Methods for Obtaining Better Diffractive Protein Crystals: From Sample Evaluation to Space Crystallization



Abstract
1. Introduction
2. Sample Quality
2.1. Evaluation Methods
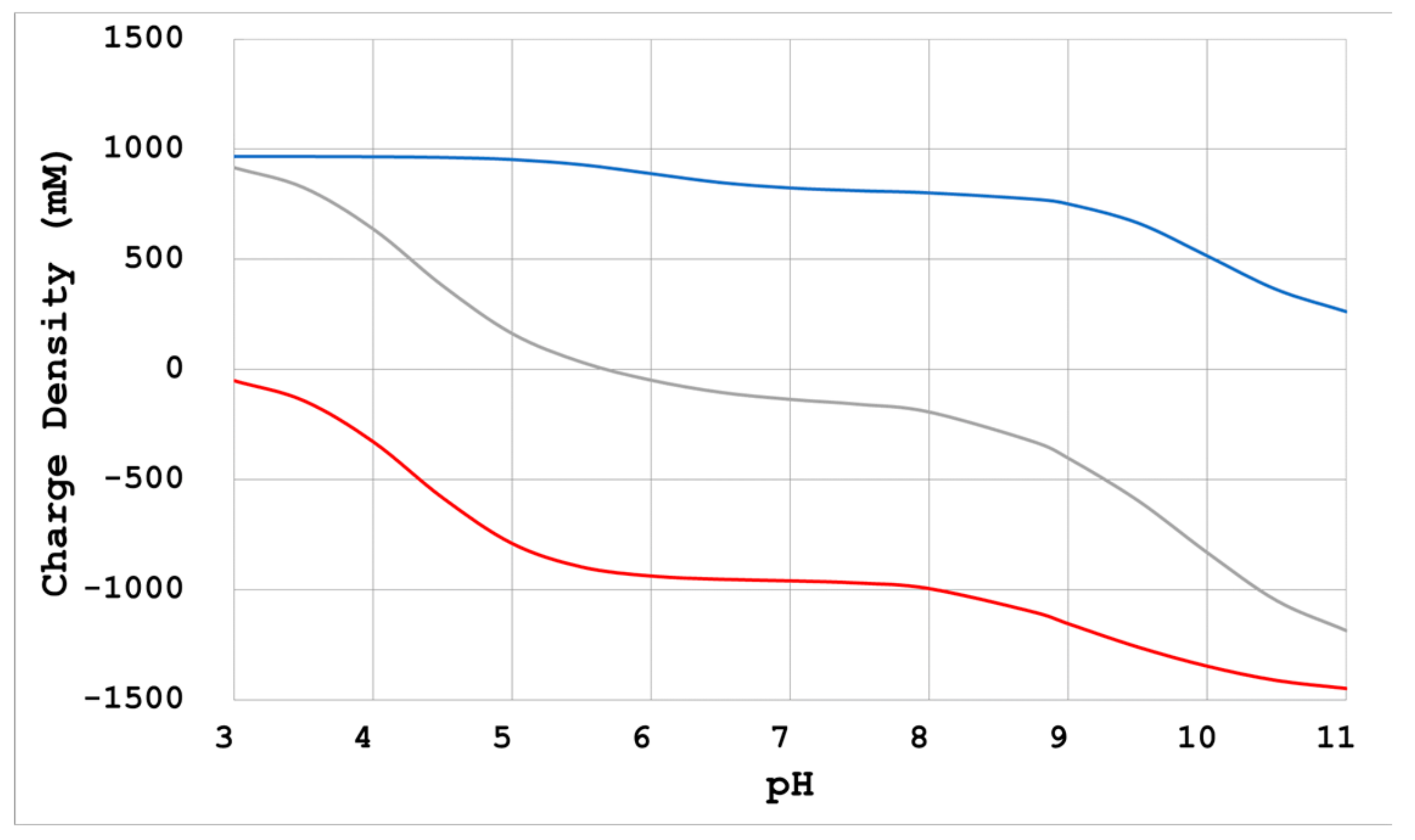
- Physicochemical calculation
- SDS-PAGE
- Native-PAGE
