU + 2·RHD |
| 1z7f [42] | P 31 2 1 | 2.10 | 3·16 | 1yrm | 2·Sr |
| 2a0p [43] | R 3 2 | 1.95 | 2·8 | 259d | S4C |
| 2fd0 [44] | C 2 2 21 | 1.80 | 2·23 | 2fcy | K + Cl + 5BU + LIV |
| 2pn4 [45] | P 21 21 21 | 2.32 | 2·(24 + 20) | 2·2pn3 | 10·Sr + 4·5BU |
| 3d2v [46] | P 21 21 2 | 2.00 | 2·77 | 2cky | 10·Mg + 2·PYI |
| 3fs0 [47] | P 31 | 2.30 | 10 + 11 | ½·3ftm | 3·Mg |
| 4enc [48] | P 21 21 2 | 2.27 | 52 | 4enb | 5·Mg + K + F |
| 5kvj [49] | R 3 | 2.26 | 16 + 16 | 2·3nd3 | ARG |
| 5l4o [50] | P 32 1 2 | 2.80 | 77 | 3cw5 | Na + PSU + OMC + 4SU + 5MU + H2U |
| 5nz6 [51] | P 32 1 2 | 2.94 | 41 | ½·5nwq | 2·CBV + GAI |
| 5tgp [52] | P 61 | 1.60 | 2·8 | 2·1dns | 4·US3 |
| 5uz6 [53] | C 2 | 2.10 | 3·(25 + 8) | 3·5ux3 | 8OS + LCC |
| 6az4 [53] | P 41 21 2 | 2.98 | 32 + 9 | 4fnj | GP3 |
| Ligand Information | |||||
| Code | Formula | Code | Formula | Code | Formula |
| 1W5 | C10 H14 N3 O9 P | BRU | C9 H12 Br N2 O8 P | MES | C6 H13 N O4 S |
| 1WA | C10 H16 N5 O7 P | CAC | C2 H6 As O2 | MOH | C H4 O |
| 2OP | C3 H6 O3 | CBV | C9 H13 Br N3 O8 P | OMC | C10 H16 N3 O8 P |
| 4SU | C9 H13 N2 O8 P S | DAP | C16 H15 N5 | PSU | C9 H13 N2 O9 P |
| 5BU | C9 H12 Br N2 O9 P | GAI | C H5 N3 | PYI | C14 H21 N4 O7 P2 |
| 5MU | C10 H15 N2 O9 P | GP3 | C20 H27 N10 O18 P3 | RHD | Rh3 H18 N6 |
| 8OS | C14 H18 N7 O7 P | H2U | C9 H15 N2 O9 P | RKF | C38 H20 F2 N13 Ru |
| ARG | C6 H15 N4 O2 | HT | C25 H24 N6 O | S4C | C9 H14 N3 O7 P S |
