Novel Sample-Stage for Combined Near Ambient Pressure X-ray Photoelectron Spectroscopy, Catalytic Characterization and Electrochemical Impedance Spectroscopy

 , , ,
, , ,
Abstract
1. Introduction
2. Experimental
2.1. General Considerations
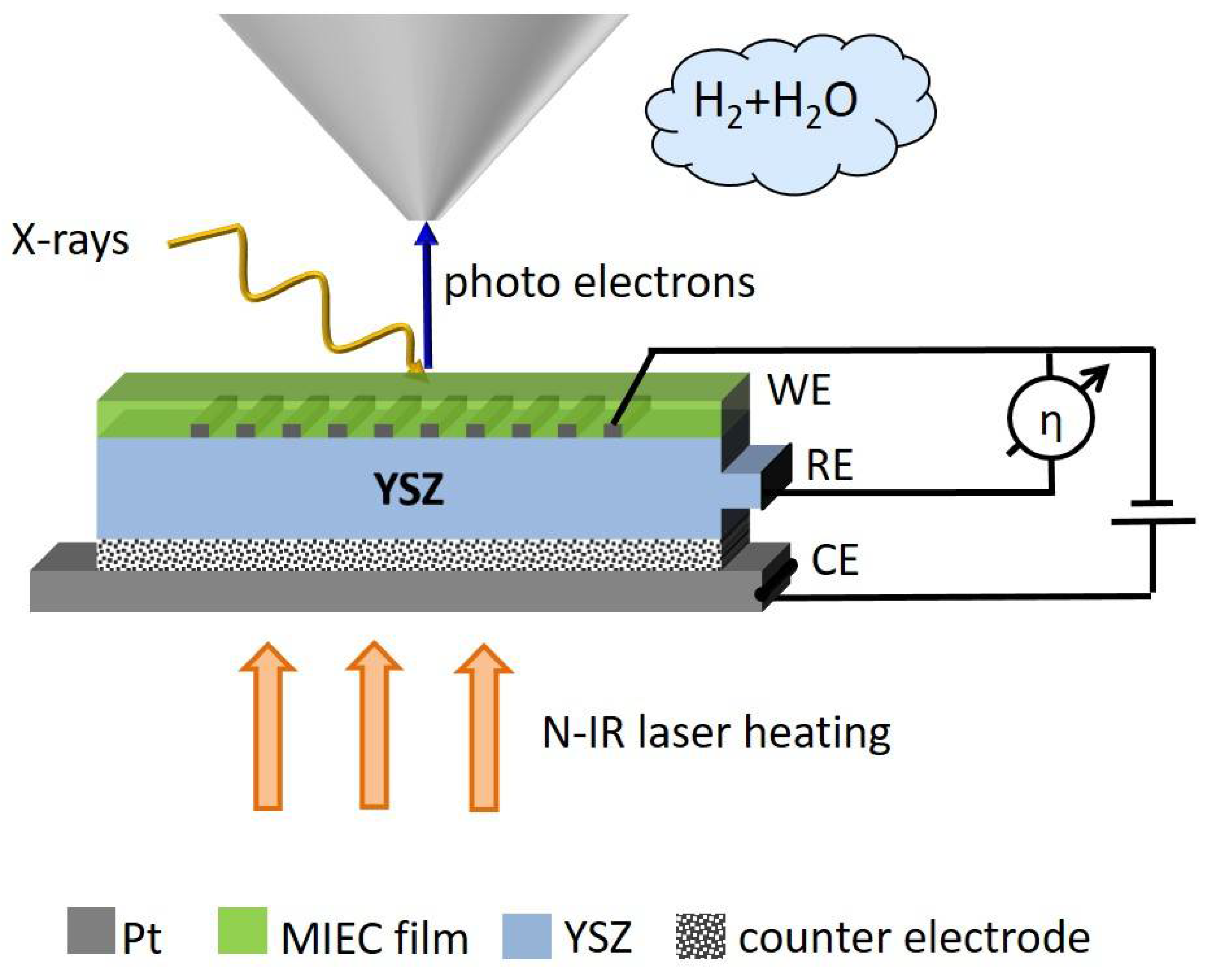
2.2. Sample-Stage and Sample Mounting
2.3. Fabrication of the Electrochemical Model Cell
3. Proof of Concept
3.1. Layout of Experiments
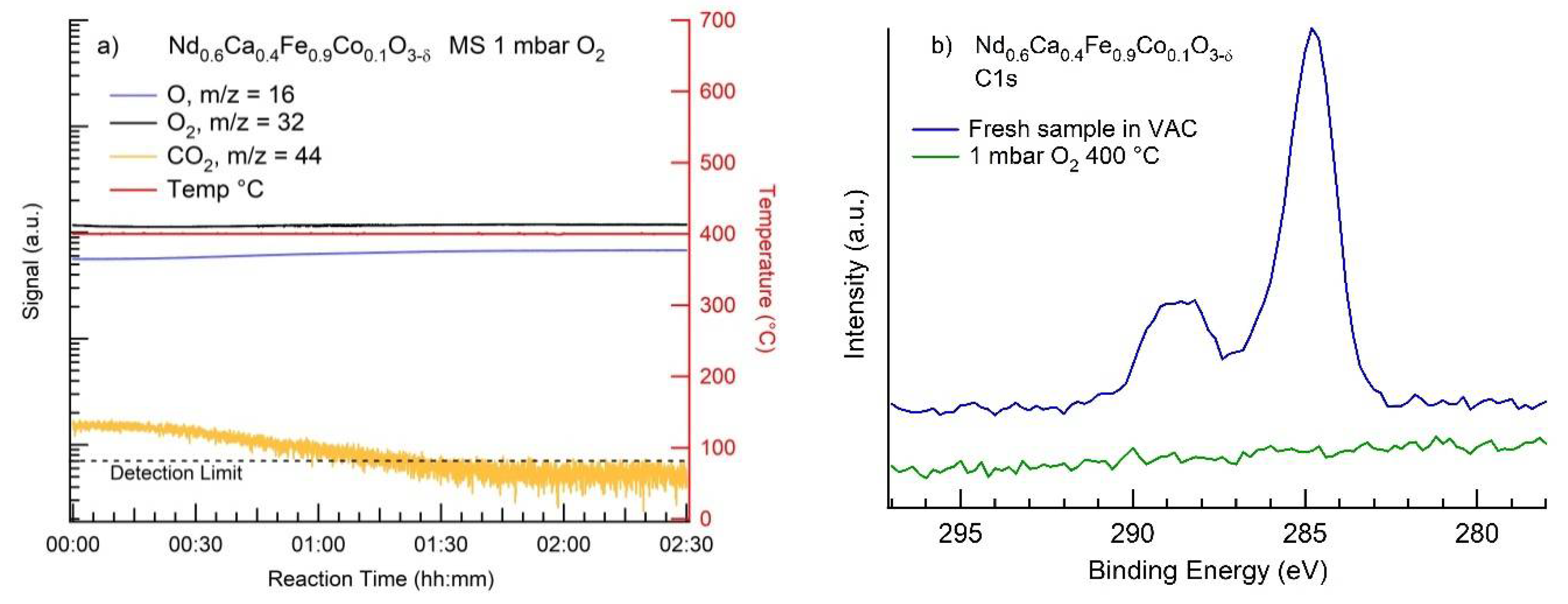
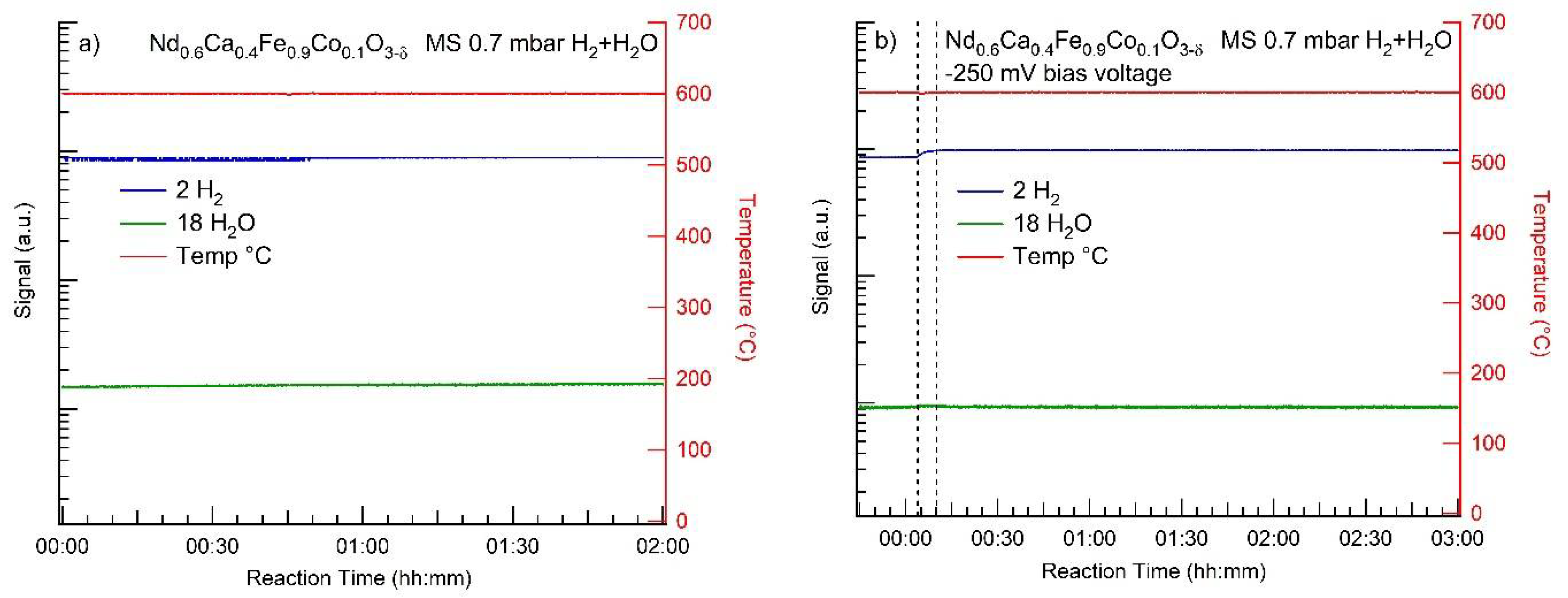
3.2. MS Results
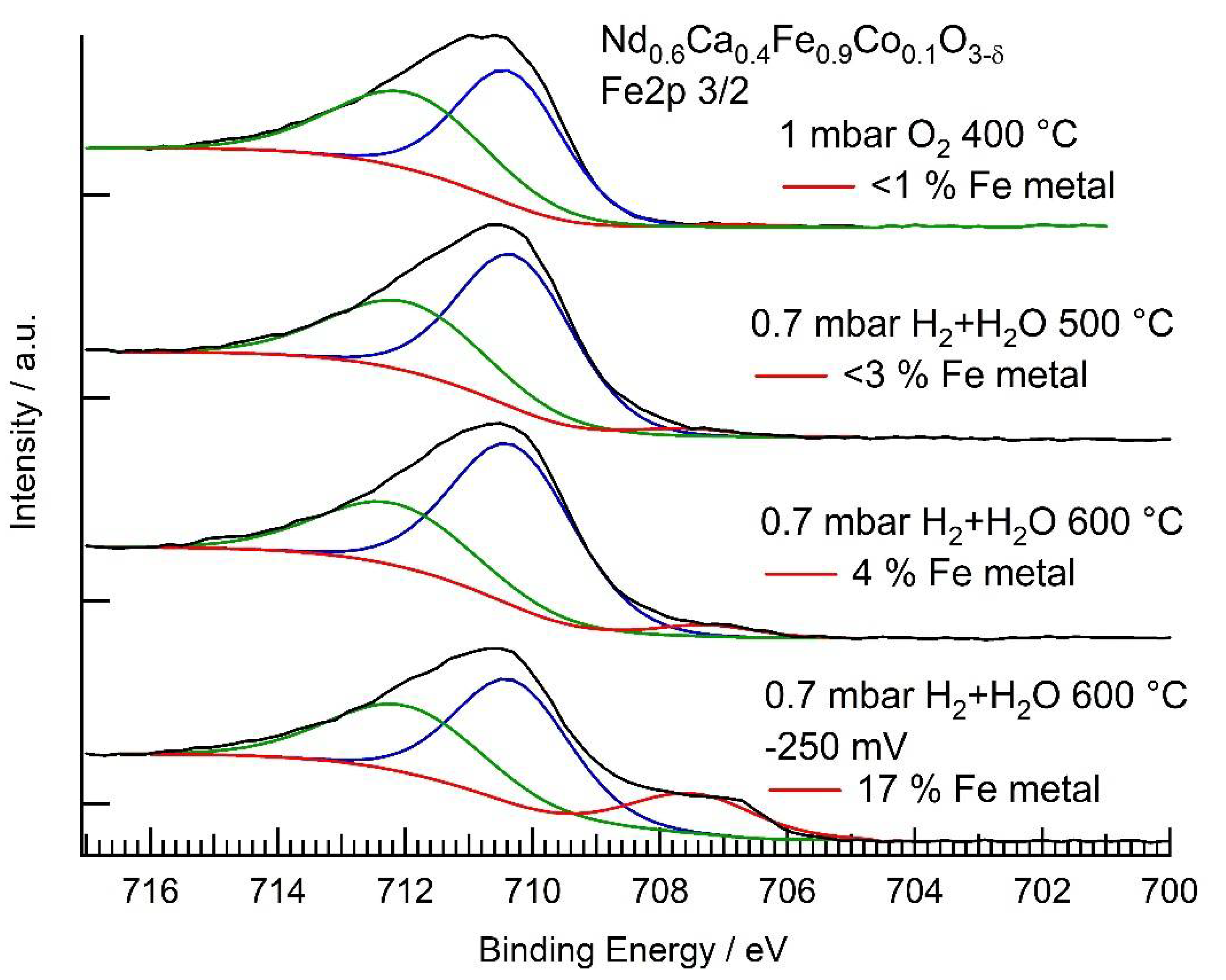
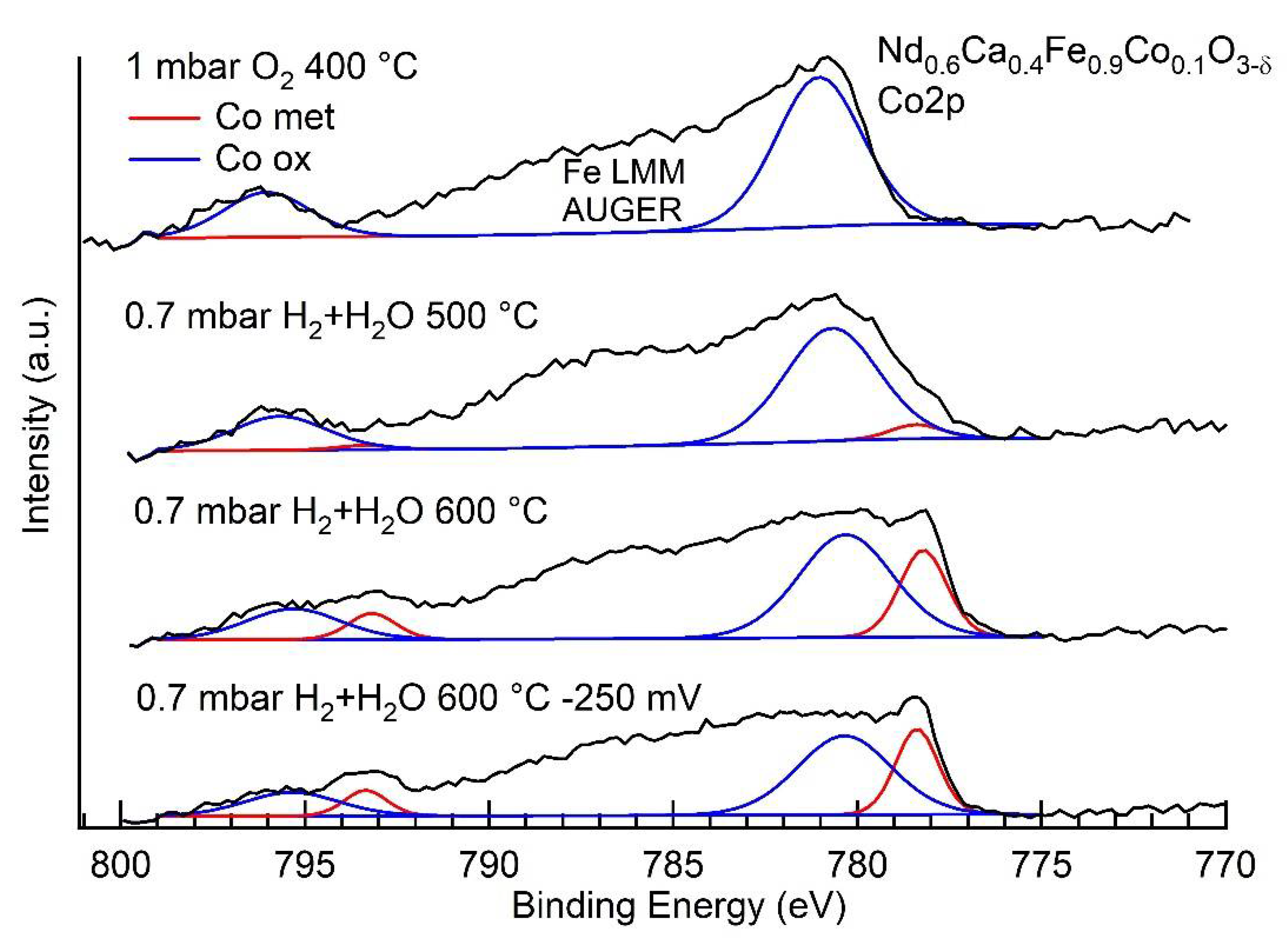
