Hidden Solvates and Transient Forms of Trimesic Acid



Abstract

1. Introduction
2. Materials and Methods
2.1. Slurrying
2.2. Liquid Assisted Grinding (LAG)
2.3. Single Crystal Preparation
2.4. Single Crystal Diffraction
2.5. X-ray Powder Diffraction
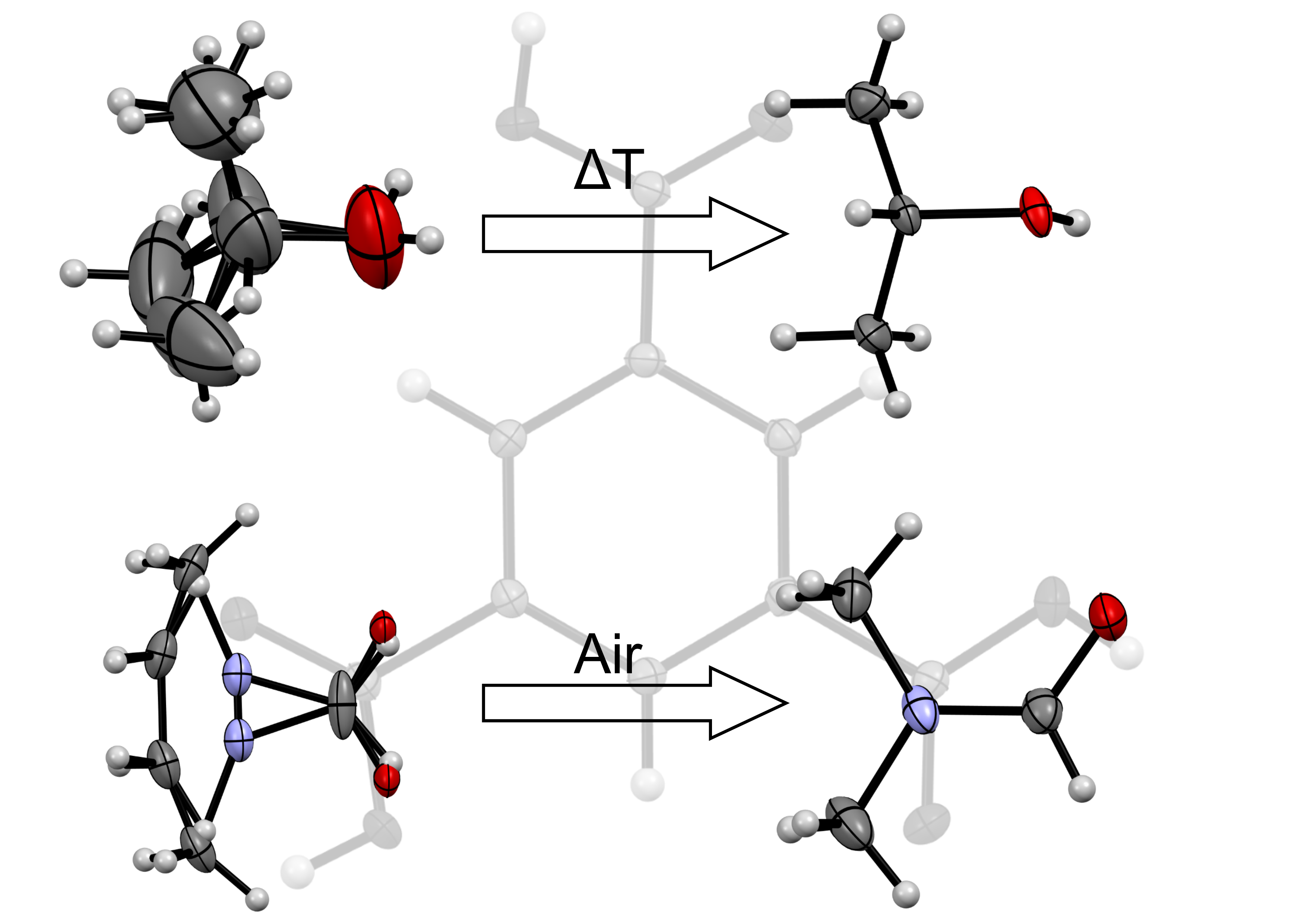
3. Results and Discussion
3.1. Solid Form Screen
3.2. TMA:EtOH Solid
3.3. TMA:IPA Solid
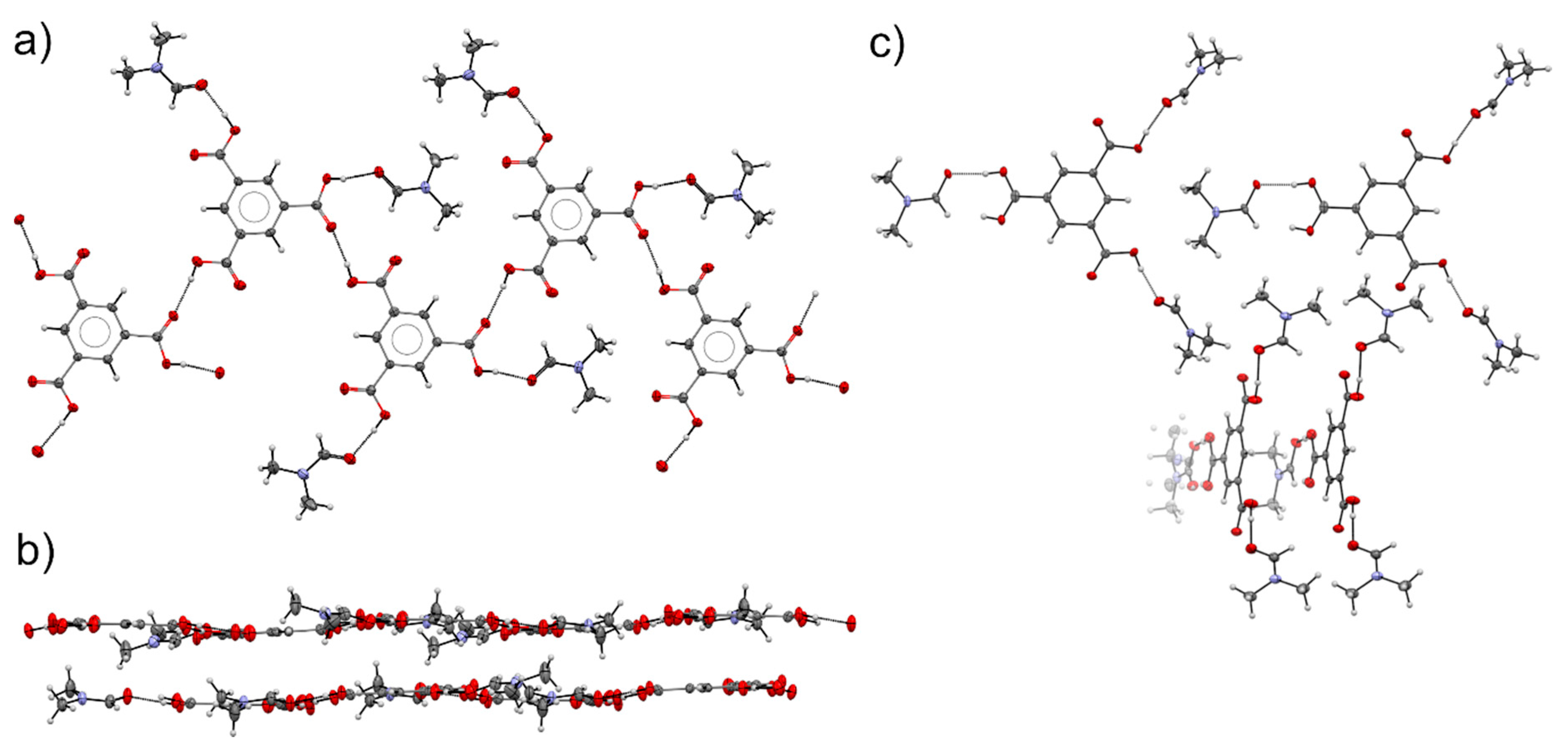
3.4. Comparison of Alcohol Structures
3.5. DMF Solvate
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Du, M.; Zhang, Z.H.; Zhao, X.J. Cocrystallization of trimesic acid and pyromellitic acid with bent dipyridines. Cryst. Growth Des. 2005, 5, 1247–1254. [Google Scholar] [CrossRef]
- Melendez, R.E.; Sharma, C.V.K.; Zaworotko, M.J.; Bauer, C.; Rogers, R.D. Toward the design of porous organic solids: Modular honeycomb grids sustained by anions of trimesic acid. Angew. Chem. 1996, 35, 2213–2215. [Google Scholar] [CrossRef]
- Tothadi, S.; Koner, K.; Dey, K.; Addicoat, M.; Banerjee, R. Morphological Evolution of Two-Dimensional Porous Hexagonal Trimesic Acid Framework. ACS Appl. Mater. Interfaces 2020, 12, 15588–15594. [Google Scholar] [CrossRef] [PubMed]
- Sel, K.; Demirci, S.; Meydan, E.; Yildiz, S.; Ozturk, O.F.; Al-Lohedan, H.; Sahiner, N. Benign preparation of metal-organic frameworks of trimesic acid and Cu, Co or Ni for potential sensor applications. J. Electron. Mater. 2015, 44, 136–143. [Google Scholar] [CrossRef]
- Sahiner, N.; Sel, K.; Ozturk, O.F.; Demirci, S.; Terzi, G. Facile synthesis and characterization of trimesic acid-Cu based metal organic frameworks. Appl. Surf. Sci. 2014, 314, 663–669. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Nath, K.G.; Ivasenko, O.; MacLeod, J.M.; Miwa, J.A.; Wuest, J.D.; Nanci, A.; Perepichka, D.F.; Rosei, F. Crystal engineering in two dimensions: An approach to molecular nanopatterning. J. Phys. Chem. C 2007, 111, 16996–17007. [Google Scholar] [CrossRef]