- Two-dimensional electrophoresis
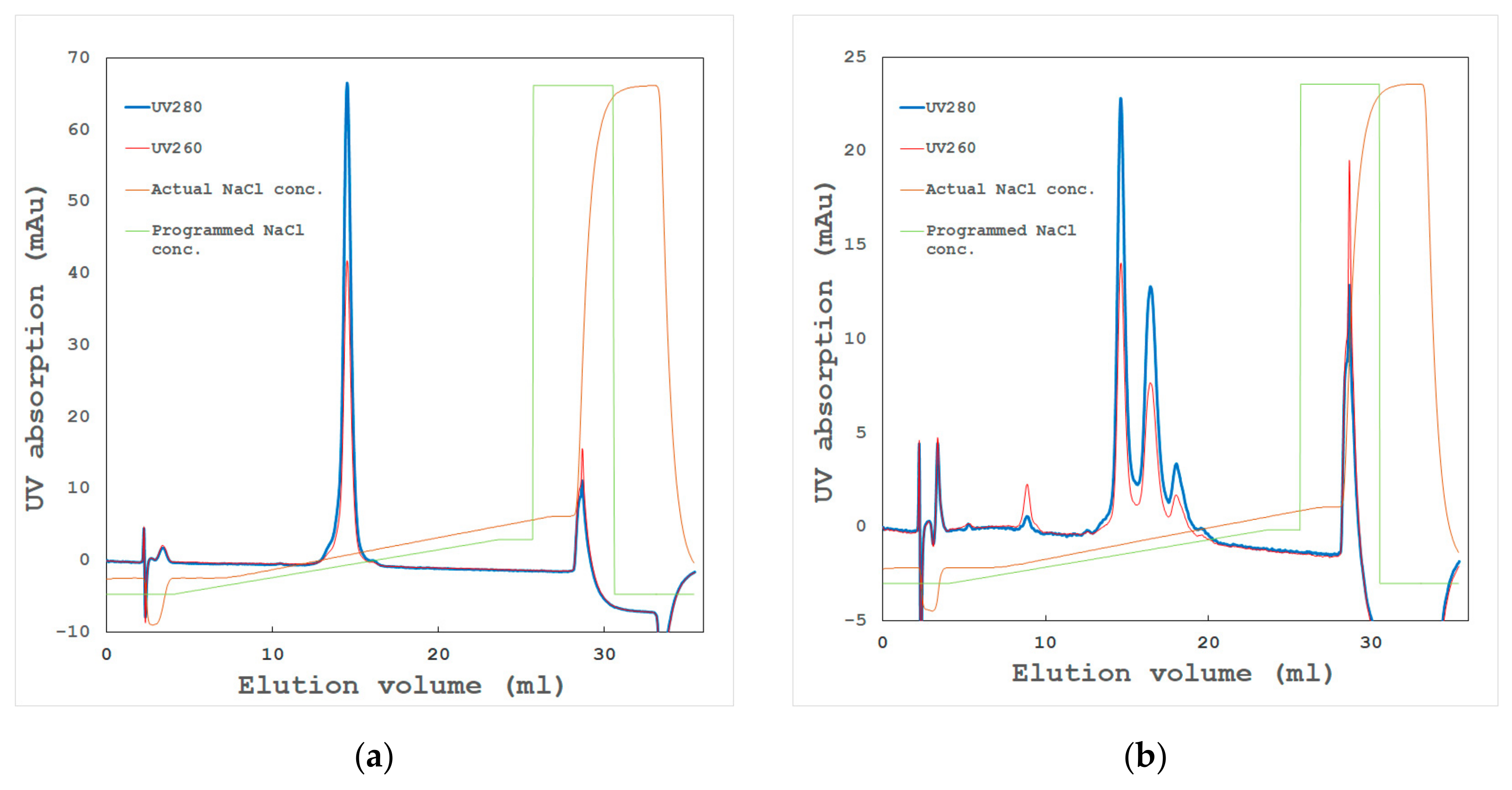
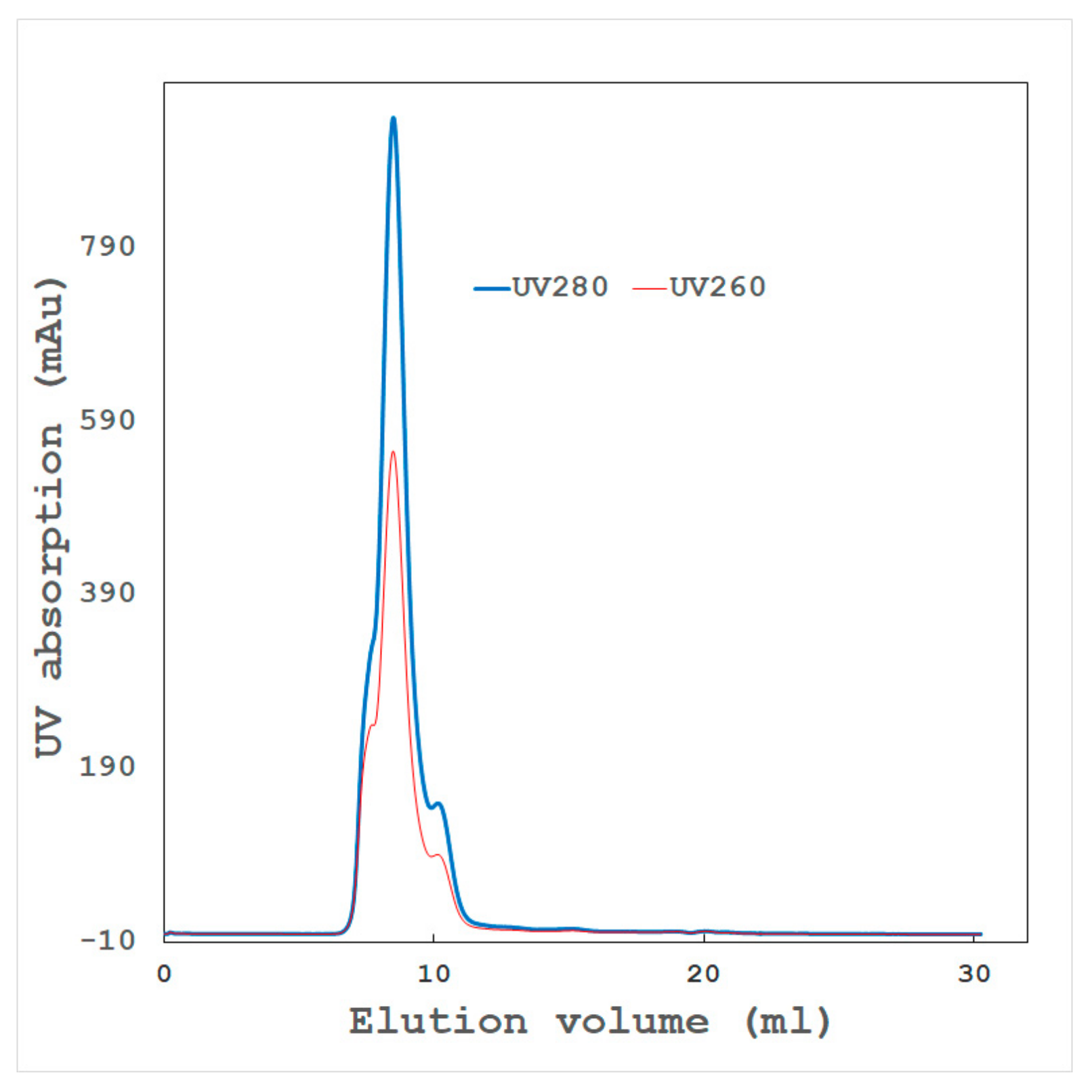
- High-resolution ion exchange chromatography
- High-resolution gel filtration chromatography
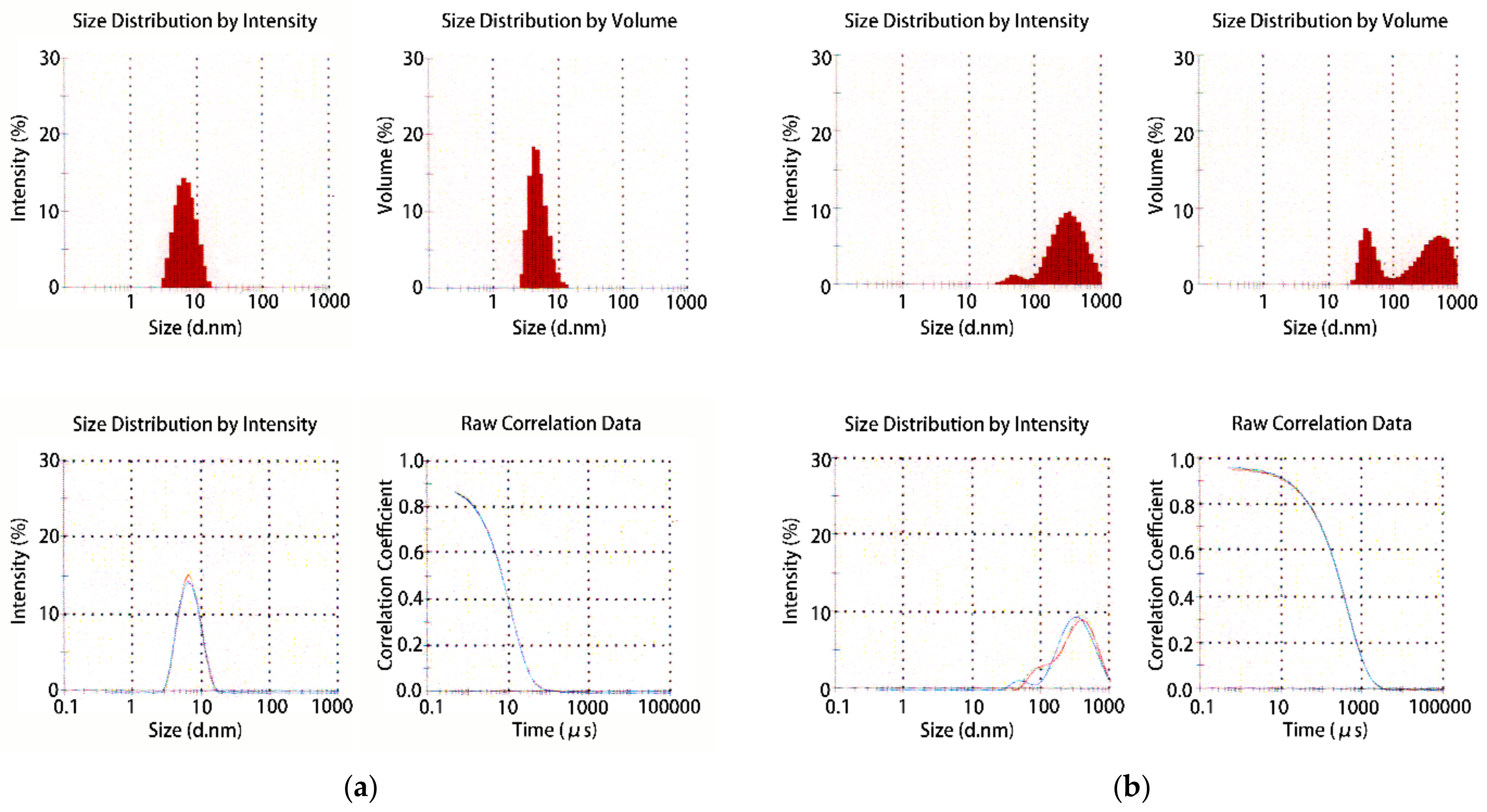
- Dynamic light scattering
2.2. Measures to Improve Protein Samples
2.2.1. Uniformity Improvement
- Other proteins, lipids, etc., which have not been removed by other purification steps;
- Proteins whose N or C terminal ends have not been processed correctly;
- Proteins including irregularly modified residues.
