A Short Review of Current Computational Concepts for High-Pressure Phase Transition Studies in Molecular Crystals


Abstract
1. Introduction
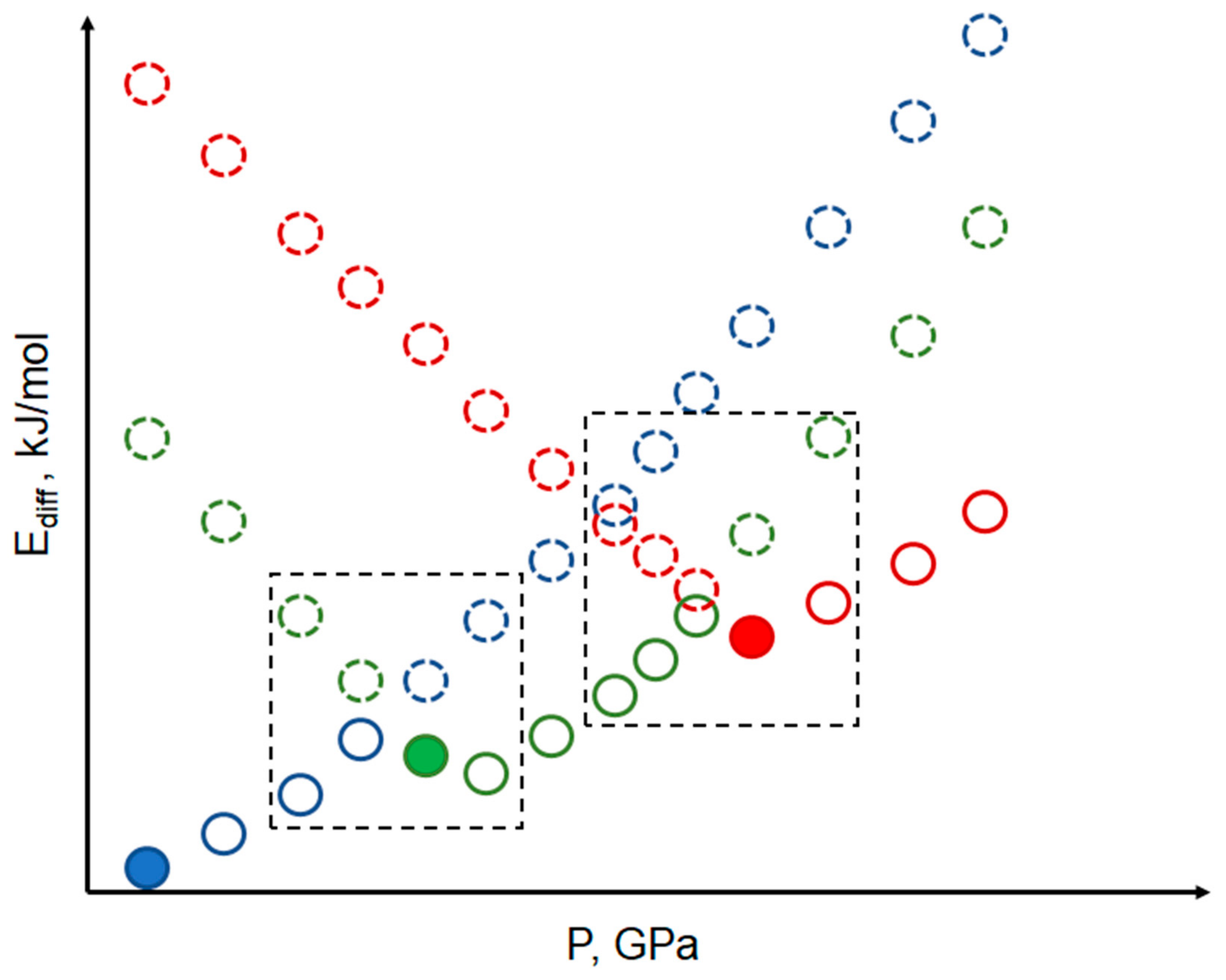
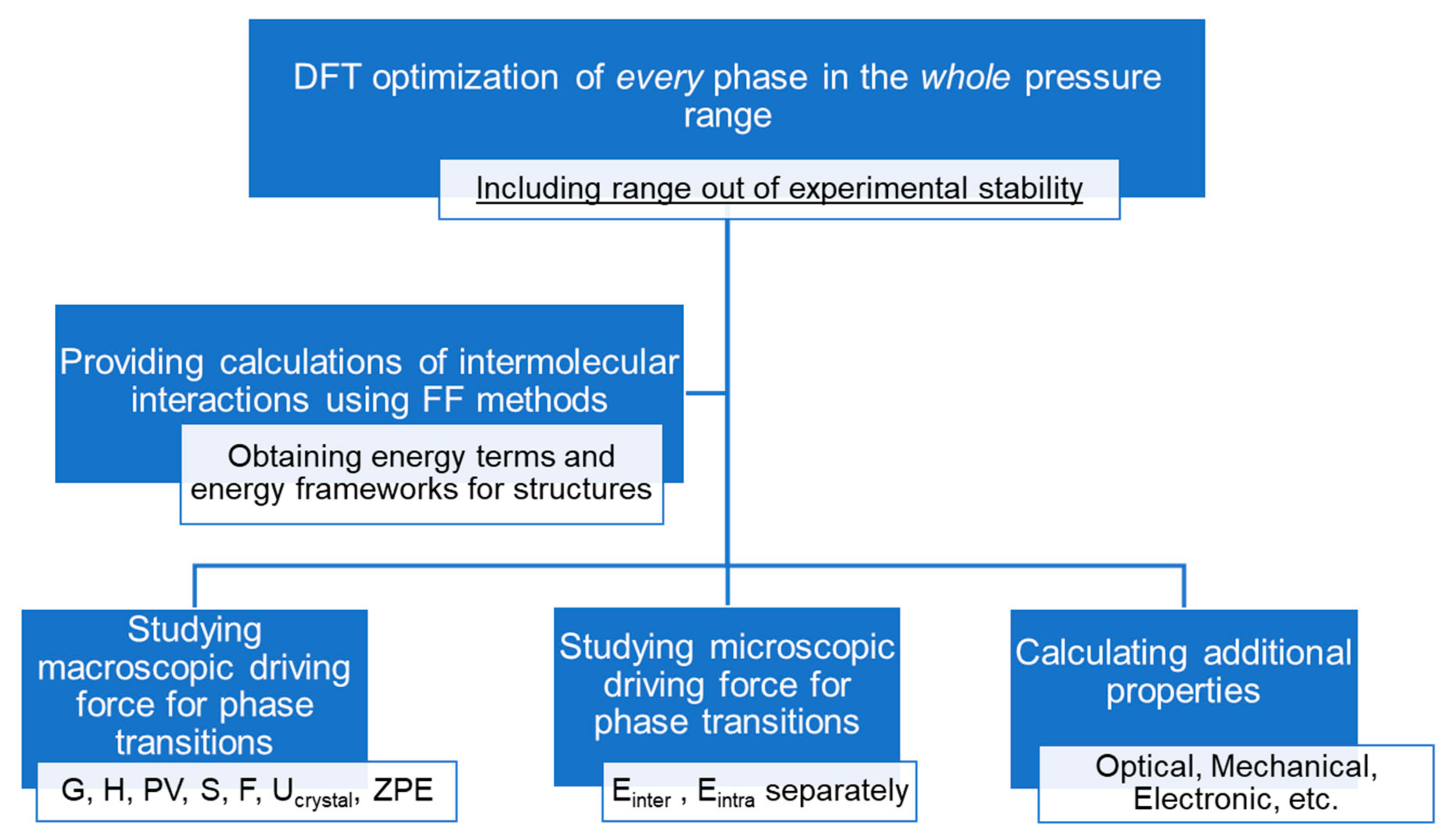
2. The Main Research Directions
- Experimental techniques usually provided in diamond anvil cells (DAC) by X-Ray diffraction and Raman spectroscopy [1] but not limited to these methods [34,35]. Significant progress in engineering made these experiments possible and even routine in some sense, but still, very time consuming and complicated. These experiments give atomic coordinates and unit cell parameters of molecular crystal and rarely some information on atom/functional groups motion, which can be interpreted in terms of energetic characteristics.
- Computational methods, which can be divided into DFT and Force Field (FF) groups, provide direct energetic characteristics of studied materials.
- DFT methods have many implementations for periodic systems, most popular are VASP [41,42,43], QuantumEspresso [44,45], CPMD [46], CASTEP [47] and CRYSTAL [48] codes, but researcher’s choice is not limited to abovementioned software. Reasonable choice of level of theory (LOT), which combines many parameters, gives very accurate energies of investigated systems but requires much more resources and time in comparison to FF methods.