- Cui, P.; Mcmahon, D.P.; Spackman, P.R.; Alston, B.M.; Little, M.A.; Day, G.M.; Cooper, A.I. Mining predicted crystal structure landscapes with high throughput crystallisation: Old molecules, new insights. Chem. Sci. 2019, 10, 9988–9997. [Google Scholar] [CrossRef]
- Dale, S.H.; Elsegood, M.R.J.; Richards, S.J. Step-wise dis-assembly of trimesic acid: Mono- and bis(methanol) solvates. Chem. Commun. 2004, 11, 1278–1279. [Google Scholar] [CrossRef]
- Cosier, J.; Glazer, A.M. A nitrogen-gas-stream cryostat for general X-ray diffraction studies. J. Appl. Crystallogr. 1986, 19, 105–107. [Google Scholar] [CrossRef]
- Bruker APEX3, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2015.
- Sheldrick, G.M. SADABS; University of Gottingen: Gottingen, Germany; Bruker AXS: Madison, WI, USA, 2001. [Google Scholar]
- Coles, S.J.; Gale, P.A. Changing and challenging times for service crystallography. Chem. Sci. 2012, 3, 683–689. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction CrysAlisPro. 2019. Available online: https://www.rigaku.com/products/smc/crysalis (accessed on 30 November 2020).
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Dale, S.H.; Elsegood, M.R.J. Trimesic acid bis(N,N-dimethylformamide) solvate at 150 K. Acta Crystallogr. Sect. E Struct. Rep. Online 2003, 59, o127–o128. [Google Scholar] [CrossRef]
- Fan, Z.-Z.; Li, X.-H.; Wang, G.-P. Trimesic acid dihydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2005, 61, o1607–o1608. [Google Scholar] [CrossRef]
- Herbstein, F.H.; Marsh, R.E. The crystal structures of trimesic acid, its hydrates and complexes. II. Trimesic acid monohydrate-2/9 picric acid and trimesic acid 5/6 hydrate. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1977, 33, 2358–2367. [Google Scholar] [CrossRef]
- Herbstein, F.H.; Kapon, M.; Wasserman, S.; Herbstein, F.H.; Kapon, M. The Crystal Structures of Trimesic Acid, its Hydrates and Complexes. Acta Crystallogr. Sect. B 1978, 34, 1608–1612. [Google Scholar] [CrossRef]