2.2.2. Aggregate Removal
- Denatured proteins caused by the concentration process in crude state or ammonium sulfate precipitation. Avoiding these processes might improve the sample, and
- Aggregated proteins associated with contaminants or isozymes with different isoelectric points (pIs) through hydrophobic or hydrophilic interactions. Dialysis, gel filtration, or high-resolution chromatography sometimes remove such aggregates, which might dramatically improve the sample quality.
2.2.3. Prevent Deterioration over Time
- For unstable proteins, the construction of mutants is advised to improve their stability [19];
- In the case of protease degradation, adding protease inhibitors, followed by removing proteases in the subsequent chromatography step, is usually effective;
- In the case of damage caused by oxidation, purifying, storing, and crystallizing the samples under a deoxygenated state is advised.
3. Crystallization
3.1. Crystallization Reagent
- Salts—a combination of mono- or multi-valent anions and cations, for example, ammonium sulfate, sodium malonate, etc. The tendency for lowering solubility is listed in the Hofmeister Series. In general, the protein solubility increases with the addition of salt, until a certain point (salting-in), after which it decreases (salting-out). Anions and cations have various tendencies for salting-in and -out effects [21]. Therefore, determining a proper salt and its concentration for crystallization is accompanied by some difficulties;
- Polymers—high-molecular-weight polymers, for example, polyethylene glycol (PEG). The mechanism for reducing solubility is explained as an excluding-volume effect [1]. In general, the preferable molecular weight of PEG is related to the target protein [22], although a molecular weight of 400 to 20,000 is frequently used. Lower-molecular-weight PEG, such as PEG 400, has the ability to denature proteins, which is similar to the effects of an alcohol. However, higher-molecular-weight PEG does not have significant side-effects other than reducing solubility. Therefore, high-molecular-weight PEG is easier for controlling the crystallization process and is frequently used in crystallization;
- Organic solvents and alcohols—for example, 2-methyl-2,4-pentanediol (MPD), isopropanol, etc. This mechanism is explained as reducing the dielectric constant of the solution [1]. Some hydrophobic proteins sometimes prefer organic solvents.
- In the case of PEG, as the concentration increases, the number of grown crystals increases until a certain point, after which it decreases as the concentration of PEG further increases. It is thought that the nucleus formation probability decreases as the viscosity increases [25,26,27] (see 3.3.2). Furthermore, in a state where the nucleation formation probability is lowered, the degree of supersaturation is high, so secondary nucleation on the crystal surface is likely to occur and cluster crystals are likely to be formed;
- When there is no reagent that enhances intermolecular interactions, a reduction of electrostatic repulsion is necessary for crystallization. Neutralization by Na+ and Cl− as counterions of divergent groups (-COO−, -NH3+) of proteins is a method that can be used for such a purpose. In this case, adding sodium chloride at a concentration in relation to the electric charge density is advised [10,21];
- Ions such as Na+ and Cl- not only interact with dissociation groups on the protein surface, but also interact with other acids and bases and affect their effects. Therefore, when these ions coexist with a reagent that enhances intermolecular interactions, conversely, the effect is diminished.
3.2. Crystallization Method
3.2.1. Batch Method
3.2.2. Vapor-Diffusion Method
3.2.3. Counter-Diffusion Method
3.2.4. Dialysis Method
3.3. Measures for the Improvement of Crystallization
3.3.1. Reproducibility of Crystallization
3.3.2. Nucleation Rate
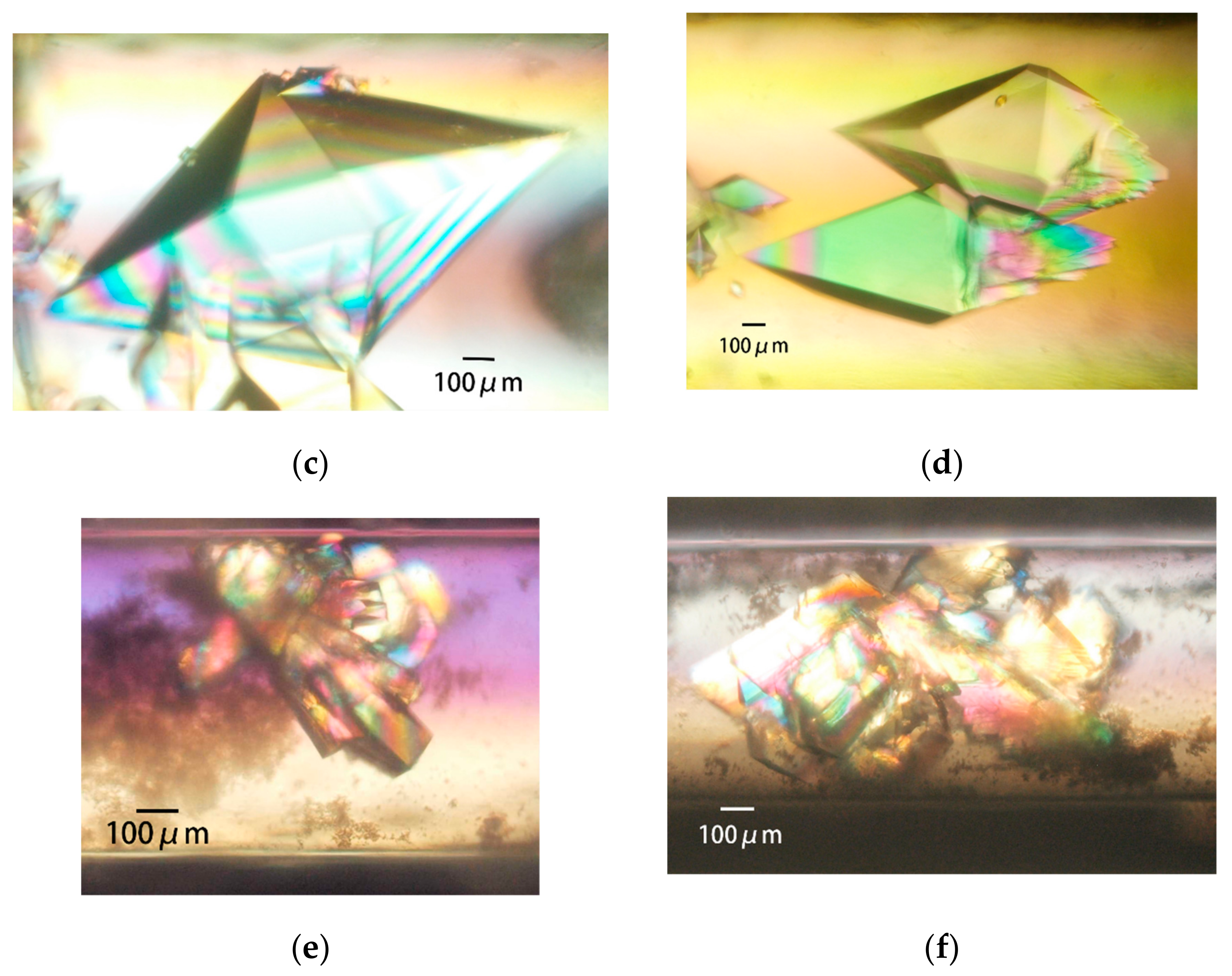
3.3.3. Obtaining Appropriately Shaped Crystals
3.3.4. Avoiding Crystal Clustering
3.3.5. Resolution and Molecular Packing