| B7C | C12 H16 N3 O7 P | LCC | C11 H16 N3 O8 P | SIN | C4 H6 O4 |
| BRA | C35 H43 N7 O2 | LIV | C29 H55 N5 O18 | UCL | C9 H12 Cl N2 O8 P |
| US3 | C10 H15 N2 O7 P Se | ||||
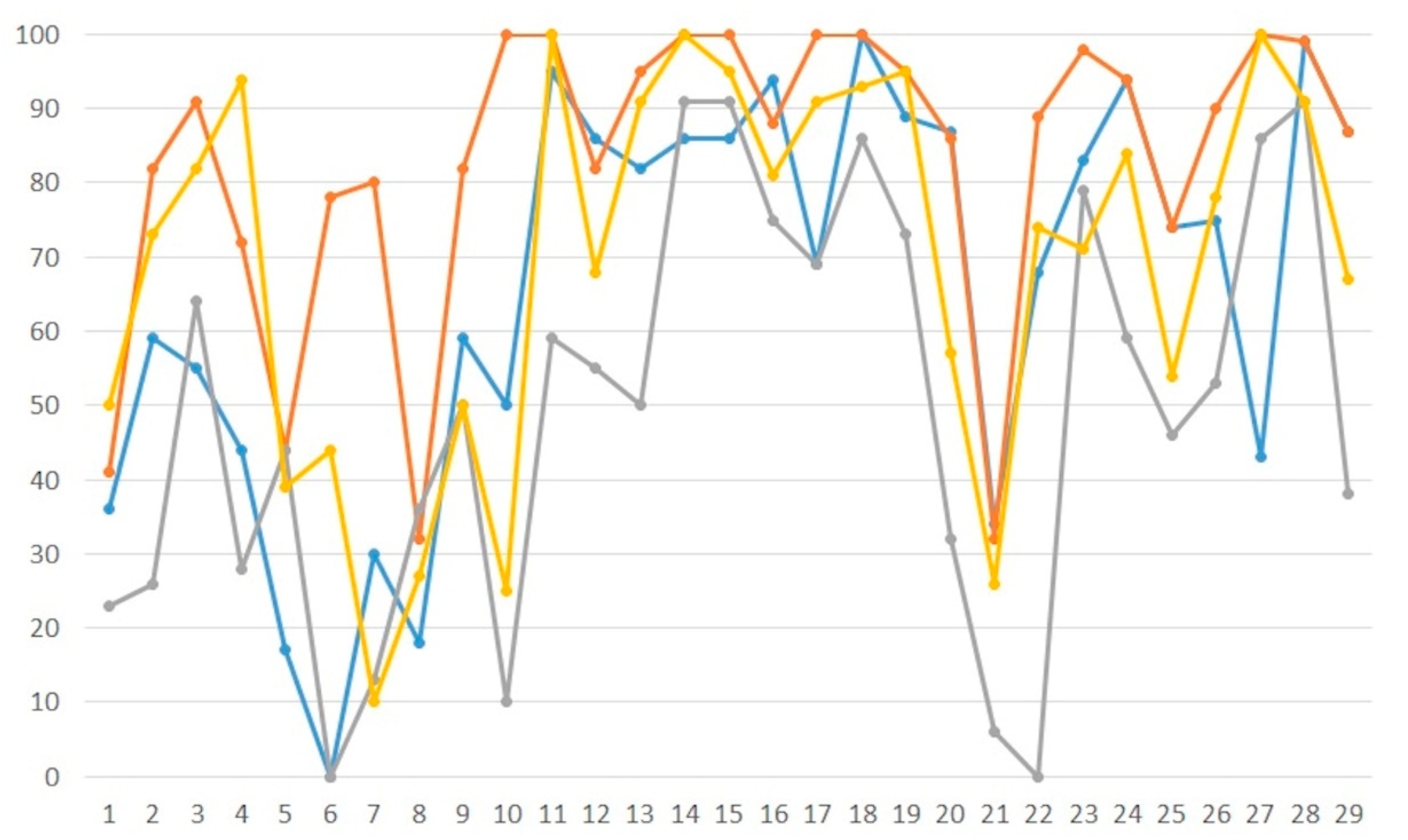
| PDB | <|Δϕr|>° | Rr | Rf | RN | RfN | MAN | MAMN | RC | RfC | MAC | MAMC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3ce5 | 50 | 41 | 43 | 54 | 59 | 36 | 16 | 52 | 53 | 41 | 18 |
| 3eil | 46 | 31 | 36 | 47 | 50 | 59 | 43 | 36 | 38 | 82 | 76 |
| 3n4o | 33 | 23 | 26 | 44 | 45 | 55 | 36 | 23 | 26 | 91 | 69 |
| 3tok | 49 | 35 | 35 | 57 | 58 | 44 | 15 | 52 | 56 | 72 | 24 |
| 4gsg | 53 | 34 | 38 | 45 | 45 | 17 | 9 | 42 | 46 | 44 | 17 |
| 4ms5 | 59 | 46 | 64 | 56 | 57 | 0 | 4 | 37 | 41 | 78 | 57 |
| 4xqz | 48 | 32 | 35 | 58 | 58 | 30 | 22 | 27 | 30 | 80 | 94 |
| 5dwx | 58 | 41 | 44 | 57 | 58 | 18 | 5 | 48 | 59 | 32 | 25 |
| 5i4s | 35 | 25 | 29 | 36 | 37 | 59 | 49 | 35 | 34 | 82 | 51 |
| 5ihd | 39 | 34 | 36 | 51 | 52 | 50 | 39 | 25 | 29 | 100 | 92 |
| 5ju4 | 26 | 26 | 28 | 37 | 37 | 95 | 83 | 26 | 28 | 100 | 100 |
| 5lj4 | 29 | 25 | 29 | 44 | 48 | 86 | 58 | 41 | 45 | 82 | 58 |
| 5mvt | 28 | 29 | 28 | 38 | 37 | 82 | 79 | 31 | 31 | 95 | 92 |
| 5nt5 | 24 | 27 | 28 | 46 | 47 | 86 | 64 | 27 | 28 | 100 | 99 |
| 5t4w | 25 | 25 | 29 | 43 | 42 | 86 | 64 | 25 | 29 | 100 | 96 |
| 1iha | 41 | 34 | 35 | 36 | 37 | 94 | 77 | 23 | 25 | 88 | 81 |