- Davey, R.J.; Brychczynska, M.; Sadiq, G.; Dent, G.; Pritchard, R.G. Crystallising trimesic acid from DMSO solutions—Can crystallography teach us anything about the process of crystal nucleation? CrystEngComm 2013, 15, 856–859. [Google Scholar] [CrossRef]
- Bernè, S.; Hernández, G.; Portillo, R.; Gutiérrez, R. Trimesic acid dimethyl sulfoxide solvate: Space group revision. Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64, o1366. [Google Scholar] [CrossRef]
- Hasa, D.; Pastore, M.; Arhangelskis, M.; Gabriele, B.; Cruz-Cabeza, A.J.; Rauber, G.S.; Bond, A.D.; Jones, W. On the kinetics of solvate formation through mechanochemistry. CrystEngComm 2019, 21, 2097–2104. [Google Scholar] [CrossRef]
- Gavezzotti, A. Efficient computer modeling of organic materials. The atom-atom, Coulomb-London-Pauli (AA-CLP) model for intermolecular electrostatic-polarization, dispersion and repulsion energies. New J. Chem. 2011, 35, 1360–1368. [Google Scholar] [CrossRef]
- Gavezzotti, A. Non-Conventional Bonding between Organic Molecules. The “Halogen Bond” in Crystalline Systems. Mol. Phys. 2008, 106, 1473–1485. [Google Scholar] [CrossRef]
- Reeves, M.G.; Wood, P.A.; Parsons, S. MrPIXEL: Automated execution of Pixel calculations via the Mercury interface. J. Appl. Crystallogr. 2020, 53, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Pedireddi, V.R.; Ranganathan, A.; Rao, C.N.R. Self-assembled four-membered networks of trimesic acid forming organic channel structures. J. Mol. Struct. 2000, 520, 107–115. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvate | Distance | Coulombic | Polarisation | Dispersion | Repulsion | Total | Interaction |
|---|---|---|---|---|---|---|---|
| MeOH | 9.513 | −140.1 | −74.5 | −23.7 | 169.4 | −68.9 | (a) |
| EtOH | 9.515 | −123.2 | −58.6 | −22.2 | 136.4 | −67.6 | |
| PrOH | 9.527 | −136.8 | −69.6 | −23.1 | 166.0 | −63.5 | |
| BuOH | 9.555 | −134.9 | −71.8 | −24.0 | 156.4 | −74.2 | |
| PenOH | 9.517 | −143.5 | −81.1 | −24.3 | 173.9 | −75.0 | |
| HexOH | 9.526 | −148.8 | −76.3 | −23.1 | 174.3 | −73.8 | |
| MeOH | 9.631 | −55.5 | −25.1 | −10.6 | 64.0 | −27.2 | (b) |