- Increasing the concentration of the main crystallization reagent: The interfacial tension increases, and a bulk force from the crystal’s surface is applied between the protein molecules isotropically, bringing them closer together. As a result, the interaction between the neighboring protein molecules increases, the disorder is reduced, and the resolution increases;
- Reducing the counter ions: The electrostatic repulsion force increases, which is the micro anisotropic force between the molecules. As a result, a protein’s molecular alignment is more sensitive to its surface charge distribution, and the molecules are aligned in a more uniform direction;
- Adding metal ions, organic acids, and organic bases: Some of these have an attractive function between protein molecules to align the molecules with the micro anisotropic forces between them;
- Changing the pH of the crystallization condition to the opposite side of the protein’s pI: This changes the polarity of the electrostatic repulsive force acting between the protein molecules and changes the micro anisotropic force acting between them, so the packing may change greatly.
3.4. Growing Large Crystals
4. Harvesting Crystals and Cryo-protection
4.1. Harvest Solution
4.2. Manipulation of Crystals Grown in a Capillary
4.3. Cryo-Protection
- Typical cryoprotectants are glycerol and PEG;
- It should be ensured that the drops of cryoprotectant solution can be flash-cooled into a glassy form in advance;
- In the case of a PEG type of a lower concentration in the harvest solution, it is preferable that PEG or glycerol is added to a total concentration of about 35% or more;
- In the case of salt as the main precipitant, it is preferable to add glycerol to about 35%;
- However, as the amount of glycerol to be added increases, the osmotic pressure difference becomes large, so one must be careful with this process. Gradually increasing the glycerol concentration may be advisable;
- In the case of salt in the harvest solution, when glycerol cannot be added, sucrose or trehalose is the next choice.
5. Microgravity Environment
5.1. Introduction
5.2. Numerical Model
5.3. Enhancing the Effects of Microgravity
5.4. Transient Growth Process
5.5. Other Phenomena
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chernov, A.A. Protein crystals and their growth. J. Struct. Biol. 2003, 142, 3–21. [Google Scholar] [CrossRef]
- McPherson, A.; Gavira, J.A. Introduction to protein crystallization. Acta Cryst. 2014, F70, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Giege, R. A historical perspective on protein crystallization from 1840 to the present day. FEBS J. 2013, 280, 6456–6497. [Google Scholar] [CrossRef] [PubMed]
- Candoni, N.; Grossier, R.; Hammadi, Z.; Morin, R.; Veesler, S. Practical Physics Behind Growing Crystals of Biological Macromolecules. Protein Peptide Lett. 2012, 19, 714–724. [Google Scholar]
- Matsuura, Y.; Chernov, A.A. Morphology and the strength of intermolecular contacts in protein crystals. Acta Cryst. 2003, D59, 1347–1356. [Google Scholar] [CrossRef]
- Thomas, B.R.; Carter, D.; Rosenberger, F. Effect of microheterogeneity on horse spleen apoferritin crystallization. J. Cryst. Growth 1998, 187, 499–510. [Google Scholar] [CrossRef]
- Thomas, B.R.; Chernov, A.A. Acetylated lysozyme as impurity in lysozyme crystals: constant distribution coefficient. J. Cryst. Growth 2001, 232, 237–243. [Google Scholar] [CrossRef]
- Vekilov, P.G. Elementary processes of protein crystal growth. Prog. Crystal Growth Charact. 1993, 26, 25–49. [Google Scholar] [CrossRef]
- Niedzialkowska, E.; Gasiorowska, O.; Handing, K.B.; Majorek, K.A.; Porebski, P.J.; Shabalin, I.G.; Zasadzinska, E.; Cymborowski, M.; Minor, W. Protein purification and crystallization artifacts: The tale usually not told. Protein Sci. 2016, 25, 720–733. [Google Scholar] [CrossRef]
- Yamanaka, M.; Inaka, K.; Furubayashi, N.; Matsushima, M.; Takahashi, S.; Tanaka, H.; Sano, S.; Sato, M.; Kobayashi, T.; Tanaka, T. Optimization of salt concentration in PEG-based crystallization solutions. J. Synchrotron Rad. 2010, 18, 84–87. [Google Scholar] [CrossRef]
- ExPASy Bioinformatics Resource Portal. Available online: https://www.expasy.org/ (accessed on 30 January 2020).