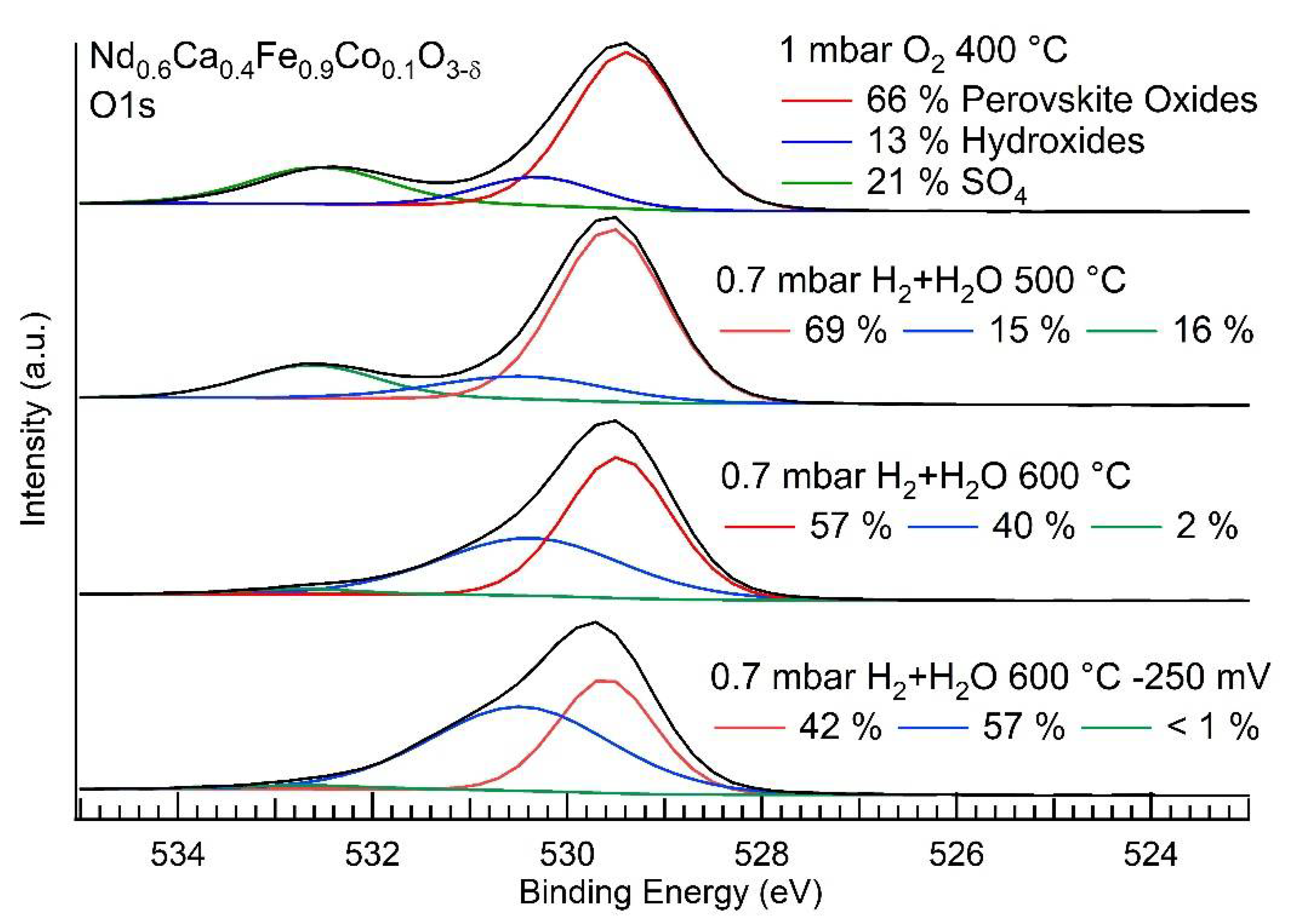
3.3. NAP-XPS Results
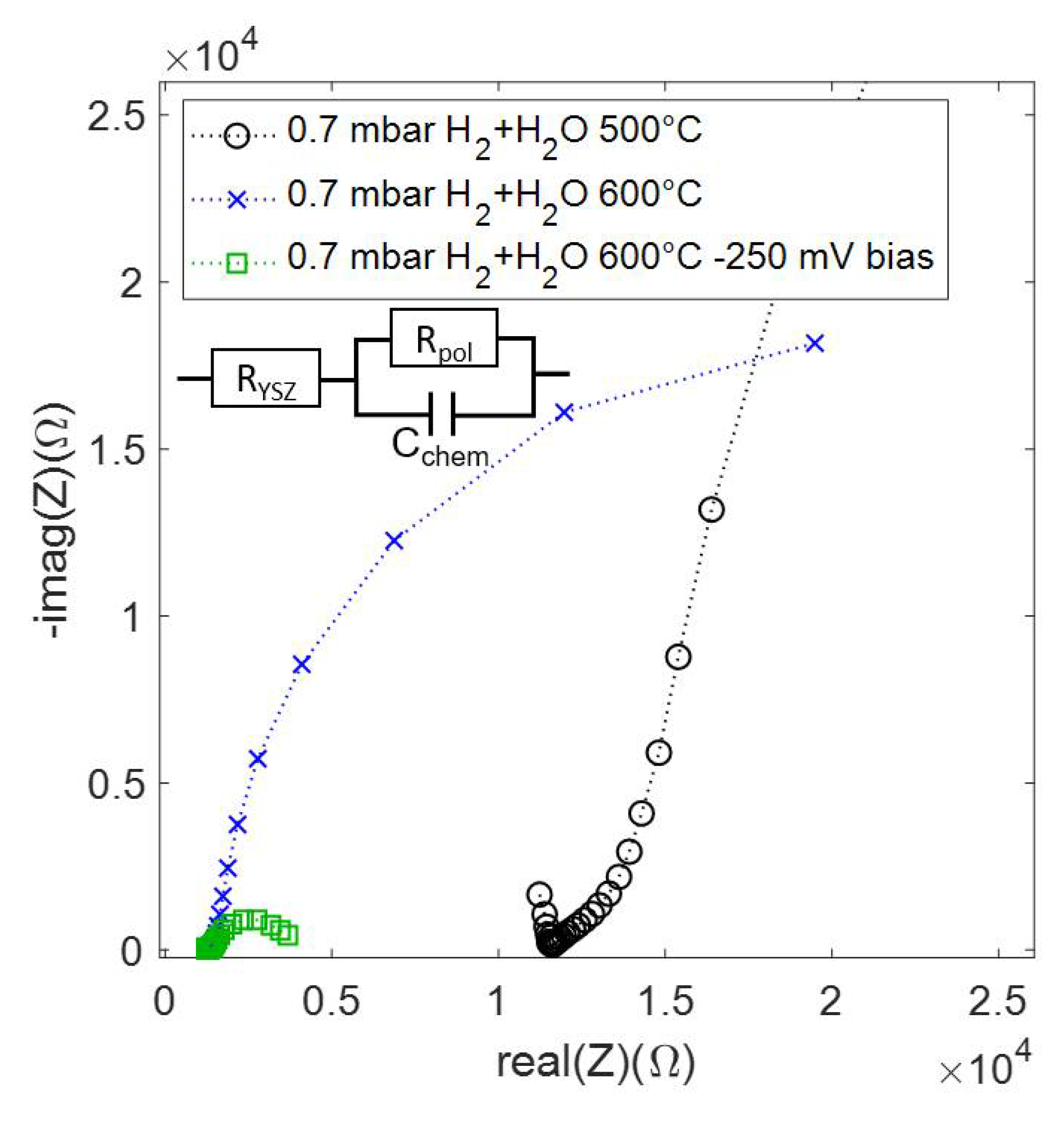
3.4. Electrochemical Characterization
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hwang, J.; Rao, R.R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [CrossRef]
- Zhu, M.; Wachs, I.E. Iron-Based Catalysts for the High-Temperature Water–Gas Shift (HT-WGS) Reaction: A Review. ACS Catal. 2015, 6, 722–732. [Google Scholar] [CrossRef]
- Popovic, J.; Lindenthal, L.; Rameshan, R.; Ruh, T.; Nenning, A.; Löffler, S.; Opitz, A.K.; Rameshan, C. High Temperature Water Gas Shift