2.1. Force Field Methods
2.2. DFT Methods
2.3. Combined Techniques
3. Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- Katrusiak, A. High-pressure crystallography. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.-K.; Chen, B.; Chen, J.; Li, K.; Lin, J.-F.; Yang, W.; Zheng, H. Recent advances in high-pressure science and technology. Matter Radiat. Extrem. 2016, 1, 59–75. [Google Scholar] [CrossRef]
- Tse, J.S. A Chemical Perspective on High Pressure Crystal Structures and Properties. Natl. Sci. Rev. 2019, 1, 53. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Y.; Duffy, T.S.; Shen, G.; Dobrzhinetskaya, L.F. (Eds.) Advances in High-Pressure Technology Geophysical Application; Elsevier: Amsterdam, The Netherlands, 2005; ISBN 9780444519795. [Google Scholar]
- Gonzalez-Platas, J.; Rodriguez-Hernandez, P.; Muñoz, A.; Rodríguez-Mendoza, U.R.; Nénert, G.; Errandonea, D. A High-Pressure Investigation of the Synthetic Analogue of Chalcomenite, CuSeO3∙2H2O. Crystals 2019, 9, 643. [Google Scholar] [CrossRef]
- Oganov, A.R.; Price, G.D.; Scandolo, S. Ab initio theory of planetary materials. Zeitschrift für Krist. Cryst. Mater. 2005, 220, 531–548. [Google Scholar] [CrossRef]
- Angel, R.J. Equations of State. Rev. Mineral. Geochem. 2000, 41, 35–59. [Google Scholar] [CrossRef]
- Angel, R.J. High-Pressure Structural Phase Transitions. Rev. Mineral. Geochem. 2000, 39, 85–104. [Google Scholar] [CrossRef]
- Angel, R.J.; Alvaro, M.; Gonzalez-Platas, J. EosFit7c and a Fortran module (library) for equation of state calculations. Zeitschrift für Krist.Cryst. Mater. 2014, 229, 405–419. [Google Scholar] [CrossRef]
- Gonzalez-Platas, J.; Alvaro, M.; Nestola, F.; Angel, R. EosFit7-GUI: A new graphical user interface for equation of state calculations, analyses and teaching. J. Appl. Crystallogr. 2016, 49, 1377–1382. [Google Scholar] [CrossRef]
- Bull, C.L.; Playford, H.Y.; Knight, K.S.; Marshall, W.G.; Stenning, G.B.G.; Smith, R.I.; Hart, Z. New insights into the phase diagram of a magnetic perovskite, LaCo 1/3 Mn 2/3 O 3. J. Phys. Condens. Matter 2015, 27, 165401. [Google Scholar] [CrossRef]
- Errandonea, D.; Muñoz, A.; Gonzalez-Platas, J. Comment on “High-pressure x-ray diffraction study of YBO 3 /Eu 3+, GdBO 3, and EuBO 3: Pressure-induced amorphization in GdBO 3”. J. Appl. Phys. 2014, 115, 216101. [Google Scholar] [CrossRef]
- Hunter, S.; Sutinen, T.; Parker, S.F.; Morrison, C.A.; Williamson, D.M.; Thompson, S.; Gould, P.J.; Pulham, C.R. Experimental and DFT-D Studies of the Molecular Organic Energetic Material RDX. J. Phys. Chem. C 2013, 117, 8062–8071. [Google Scholar] [CrossRef]
- Sharma, S.M.; Garg, N. Material Studies at High Pressure. In Materials Under Extreme Conditions; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–47. ISBN 9780128014424. [Google Scholar]
- Millar, D.I.A. Energetic Materials at Extreme Conditions; Springer Berlin Heidelberg: Berlin, Heidelberg, 2012; ISBN 978-3-642-23131-5. [Google Scholar]
- Dubrovinsky, L.; Dubrovinskaia, N.; Bykova, E.; Bykov, M.; Prakapenka, V.; Prescher, C.; Glazyrin, K.; Liermann, H.-P.; Hanfland, M.; Ekholm, M.; et al. The most incompressible metal osmium at static pressures above 750 gigapascals. Nature 2015, 525, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Oganov, A.R.; Ma, Y.; Wang, H.; Li, P.; Li, Y.; Iitaka, T.; Zou, G. Dissociation of methane under high pressure. J. Chem. Phys. 2010, 133, 144508. [Google Scholar] [CrossRef]
- Zhang, W.; Oganov, A.R.; Goncharov, A.F.; Zhu, Q.; Boulfelfel, S.E.; Lyakhov, A.O.; Stavrou, E.; Somayazulu, M.; Prakapenka, V.B.; Konopkova, Z. Unexpected Stable Stoichiometries of Sodium Chlorides. Science 2013, 342, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Zentkova, M.; Mihalik, M. The Effect of Pressure on Magnetic Properties of Prussian Blue Analogues. Crystals 2019, 9, 112. [Google Scholar] [CrossRef]