| EtOH | 9.600 | −56.6 | −26.1 | −10.7 | 68.0 | −25.4 | |
| PrOH | 9.618 | −59.6 | −28.0 | −10.8 | 70.3 | −28.1 | |
| BuOH | 9.611 | −55.5 | −26.7 | −10.8 | 63.5 | −29.4 | |
| PenOH | 9.547 | −58.7 | −28.5 | −11.0 | 67.5 | −30.8 | |
| HexOH | 9.588 | −58.7 | −28.3 | −10.7 | 67.7 | −30.0 | |
| MeOH | 6.358 | −81.3 | −37.9 | −13.8 | 93.9 | −39.0 | (c) |
| EtOH | 6.849 | −85.7 | −42.3 | −14.1 | 107.2 | −34.9 | |
| PrOH | 6.965 | −85.5 | −40.9 | −16.2 | 104.5 | −38.1 | |
| BuOH | 6.961 | −84.3 | −41.9 | −15.4 | 105.4 | −36.3 | |
| PenOH | 8.060 | −83.9 | −42.4 | −14.9 | 101.3 | −40.0 | |
| HexOH | 8.481 | −82.7 | −40.4 | −14.7 | 101.6 | −36.3 | |
| MeOH | 6.426 | −39.8 | −15.2 | −11.0 | 45.5 | −20.5 | (d) |
| EtOH | 6.889 | −41.8 | −15.1 | −10.0 | 45.5 | −21.3 | |
| PrOH | 7.823 | −35.5 | −12.9 | −9.3 | 37.7 | −20.0 | |
| BuOH | 7.399 | −41.6 | −16.0 | −11.3 | 46.7 | −22.1 | |
| PenOH | 8.909 | −39.1 | −14.7 | −9.5 | 42.8 | −20.5 | |
| HexOH | 9.423 | −39.0 | −14.1 | −9.3 | 42.9 | −19.5 | |
| MeOH | 3.465 | −15.3 | −4.5 | −48.4 | 37.2 | −31.0 | (e) |
| EtOH | 3.458 | −22.6 | −6.4 | −52.9 | 47.0 | −35.0 | |
| PrOH | 4.931 | −4.5 | −2.9 | −31.2 | 18.0 | −20.6 | |
| BuOH | 3.690 | −11.9 | −2.8 | −37.5 | 22.2 | −29.9 | |
| PenOH | 5.235 | −5.2 | −4.4 | −32.5 | 24.0 | −18.0 | |
| HexOH | 5.321 | −7.0 | −3.8 | −31.1 | 22.4 | −19.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, M.R.; Oswald, I.D.H. Hidden Solvates and Transient Forms of Trimesic Acid. Crystals 2020, 10, 1098. https://doi.org/10.3390/cryst10121098
Ward MR, Oswald IDH. Hidden Solvates and Transient Forms of Trimesic Acid. Crystals. 2020; 10(12):1098. https://doi.org/10.3390/cryst10121098
Chicago/Turabian StyleWard, Martin R., and Iain D. H. Oswald. 2020. "Hidden Solvates and Transient Forms of Trimesic Acid" Crystals 10, no. 12: 1098. https://doi.org/10.3390/cryst10121098
APA StyleWard, M. R., & Oswald, I. D. H. (2020). Hidden Solvates and Transient Forms of Trimesic Acid. Crystals, 10(12), 1098. https://doi.org/10.3390/cryst10121098