- Garfin, D.E. One-dimensional gel electrophoresis. Methods Enzymol. 2009, 463, 497–513. [Google Scholar] [PubMed]
- Van Der Laan, J.M.; Swarte, M.B.A.; Groendijk, H.; Hol, W.G.J.; Drenth, J. The influence of purification and protein heterogeneity on the crystallization of p-hydroxybenzoate hydroxylase. Eur. J. Biochem. 1989, 179, 715–724. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [PubMed]
- Jungbauer, A.; Hahn, R. Ion-exchange chromatography. Methods Enzymol. 2009, 463, 349–371. [Google Scholar]
- Cummins, P.M.; Rochfort, K.D.; O’Connor, B.F. Ion-exchange chromatography: Basic principles and application. In Protein Chromatography. Methods in Molecular Biology; Walls, D., Loughran, S., Eds.; Human Press: New York, NY, USA, 2017; p. 1485. [Google Scholar]
- Proteau, A.; Shi, R.; Cygler, M. Application of dynamic light scattering in protein crystallization. Curr. Protoc. Protein Sci. 2010, 61, 17.10.1–17.10.9. [Google Scholar] [CrossRef]
- Vorontsova, M.A.; Maes, M.; Vekilov, P.G. Recent advances in the understanding of two-step nucleation of protein crystals. Faraday Discuss. 2015, 179, 27–40. [Google Scholar] [CrossRef]
- Tyler-Cross, R.; Schirch, V. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J. Biol. Chem. 1991, 266, 22549–22556. [Google Scholar]
- Majeed, S.; Ofek, G.; Belachew, A.; Huang, C.; Zhou, T.; Kwong, P.D. Enhancing protein crystallization through precipitant synergy. Structure 2003, 11, 1061–1070. [Google Scholar] [CrossRef]
- Collins, K.D. Ions from the Hofmeister series and osmolytes: effects on proteins in solution and in the crystallization process. Methods 2004, 34, 300–311. [Google Scholar] [CrossRef]
- Tanaka, S.; Ataka, M. Protein crystallization induced by polyethylene glycol: A model study using apoferritin. J. Chem. Phys. 2002, 117, 3504–3510. [Google Scholar] [CrossRef]
- Newstead, S.; Ferrandon, S.; Iwata, S. Rationalizing alpha-helical membrane protein crystallization. Protein Sci. 2008, 17, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Stetsenko, A.; Guskov, A. An Overview of the Top Ten Detergents Used for Membrane Protein Crystallization. Crystals 2017, 7, 197. [Google Scholar] [CrossRef]
- García-Ruiz, J.M. Nucleation of protein crystals. J. Struct. Biol. 2003, 142, 22–31. [Google Scholar] [CrossRef]
- Yoshizaki, I.; Nakamura, H.; Fukuyama, S.; Komatsu, H.; Yoda, S. Estimation of crystallization boundary of a model protein as a function of solute concentration and experiment time. Int. J. Microgravity Sci. Appl. 2002, 19, 30–33. [Google Scholar]
- Takahashi, S.; Yan, B.; Furubayashi, N.; Inaka, K.; Tanaka, H. Effects of polyethylene glycol 4000 and Sodium chloride: 3-dimensional phase diagram for a protein crystallization. Mater. Struct. 2016, 23, 154. [Google Scholar]
- Jakoby, W.B. A technique for the crystallization of proteins. Anal. Biochem. 1968, 26, 295–298. [Google Scholar] [CrossRef]
- Nakamura, H.; Fukuyama, S.; Yoshizaki, I.; Yoda, S.-I. Theory of vapor diffusion in the high-density protein crystal growth device and its application to the measurement of the vapor pressures of aqueous solutions. J. Cryst. Growth 2003, 259, 149–159. [Google Scholar] [CrossRef]
- Benvenuti, M.; Mangani, S. Crystallization of soluble proteins in vapor diffusion for x-ray crystallography. Nat. Protoc. 2007, 2, 1633–1651. [Google Scholar] [CrossRef]
- Takahashi, S.; Koga, M.; Yan, B.; Furubayashi, N.; Kamo, M.; Inaka, K.; Tanaka, H. JCB-SGT crystallization devices applicable to PCG experiments and their crystallization conditions. Int. J. Microgravity Sci. Appl. 2019, 36, 360107. [Google Scholar]
- García-Ruiz, J.M.; González-Ramírez, L.A.; Gavira, J.A.; Otálora, F. Granada Crystallization Box: a new device for protein crystallization by counter-diffusion techniques. Acta Cryst. 2002, D58, 1638–1642. [Google Scholar]
- Otálora, F.; Gavira, J.A.; Ng, J.D.; García-Ruiz, J.M. Counterdiffusion methods applied to protein crystallization. Prog. Biophys. Mol. Biol. 2009, 101, 26–37. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, J.M. Counterdiffusion method for macromolecular crystallization. Methods Enzymol. 2003, 368, 130–154. [Google Scholar] [PubMed]