- Manjón, F.J.; Sans, J.A.; Ibáñez, J.; Pereira, A.L.J. Pressure-Induced Phase Transitions in Sesquioxides. Crystals 2019, 9, 630. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Wang, Y.; Li, K.; Zheng, H. From Molecules to Carbon Materials—High Pressure Induced Polymerization and Bonding Mechanisms of Unsaturated Compounds. Crystals 2019, 9, 490. [Google Scholar] [CrossRef]
- Cazorla, C.; MacLeod, S.G.; Errandonea, D.; Munro, K.A.; McMahon, M.I.; Popescu, C. Thallium under extreme compression. J. Phys. Condens. Matter 2016, 28, 445401. [Google Scholar] [CrossRef]
- Barrio, M.; Maccaroni, E.; Rietveld, I.B.; Malpezzi, L.; Masciocchi, N.; Céolin, R.; Tamarit, J.-L. Pressure-temperature state diagram for the phase relationships between benfluorex hydrochloride forms I and II: A case of enantiotropic behavior. J. Pharm. Sci. 2012, 101, 1073–1078. [Google Scholar] [CrossRef]
- Neumann, M.A.; van de Streek, J.; Fabbiani, F.P.A.; Hidber, P.; Grassmann, O. Combined crystal structure prediction and high-pressure crystallization in rational pharmaceutical polymorph screening. Nat. Commun. 2015, 6, 7793. [Google Scholar] [CrossRef]
- Fabbiani, F.P.A.; Pulham, C.R. High-pressure studies of pharmaceutical compounds and energetic materials. Chem. Soc. Rev. 2006, 35, 932. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J. Polymorphism—A Perspective. Cryst. Growth Des. 2011, 11, 632–650. [Google Scholar] [CrossRef]
- Bučar, D.-K.; Lancaster, R.W.; Bernstein, J. Disappearing Polymorphs Revisited. Angew. Chemie Int. Ed. 2015, 54, 6972–6993. [Google Scholar] [CrossRef]
- Konieczny, K.; Ciesielski, A.; Bąkowicz, J.; Galica, T.; Turowska-Tyrk, I. Structural Transformations in Crystals Induced by Radiation and Pressure. Part 7. Molecular and Crystal Geometries as Factors Deciding about Photochemical Reactivity under Ambient and High Pressures. Crystals 2018, 8, 299. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: New York, NY, USA, 2002; Volume 14, ISBN 978-0-19-923656-5. [Google Scholar]
- Boldyreva, E.V.; Shakhtshneider, T.P.; Ahsbahs, H.; Sowa, H.; Uchtmann, H. Effect of high pressure on the polymorphs of paracetamol. J. Therm. Anal. Calori. 2002, 68, 437–452. [Google Scholar]
- Seryotkin, Y.V.; Drebushchak, T.N.; Boldyreva, E.V. A high-pressure polymorph of chlorpropamide formed on hydrostatic compression of the α-form in saturated ethanol solution. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 77–85. [Google Scholar] [CrossRef]
- Görbitz, C.H. Crystal structures of amino acids: from bond lengths in glycine to metal complexes and high-pressure polymorphs. Crystallogr. Rev. 2015, 21, 160–212. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Anzellini, S.; Errandonea, D.; Cazorla, C.; MacLeod, S.; Monteseguro, V.; Boccato, S.; Bandiello, E.; Anichtchenko, D.D.; Popescu, C.; Beavers, C.M. Thermal equation of state of ruthenium characterized by resistively heated diamond anvil cell. Sci. Rep. 2019, 9, 14459. [Google Scholar] [CrossRef]
- Knudson, M.D.; Desjarlais, M.P.; Dolan, D.H. Shock-Wave Exploration of the High-Pressure Phases of Carbon. Science 2008, 322, 1822–1825. [Google Scholar] [CrossRef] [PubMed]
- Gavezzotti, A. Efficient computer modeling of organic materials. The atom–atom, Coulomb–London–Pauli (AA-CLP) model for intermolecular electrostatic-polarization, dispersion and repulsion energies. New J. Chem. 2011, 35, 1360. [Google Scholar] [CrossRef]
- Gavezzotti, A. Calculation of lattice energies of organic crystals: the PIXEL integration method in comparison with more traditional methods. Zeitschrift für Krist. Cryst. Mater. 2005, 220, 499–510. [Google Scholar] [CrossRef]
- Edwards, A.J.; Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Intermolecular interactions in molecular crystals: What’s in a name? Faraday Discuss. 2017, 203, 93–112. [Google Scholar] [CrossRef]
- Thomas, S.P.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Accurate Lattice Energies for Molecular Crystals from Experimental Crystal Structures. J. Chem. Theory Comput. 2018, 14, 1614–1623. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- CPMD.org. Available online: http://www.cpmd.org/ (accessed on 7 October 2015).