| 1z7f | 34 | 32 | 34 | 42 | 43 | 69 | 71 | 30 | 30 | 100 | 100 |
| 2a0p | 31 | 27 | 35 | 32 | 39 | 100 | 93 | 27 | 35 | 100 | 99 |
| 2fd0 | 33 | 32 | 36 | 37 | 38 | 89 | 78 | 32 | 36 | 95 | 85 |
| 2pn4 | 40 | 34 | 40 | 41 | 48 | 87 | 68 | 36 | 41 | 86 | 74 |
| 3d2v | 57 | 47 | 51 | 49 | 51 | 34 | 29 | 49 | 50 | 32 | 30 |
| 3fs0 | 63 | 42 | 47 | 40 | 41 | 68 | 51 | 29 | 33 | 89 | 86 |
| 4enc | 28 | 25 | 28 | 36 | 39 | 83 | 74 | 25 | 28 | 98 | 95 |
| 5kvj | 49 | 31 | 39 | 37 | 46 | 94 | 55 | 32 | 41 | 94 | 63 |
| 5l4o | 40 | 31 | 36 | 35 | 39 | 74 | 51 | 34 | 39 | 74 | 53 |
| 5nz6 | 45 | 23 | 23 | 39 | 43 | 75 | 44 | 31 | 32 | 90 | 53 |
| 5tgp | 26 | 28 | 29 | 51 | 51 | 43 | 40 | 27 | 27 | 100 | 100 |
| 5uz6 | 34 | 34 | 36 | 30 | 33 | 99 | 88 | 30 | 33 | 99 | 88 |
| 6az4 | 51 | 36 | 40 | 28 | 30 | 87 | 63 | 28 | 30 | 87 | 63 |
| With | Without | |||
|---|---|---|---|---|
| PDB | MAC | MAMC | MAC | MAMC |
| 3n4o | 91 | 69 | 77 | 38 |
| 4xqz | 80 | 94 | 43 | 30 |
| 5ihd | 100 | 92 | 70 | 47 |
| 5ju4 | 100 | 100 | 95 | 83 |
| 5nt5 | 100 | 99 | 100 | 87 |
| 5t4w | 100 | 96 | 91 | 64 |
| 1iha | 88 | 81 | 81 | 77 |
| 2a0p | 100 | 99 | 100 | 99 |
| 2fd0 | 95 | 85 | 95 | 81 |
| 4enc | 98 | 95 | 83 | 78 |
| 5tgp | 100 | 100 | 100 | 75 |
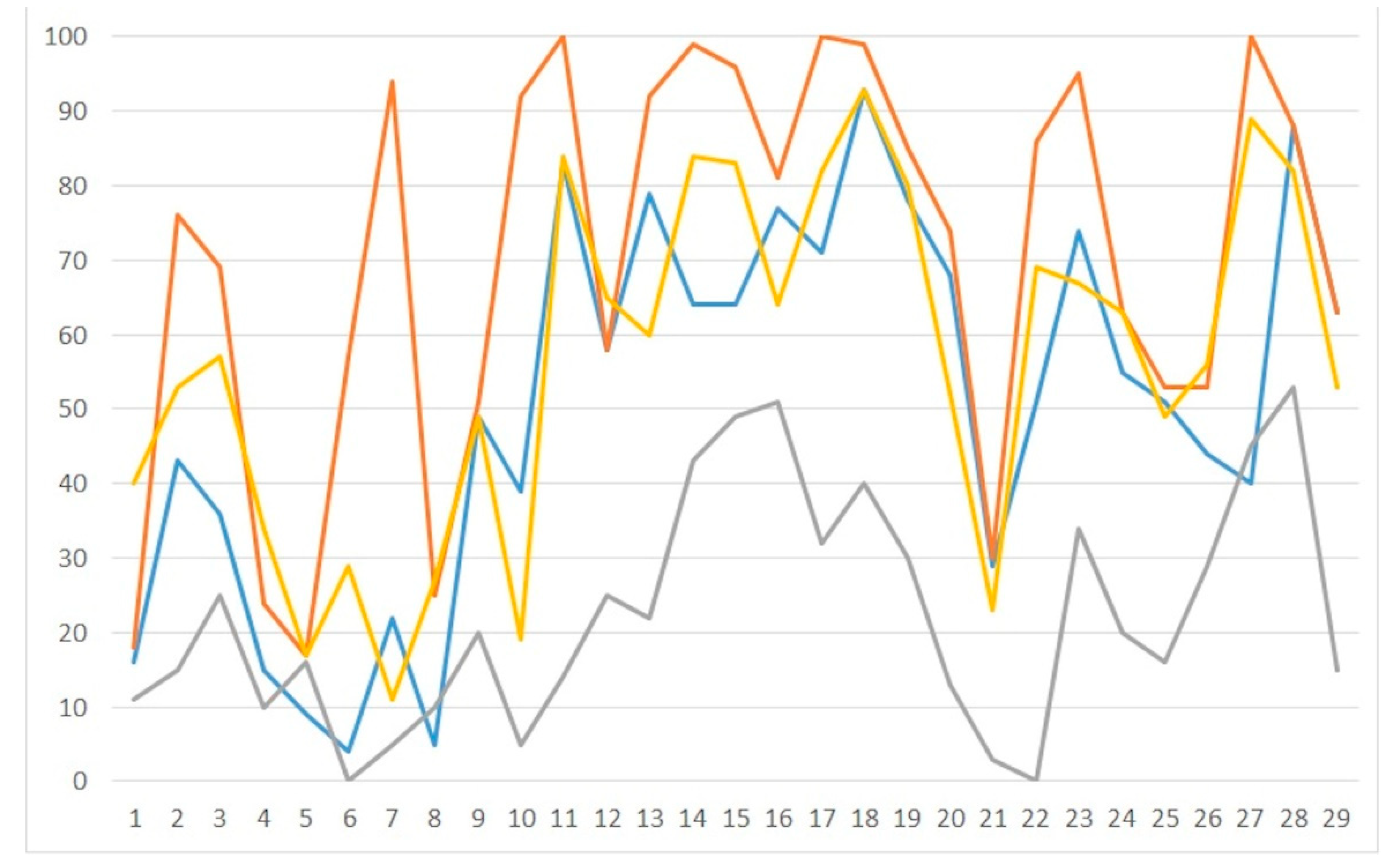
| ARP/wARP | PHENIX.AUTOBUILD | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PDB | <|Δϕr|>° | RA | RfA | MAA | MAMA | RP | RfP | MAP | MAMP |