- Tanaka, H.; Inaka, K.; Sugiyama, S.; Takahashi, S.; Sano, S.; Sato, M.; Yoshitomi, S. A simplified counter diffusion method combined with a 1D simulation program for optimizing crystallization conditions. J. Synchrotron Rad. 2004, 11, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Yoshizaki, I.; Takahashi, S.; Yamanaka, M.; Fukuyama, S.; Sato, M.; Sano, S.; Motohara, M.; Kobayashi, T.; Yoshitomi, S.; et al. Diffusion coefficient of the protein in various crystallization solutions: The Key to growing high-quality crystals in space. Microgravity Sci. Technol. 2006, XVIII-3/4, 91–94. [Google Scholar] [CrossRef]
- McPherson, A. Practical procedures for macromolecular crystallization. In Crystallization of Biological Macromolecules; Bianco, C., Ed.; Cold Spring Harbor: New York, NY, USA, 1999. [Google Scholar]
- Maeda, M.; Chatake, T.; Tanaka, I.; Ostermann, A.; Niimura, N. Crystallization of a large single crystal of cubic insulin for neutron protein crystallography. J. Synchrotron Rad. 2004, 11, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.D.; Baird, J.K.; Coates, L.; García-Ruiz, J.M.; Hodge, T.A.; Huang, S. Large-volume protein crystal growth for neutron macromolecular crystallography. Acta Cryst. 2015, F71, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Ishida, T.; Fushinobu, S.; Kusaka, K.; Tanaka, I.; Inaka, K.; Higuchi, Y.; Masaki, M.; Ohta, K.; Kaneko, S.; et al. Phase-diagram-guided method for growth of a large crystal of glycoside hydrolase family 45 inverting cellulose suitable for neutron structural analysis. J. Synchrotron Rad. 2013, 20, 859–863. [Google Scholar] [CrossRef]
- Crystal growth apparatus for biological macromolecules. Japan Patent JP4161101B.
- Takahashi, S.; Tanaka, H. Background of protein crystallization technologies in space. Int. J. Microgravity Sci. Appl. 2017, 34, 340103. [Google Scholar]
- Garman, E.F.; Schneider, T.R. Macromolecular Cryocrystallography. J. Appl. Cryst. 1997, 30, 211–237. [Google Scholar] [CrossRef]
- Day, J.; McPherson, A. Macromolecular crystal growth experiments on International Microgravity Laboratory-1. Protein Sci. 1992, 1, 1254–1268. [Google Scholar] [CrossRef]
- Ng, J.D.; Sauter, C.; Lorber, B.; Kirkland, N.; Arnez, J.; Giege, R. Comparative analysis of space-grown and earth-grown crystals of an aminoacyl-tRNA synthetase: space-grown crystals are more useful for structural determination. Acta Cryst. 2002, D58, 645–652. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; DeLucas, L.J. Microgravity protein crystallization. npj Microgravity 2015, 1, 15010. [Google Scholar] [CrossRef] [PubMed]
- Snell, E.H.; Helliwell, J.R. Macromolecular crystallization in microgravity. Rep. Prog. Phys. 2005, 68, 799–853. [Google Scholar] [CrossRef]
- Mares, D.; Evrard, C.; Gavira, J.A.; Sleutel, M.; Van de Weerdt, C.; Otalora, F.; Garcia-Ruiz, J.M.; Nicolis, G.; Martial, J.; Decanniere, K. Toward a Definition of X-ray Crystal Quality. Cryst. Growth Des. 2008, 8, 4284–4290. [Google Scholar] [CrossRef]
- Otálora, F.; Novella, M.L.; Gavira, J.A.; Thomas, B.R.; García-Ruiz, J.M. Experimental evidence for the stability of the depletion zone around a growing protein crystal under microgravity. Acta Cryst. 2001, D57, 412–417. [Google Scholar] [CrossRef]
- Thomas, B.R.; Chernov, A.A.; Vekilov, P.G.; Carter, D.C. Distribution coefficients of protein impurities in ferritin and lysozyme crystals Self-Purification in microgravity. J. Cryst. Growth 2000, 211, 149–156. [Google Scholar] [CrossRef]
- Vekilov, P.G.; Thomas, B.R.; Rosenberger, F. Effects of convective solute and impurity transport in protein crystal growth. J. Phys. Chem. B 1998, 102, 5208–5216. [Google Scholar] [CrossRef]
- Adachi, H.; Takano, K.; Matsumura, H.; Inoue, T.; Mori, Y.; Sasaki, T. Protein crystal growth with a two-liquid system and stirring solution. J. Synchrotron Rad. 2004, 11, 121–124. [Google Scholar] [CrossRef]
- Chernov, A.A. Crystal growth and crystallography. Acta Cryst. 1998, A54, 859–872. [Google Scholar] [CrossRef]
- Inaka, K.; Tanaka, H.; Takahashi, S.; Sano, S.; Sato, M.; Shirakawa, M.; Yoshimura, Y. Numerical Analysis of the diffusive field around a growing protein crystal in microgravity. Defect and Diffusion Forum 2012, 323–325, 565–569. [Google Scholar] [CrossRef]