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Zeitschrift für Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Macchi, P.; Casati, N.; Kleppe, A.; Jephcoat, A. Electron density of molecular crystals at high pressure from synchrotron data. Acta Crystallogr. Sect. A Found. Adv. 2014, 70, C1340. [Google Scholar] [CrossRef]
- Casati, N.; Kleppe, A.; Jephcoat, A.P.; Macchi, P. Putting pressure on aromaticity along with in situ experimental electron density of a molecular crystal. Nat. Commun. 2016, 7, 10901. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A. How Reliable Are Intermolecular Interaction Energies Estimated from Topological Analysis of Experimental Electron Densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Kumar, P.; Cabaj, M.K.; Dominiak, P.M. Intermolecular Interactions in Ionic Crystals of Nucleobase Chlorides—Combining Topological Analysis of Electron Densities with Energies of Electrostatic Interactions. Crystals 2019, 9, 668. [Google Scholar] [CrossRef]
- Mahesta, R.; Mochizuki, K. Stepwise Homogeneous Melting of Benzene Phase I at High Pressure. Crystals 2019, 9, 279. [Google Scholar] [CrossRef]
- Nemkevich, A.; Bürgi, H.-B.; Spackman, M.A.; Corry, B. Molecular dynamics simulations of structure and dynamics of organic molecular crystals. Phys. Chem. Chem. Phys. 2010, 12, 14916. [Google Scholar] [CrossRef]
- Iyer, S.; Slagg, N. Energetic Materials; Boddu, V., Redner, P., Eds.; CRC Press: Boca Raton, FL, USA, 2010; Volume 2, ISBN 978-1-4398-3513-5. [Google Scholar]
- Musil, F.; De, S.; Yang, J.; Campbell, J.E.; Day, G.M.; Ceriotti, M. Machine learning for the structure-energy-property landscapes of molecular crystals. Chem. Sci. 2018, 9, 1289–1300. [Google Scholar] [CrossRef]
- Wicker, J.G.P.; Cooper, R.I. Will it crystallise? Predicting crystallinity of molecular materials. CrystEngComm 2015, 17, 1927–1934. [Google Scholar] [CrossRef]
- Wicker, J.G.P.; Crowley, L.M.; Robshaw, O.; Little, E.J.; Stokes, S.P.; Cooper, R.I.; Lawrence, S.E. Will they co-crystallize? CrystEngComm 2017, 19, 5336–5340. [Google Scholar] [CrossRef]
- Podryabinkin, E.V.; Tikhonov, E.V.; Shapeev, A.V.; Oganov, A.R. Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning. Phys. Rev. B 2019, 99, 064114. [Google Scholar] [CrossRef]
- Evans, J.D.; Coudert, F.-X. Predicting the Mechanical Properties of Zeolite Frameworks by Machine Learning. Chem. Mater. 2017, 29, 7833–7839. [Google Scholar] [CrossRef]
- Rychkov, D.A.; Stare, J.; Boldyreva, E.V. Pressure-driven phase transition mechanisms revealed by quantum chemistry: l -serine polymorphs. Phys. Chem. Chem. Phys. 2017, 19, 6671–6676. [Google Scholar] [CrossRef]
- Kolesnik, E.N.; Goryainov, S.V.; Boldyreva, E.V. Different behavior of L- and DL-serine crystals at high pressures: Phase transitions in L-serine and stability of the DL-serine structure. Dokl. Phys. Chem. 2005, 404, 169–172. [Google Scholar] [CrossRef]
- Moggach, S.A.; Allan, D.R.; Morrison, C.A.; Parsons, S.; Sawyer, L. Effect of pressure on the crystal structure of L-serine-I and the crystal structure of L-serine-II at 5.4 GPa. Acta Crystallogr. Sect. B Struct. Sci. 2005, 61, 58–68. [Google Scholar] [CrossRef]
- Drebushchak, T.N.; Sowa, H.; Seryotkin, Y.V.; Boldyreva, E.V.; Ahsbahs, H. L-Serine III at 8.0 GPa. Acta Crystallogr. Sect. E Struct. Rep. Online 2006, 62, 4052–4054. [Google Scholar] [CrossRef]
- Boldyreva, E.V.; Sowa, H.; Seryotkin, Y.V.; Drebushchak, T.N.; Ahsbahs, H.; Chernyshev, V.; Dmitriev, V. Pressure-induced phase transitions in crystalline l-serine studied by single-crystal and high-resolution powder X-ray diffraction. Chem. Phys. Lett. 2006, 429, 474–478. [Google Scholar] [CrossRef]