Reactivity of Novel Perovskite Catalysts. Catalysts 2020, 10, 582. [Google Scholar] [CrossRef]
- Aramouni, N.A.K.; Touma, J.G.; Abu Tarboush, B.; Zeaiter, J.; Ahmad, M.N. Catalyst design for dry reforming of methane: Analysis review. Renew. Sustain. Energy Rev. 2018, 82, 2570–2585. [Google Scholar] [CrossRef]
- Kubicek, M.; Bork, A.H.; Rupp, J.L.M. Perovskite oxides—A review on a versatile material class for solar-to-fuel conversion processes. J. Mater. Chem. A 2017, 5, 11983–12000. [Google Scholar] [CrossRef]
- Sá, S.; Silva, H.; Brandão, L.; Sousa, J.M.; Mendes, A. Catalysts for methanol steam reforming—A review. Appl. Catal. B Environ. 2010, 99, 43–57. [Google Scholar] [CrossRef]
- Mahato, N.; Banerjee, A.; Gupta, A.; Omar, S.; Balani, K. Progress in material selection for solid oxide fuel cell technology: A review. Prog. Mater. Sci. 2015, 72, 141–337. [Google Scholar] [CrossRef]
- Yang, X.; Irvine, J.T.S. (La0.75Sr0.25)0.95Mn0.5Cr0.5O3 as the cathode of solid oxide electrolysis cells for high temperature hydrogen production from steam. J. Mater. Chem. 2008, 18, 2349–2354. [Google Scholar] [CrossRef]