| 3ce5 | 50 | 53 | 56 | 23 | 11 | 45 | 47 | 50 | 40 |
| 3eil | 46 | 48 | 56 | 26 | 15 | 43 | 47 | 73 | 53 |
| 3n4o | 33 | 33 | 52 | 64 | 25 | 33 | 37 | 82 | 57 |
| 3tok | 49 | 52 | 53 | 28 | 10 | 45 | 47 | 94 | 34 |
| 4gsg | 53 | 37 | 43 | 44 | 16 | 38 | 38 | 39 | 17 |
| 4ms5 | 59 | 0 | 0 | 0 | 0 | 48 | 53 | 44 | 29 |
| 4xqz | 48 | 53 | 54 | 13 | 5 | 57 | 60 | 10 | 11 |
| 5dwx | 58 | 40 | 47 | 36 | 10 | 49 | 53 | 27 | 27 |
| 5i4s | 35 | 34 | 44 | 50 | 20 | 36 | 39 | 50 | 49 |
| 5ihd | 39 | 51 | 51 | 10 | 5 | 52 | 56 | 25 | 19 |
| 5ju4 | 26 | 49 | 58 | 59 | 14 | 35 | 33 | 100 | 84 |
| 5lj4 | 29 | 41 | 52 | 55 | 25 | 40 | 41 | 68 | 65 |
| 5mvt | 28 | 45 | 51 | 50 | 22 | 46 | 44 | 91 | 60 |
| 5nt5 | 24 | 35 | 48 | 91 | 43 | 35 | 38 | 100 | 84 |
| 5t4w | 25 | 31 | 45 | 91 | 49 | 31 | 33 | 95 | 83 |
| 1iha | 41 | 41 | 41 | 75 | 51 | 36 | 33 | 81 | 64 |
| 1z7f | 34 | 40 | 46 | 69 | 32 | 35 | 36 | 91 | 82 |
| 2a0p | 31 | 39 | 53 | 86 | 40 | 31 | 37 | 93 | 93 |
| 2fd0 | 33 | 45 | 52 | 73 | 30 | 37 | 36 | 95 | 80 |
| 2pn4 | 40 | 47 | 55 | 32 | 13 | 42 | 48 | 57 | 52 |
| 3d2v | 57 | 56 | 57 | 6 | 3 | 47 | 48 | 26 | 23 |
| 3fs0 | 63 | 0 | 0 | 0 | 0 | 29 | 34 | 74 | 69 |
| 4enc | 28 | 33 | 46 | 79 | 34 | 40 | 41 | 71 | 67 |
| 5kvj | 49 | 39 | 55 | 59 | 20 | 35 | 40 | 84 | 63 |
| 5l4o | 40 | 44 | 53 | 46 | 16 | 45 | 50 | 54 | 49 |