- Moggach, S.A.; Marshall, W.G.; Parsons, S. High-pressure neutron diffraction study of L-serine-I and L-serine-II, and the structure of L-serine-III at 8.1 GPa. Acta Crystallogr. Sect. B Struct. Sci. 2006, 62, 815–825. [Google Scholar] [CrossRef]
- Wood, P.A.; Francis, D.; Marshall, W.G.; Moggach, S.A.; Parsons, S.; Pidcock, E.; Rohl, A.L. A study of the high-pressure polymorphs of L-serine using ab initio structures and PIXEL calculations. CrystEngComm 2008, 10, 1154. [Google Scholar] [CrossRef]
- Antila, H.S.; Salonen, E. Biomolecular Simulations; Monticelli, L., Salonen, E., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 924, ISBN 978-1-62703-016-8. [Google Scholar]
- Kukol, A. (Ed.) Molecular Modeling of Proteins; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2015; Volume 1215, ISBN 978-1-4939-1464-7. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; Wiley: Hoboken, NJ, USA, 2007; Volume 2, ISBN 9780470058046. [Google Scholar]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]
- Rychkov, D.; Arkhipov, S.; Boldyreva, E. Structure-forming units of amino acid maleates. Case study of L-valinium hydrogen maleate. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 160–163. [Google Scholar] [CrossRef]
- Binns, J.; Parsons, S.; McIntyre, G.J. Accurate hydrogen parameters for the amino acid l -leucine. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 885–892. [Google Scholar] [CrossRef]
- Moggach, S.A.; Marshall, W.G.; Rogers, D.M.; Parsons, S. How focussing on hydrogen bonding interactions in amino acids can miss the bigger picture: A high-pressure neutron powder diffraction study of ε-glycine. CrystEngComm 2015, 17, 5315–5328. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B. Gaussian 09. Expanding the limits of computational chemistry. 2009. Available online: https://gaussian.com/g09citation/ (accessed on 30 January 2020).
- Johnstone, R.D.L.; Lennie, A.R.; Parker, S.F.; Parsons, S.; Pidcock, E.; Richardson, P.R.; Warren, J.E.; Wood, P.A. High-pressure polymorphism in salicylamide. CrystEngComm 2010, 12, 1065. [Google Scholar] [CrossRef]
- Munday, L.B.; Chung, P.W.; Rice, B.M.; Solares, S.D. Simulations of High-Pressure Phases in RDX. J. Phys. Chem. B 2011, 115, 4378–4386. [Google Scholar] [CrossRef]
- Dolgonos, G.A.; Hoja, J.; Boese, A.D. Revised values for the X23 benchmark set of molecular crystals. Phys. Chem. Chem. Phys. 2019, 21, 24333–24344. [Google Scholar] [CrossRef]
- Colmenero, F. Mechanical properties of anhydrous oxalic acid and oxalic acid dihydrate. Phys. Chem. Chem. Phys. 2019, 21, 2673–2690. [Google Scholar] [CrossRef]
- Giordano, N.; Beavers, C.M.; Kamenev, K.V.; Marshall, W.G.; Moggach, S.A.; Patterson, S.D.; Teat, S.J.; Warren, J.E.; Wood, P.A.; Parsons, S. High-pressure polymorphism in l-threonine between ambient pressure and 22 GPa. CrystEngComm 2019, 21, 4444–4456. [Google Scholar] [CrossRef]
- Giordano, N.; Afanasjevs, S.; Beavers, C.M.; Hobday, C.L.; Kamenev, K.V.; O’Bannon, E.F.; Ruiz-Fuertes, J.; Teat, S.J.; Valiente, R.; Parsons, S. The Effect of Pressure on Halogen Bonding in 4-Iodobenzonitrile. Molecules 2019, 24, 2018. [Google Scholar] [CrossRef]
- Abraham, B.M.; Vaitheeswaran, G. First principles study of pressure induced polymorphic phase transition in trimethylamine. In Proceedings of the AIP Conference Proceedings. AIP Conf. Proc. 2018, 1942, 030006. [Google Scholar]