- Arble, C.; Jia, M.; Newberg, J.T. Lab-based ambient pressure X-ray photoelectron spectroscopy from past to present. Surf. Sci. Rep. 2018, 73, 37–57. [Google Scholar] [CrossRef]
- Starr, D.E.; Liu, Z.; Hävecker, M.; Knop-Gericke, A.; Bluhm, H. Investigation of solid/vapor interfaces using ambient pressure X-ray photoelectron spectroscopy. Chem. Soc. Rev. 2013, 42, 5833–5857. [Google Scholar] [CrossRef]
- Yamamoto, S.; Bluhm, H.; Andersson, K.; Ketteler, G.; Ogasawara, H.; Salmeron, M.; Nilsson, A. In situx-ray photoelectron spectroscopy studies of water on metals and oxides at ambient conditions. J. Phys. Condens. Matter 2008, 20. [Google Scholar] [CrossRef]
- Feng, Z.A.; Gopal, C.B.; Ye, X.; Guan, Z.; Jeong, B.; Crumlin, E.; Chueh, W.C. Origin of Overpotential-Dependent Surface Dipole at CeO2−x/Gas Interface During Electrochemical Oxygen Insertion Reactions. Chem. Mater. 2016, 28, 6233–6242. [Google Scholar] [CrossRef]
- Chueh, W.C.; Hao, Y.; Jung, W.; Haile, S.M. High electrochemical activity of the oxide phase in model ceria–Pt and ceria–Ni composite anodes. Nat. Mater. 2011, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.A.; Machala, M.L.; Chueh, W.C. Surface electrochemistry of CO2 reduction and CO oxidation on Sm-doped