- Tanaka, H.; Inaka, K.; Sugiyama, S.; Takahashi, S.; Sano, S.; Sato, M.; Yoshitomi, S. Numerical Analysis of the depletion zone formation around a growing protein crystal. Ann. N.Y. Acad. Sci. 2004, 1027, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Inaka, K.; Furubayashi, N.; Yamanaka, M.; Takahashi, S.; Sano, S.; Sato, M.; Shirakawa, M.; Yoshimura, Y. Controlling the diffusive field to grown a higher quality protein crystal in microgravity. Defect Diffus. Forum 2012, 323–325, 549–554. [Google Scholar] [CrossRef]
- Takahashi, S.; Ohta, K.; Furubayashi, N.; Yan, B.; Koga, M.; Wada, Y.; Yamada, M.; Inaka, K.; Tanaka, H.; Miyoshi, H.; et al. JAXA protein crystallization in space: ongoing improvements for growing high-quality crystals. J. Synchrotron Rad. 2013, 20, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Tsurumura, T.; Aritake, K.; Furubayashi, N.; Takahashi, S.; Yamanaka, M.; Hirota, E.; Sano, S.; Sato, M.; Kobayashi, T.; et al. Improvement in the quality of hematopoietic prostaglandin D synthase crystals in a microgravity environment. J. Synchrotron Rad. 2011, 18, 88–91. [Google Scholar] [CrossRef]
- Inaka, K.; Takahashi, S.; Aritake, K.; Tsurumura, T.; Furubayashi, N.; Yan, B.; Hirota, E.; Sano, S.; Sato, M.; Kobayashi, T.; et al. High-quality protein crystal growth of mouse lipocalin-type prostaglandin D Synthase in microgravity. Cryst. Growth Des. 2011, 11, 2107–2111. [Google Scholar] [CrossRef]
- Tanaka, H.; Sasaki, S.; Takahashi, S.; Inaka, K.; Wada, Y.; Yamada, M.; Ohta, K.; Miyoshi, H.; Kobayashi, T.; Kamigaichi, S. Numerical model of protein crystal growth in a diffusive field such as the microgravity environment. J. Synchrotron Rad. 2013, 20, 1003–1009. [Google Scholar] [CrossRef]
- Takahashi, S.; Nakagawa, A.; Furubayashi, N.; Inaka, K.; Ohta, K.; Tanaka, H. Non-uniform diffractive quality of the crystals grown in microgravity and in terrestrial gravity. J. Jpn. Association Crystal Growth 2016, 43, 110–116. [Google Scholar]
- Tsukamoto, K. Growth rate of protein crystals under microgravity. J. Jpn. Soc. Microgravity Appl. 2012, 29, 106–110. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effect | Classification | Reagent and Usage Example | Explanation |
|---|---|---|---|
| Electrostatic interaction | Counter ion | 10–1000 mM sodium chloride | Reduces the electrostatic repulsion between protein molecules by creating an ion pair on the protein molecule surface [21]. Na+ and Cl− are the most conventional ones. |
| Organic solvent | 5%–20% MPD 5%–20% dioxane | Reduces the electrostatic repulsion between protein molecules by reducing the dielectric constant of the solvent [1]. | |
| Specific intermolecular interactions | Multivalent acid | 10–200 mM tartrate | Intervenes and produces attractions between protein molecules |
| Multivalent metal ion | 10–200 mM MgCl2 | ||
| Multivalent base | 10–200 mM bis tris propane | ||
| pH buffering | Weak acid | 10–100 mM acetate | Buffer pH of solution |
| Weak base | 10–100 mM tris | ||
| Solubilizing | Detergent | 0.1%–2% DDM (n-dodecyl-beta-D-maltopyranoside) | Solubilization of protein with a strong hydrophobicity of the membrane protein [23,24] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashizume, Y.; Inaka, K.; Furubayashi, N.; Kamo, M.; Takahashi, S.; Tanaka, H. Methods for Obtaining Better Diffractive Protein Crystals: From Sample Evaluation to Space Crystallization. Crystals 2020, 10, 78. https://doi.org/10.3390/cryst10020078
Hashizume Y, Inaka K, Furubayashi N, Kamo M, Takahashi S, Tanaka H. Methods for Obtaining Better Diffractive Protein Crystals: From Sample Evaluation to Space Crystallization. Crystals. 2020; 10(2):78. https://doi.org/10.3390/cryst10020078
Chicago/Turabian StyleHashizume, Yoshinobu, Koji Inaka, Naoki Furubayashi, Masayuki Kamo, Sachiko Takahashi, and Hiroaki Tanaka. 2020. "Methods for Obtaining Better Diffractive Protein Crystals: From Sample Evaluation to Space Crystallization" Crystals 10, no. 2: 78. https://doi.org/10.3390/cryst10020078
APA StyleHashizume, Y., Inaka, K., Furubayashi, N., Kamo, M., Takahashi, S., & Tanaka, H. (2020). Methods for Obtaining Better Diffractive Protein Crystals: From Sample Evaluation to Space Crystallization. Crystals, 10(2), 78. https://doi.org/10.3390/cryst10020078