- Wen, X.D.; Hoffmann, R.; Ashcroft, N.W. Benzene under high pressure: A story of molecular crystals transforming to saturated networks, with a possible intermediate metallic phase. J. Am. Chem. Soc. 2011, 133, 9023–9035. [Google Scholar] [CrossRef]
- Zucchini, A.; Prencipe, M.; Belmonte, D.; Comodi, P. Ab initio study of the dolomite to dolomite-II high-pressure phase transition. Eur. J. Mineral. 2017, 29, 227–238. [Google Scholar] [CrossRef]
- Belmonte, D.; Ottonello, G.; Zuccolini, M.V. Ab initio-assisted assessment of the CaO-SiO2 system under pressure. Calphad 2017, 59, 12–30. [Google Scholar] [CrossRef]
- Belmonte, D.; Gatti, C.; Ottonello, G.; Richet, P.; Zuccolini, M.V. Ab initio thermodynamic and thermophysical properties of sodium metasilicate, Na2SiO3, and their electron-density and electron-pair-density counterparts. J. Phys. Chem. A 2016, 120, 8881–8895. [Google Scholar] [CrossRef] [PubMed]
- Wdowik, U.D. Structural stability and thermal properties of BeO from the quasiharmonic approximation. J. Phys. Condens. Matter 2010, 22, 045404. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, D. First Principles Thermodynamics of Minerals at HP–HT Conditions: MgO as a Prototypical Material. Minerals 2017, 7, 183. [Google Scholar] [CrossRef]
- Moses Abraham, B.; Adivaiah, B.; Vaitheeswaran, G. Microscopic origin of pressure-induced phase-transitions in urea: A detailed investigation through first principles calculations. Phys. Chem. Chem. Phys. 2019, 21, 884–900. [Google Scholar] [CrossRef]
- Katrusiak, A.; McMillan, P.F. High-Pressure Crystallography; Katrusiak, A., McMillan, P., Eds.; Springer: Dordrecht, The Netherlands, 2004. [Google Scholar]
- Fedorov, A.Y.; Rychkov, D.A.; Losev, E.A.; Zakharov, B.A.; Stare, J.; Boldyreva, E.V. Effect of pressure on two polymorphs of tolazamide: Why no interconversion? CrystEngComm 2017, 19, 2243–2252. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.G.; Grimme, S. Dispersion Corrected Hartree–Fock and Density Functional Theory for Organic Crystal Structure Prediction. In Peptide-Based Materials; Springer: Dordrecht, The Netherlands, 2013; Volume 310, pp. 1–23. [Google Scholar]
- Klimeš, J.; Michaelides, A. Perspective: Advances and challenges in treating van der Waals dispersion forces in density functional theory. J. Chem. Phys. 2012, 137, 120901. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Kronik, L.; Tkatchenko, A. Understanding Molecular Crystals with Dispersion-Inclusive Density Functional Theory: Pairwise Corrections and Beyond. Acc. Chem. Res. 2014, 47, 3208–3216. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.G.; Grimme, S. Accurate Modeling of Organic Molecular Crystals by Dispersion-Corrected Density Functional Tight Binding (DFTB). J. Phys. Chem. Lett. 2014, 5, 1785–1789. [Google Scholar] [CrossRef]
- Marom, N.; DiStasio, R.A.; Atalla, V.; Levchenko, S.; Reilly, A.M.; Chelikowsky, J.R.; Leiserowitz, L.; Tkatchenko, A. Many-Body Dispersion Interactions in Molecular Crystal Polymorphism. Angew. Chemie Int. Ed. 2013, 52, 6629–6632. [Google Scholar] [CrossRef]
- Jahn, S. High-pressure phase transitions in MgSiO3 orthoenstatite studied by atomistic computer simulation. Am. Mineral. 2008, 93, 528–532. [Google Scholar] [CrossRef][Green Version]
- Giordano, N.; Beavers, C.M.; Campbell, B.J.; Eigner, V.; Gregoryanz, E.; Marshall, W.G.; Peña-Álvarez, M.; Teat, S.J.; Vennari, C.E.; Parsons, S. High-pressure polymorphism in pyridine. IUCrJ 2020, 7, 58–70. [Google Scholar] [CrossRef]