| 5nz6 | 45 | 34 | 38 | 53 | 29 | 35 | 37 | 78 | 56 |
| 5tgp | 26 | 45 | 51 | 86 | 45 | 34 | 33 | 100 | 89 |
| 5uz6 | 34 | 34 | 40 | 91 | 53 | 33 | 33 | 91 | 82 |
| 6az4 | 51 | 42 | 46 | 38 | 15 | 39 | 40 | 67 | 53 |
| SUB1 | SUB2 | SUB3 | SUB4 |
|---|---|---|---|
| NAUTILUS [10] | RCRANE [6] in COOT [54] | NAFIT, NABUILD in LAFIRE [5] | AMBER [55] |
| ARP/wARP [9] | ERRASER [56] | FARFAR [57,58] | |
| PHENIX.AUTOB [3] | ROSETTA [59] | ||
| NUT/DHL/RSR [60,61] | 3DNA [62] | ||
| CAB |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burla, M.C.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G. Cyclic Automated Model Building (CAB) Applied to Nucleic Acids. Crystals 2020, 10, 280. https://doi.org/10.3390/cryst10040280
Burla MC, Carrozzini B, Cascarano GL, Giacovazzo C, Polidori G. Cyclic Automated Model Building (CAB) Applied to Nucleic Acids. Crystals. 2020; 10(4):280. https://doi.org/10.3390/cryst10040280
Chicago/Turabian StyleBurla, Maria Cristina, Benedetta Carrozzini, Giovanni Luca Cascarano, Carmelo Giacovazzo, and Giampiero Polidori. 2020. "Cyclic Automated Model Building (CAB) Applied to Nucleic Acids" Crystals 10, no. 4: 280. https://doi.org/10.3390/cryst10040280
APA StyleBurla, M. C., Carrozzini, B., Cascarano, G. L., Giacovazzo, C., & Polidori, G. (2020). Cyclic Automated Model Building (CAB) Applied to Nucleic Acids. Crystals, 10(4), 280. https://doi.org/10.3390/cryst10040280