- Boldyreva, E. High-Pressure Polymorphs of Molecular Solids: When Are They Formed, and When Are They Not? Some Examples of the Role of Kinetic Control. Cryst. Growth Des. 2007, 7, 1662–1668. [Google Scholar]
- Fisch, M.; Lanza, A.; Boldyreva, E.; Macchi, P.; Casati, N. Kinetic Control of High-Pressure Solid-State Phase Transitions: A Case Study on l-Serine. J. Phys. Chem. C 2015, 119, 18611–18617. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Singh, S. Pressure induced structural phase transitions—A review. Open Chem. 2012, 10, 1391–1422. [Google Scholar] [CrossRef]
- Han, Y.; Liu, J.; Huang, L.; He, X.; Li, J. Predicting the phase diagram of solid carbon dioxide at high pressure from first principles. npj Quantum Mater. 2019, 4, 10. [Google Scholar] [CrossRef]
- Smith, J.C.; Sagredo, F.; Burke, K. Warming Up Density Functional Theory. In Frontiers of Quantum Chemistry; Springer: Singapore, 2018. [Google Scholar]
- Majewski, A.R.; Billman, C.R.; Cheng, H.-P.; Sullivan, N.S. DFT Calculations of Temperature-Dependent NQR Parameters in alpha-paradichlorobenzene and beta-HMX. arXiv 2019, arXiv:1903.10097. [Google Scholar]
- Hickel, T.; Grabowski, B.; Körmann, F.; Neugebauer, J. Advancing density functional theory to finite temperatures: Methods and applications in steel design. J. Phys. Condens. Matter 2012, 24, 053202. [Google Scholar] [CrossRef]
- Reilly, A.M.; Cooper, R.I.; Adjiman, C.S.; Bhattacharya, S.; Boese, A.D.; Brandenburg, J.G.; Bygrave, P.J.; Bylsma, R.; Campbell, J.E.; Car, R.; et al. Report on the sixth blind test of organic crystal structure prediction methods. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 439–459. [Google Scholar]
- Iuzzolino, L.; McCabe, P.; Price, S.L.; Brandenburg, J.G. Crystal structure prediction of flexible pharmaceutical-like molecules: Density functional tight-binding as an intermediate optimisation method and for free energy estimation. Faraday Discuss. 2018, 211, 275–296. [Google Scholar] [CrossRef]
- Price, S.L.; Reutzel-Edens, S.M. The potential of computed crystal energy landscapes to aid solid-form development. Drug Discov. Today 2016, 21, 912–923. [Google Scholar]
- Zurek, E.; Grochala, W. Predicting crystal structures and properties of matter under extreme conditions via quantum mechanics: The pressure is on. Phys. Chem. Chem. Phys. 2015, 17, 2917–2934. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Method | High-Pressure Experimental Data is Mandatory | Time and Resource-Consuming | Energy Estimation Accuracy | “Chemical” Energy Terms | Intermolecular Energies (Einter) | Intramolecular Energies (Eintra) |
|---|---|---|---|---|---|---|
| FF | yes | no | lower | yes | yes | no2 |
| DFT | yes/no1 | yes | higher | no | yes | yes |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rychkov, D.A. A Short Review of Current Computational Concepts for High-Pressure Phase Transition Studies in Molecular Crystals. Crystals 2020, 10, 81. https://doi.org/10.3390/cryst10020081
Rychkov DA. A Short Review of Current Computational Concepts for High-Pressure Phase Transition Studies in Molecular Crystals. Crystals. 2020; 10(2):81. https://doi.org/10.3390/cryst10020081
Chicago/Turabian StyleRychkov, Denis A. 2020. "A Short Review of Current Computational Concepts for High-Pressure Phase Transition Studies in Molecular Crystals" Crystals 10, no. 2: 81. https://doi.org/10.3390/cryst10020081
APA StyleRychkov, D. A. (2020). A Short Review of Current Computational Concepts for High-Pressure Phase Transition Studies in Molecular Crystals. Crystals, 10(2), 81. https://doi.org/10.3390/cryst10020081