CeO2−x: Coupling between Ce3+ and carbonate adsorbates. Phys. Chem. Chem. Phys. 2015, 17, 12273–12281. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, Y.; Grass, M.E.; Dejoie, C.; Ding, W.; Gaskell, K.; Jabeen, N.; Hong, Y.P.; Shavorskiy, A.; Bluhm, H.; et al. Mechanistic Studies of Water Electrolysis and Hydrogen Electro-Oxidation on High Temperature Ceria-Based Solid Oxide Electrochemical Cells. J. Am. Chem. Soc. 2013, 135, 11572–11579. [Google Scholar] [CrossRef] [PubMed]
- Nenning, A.; Opitz, A.K.; Rameshan, C.; Rameshan, R.; Blume, R.; Hävecker, M.; Knop-Gericke, A.; Rupprechter, G.; Klötzer, B.; Fleig, J. Ambient Pressure XPS Study of Mixed Conducting Perovskite-Type SOFC Cathode and Anode Materials under Well-Defined Electrochemical Polarization. J. Phys. Chem. C 2016, 120, 1461–1471. [Google Scholar] [CrossRef]
- Opitz, A.K.; Nenning, A.; Rameshan, C.; Rameshan, R.; Blume, R.; Hävecker, M.; Knop-Gericke, A.; Rupprechter, G.; Fleig, J.; Klötzer, B. Enhancing Electrochemical Water-Splitting Kinetics by Polarization-Driven Formation of Near-Surface Iron(0): An In Situ XPS Study on Perovskite-Type Electrodes. Angew. Chem. Int. Ed. 2014, 54, 2628–2632. [Google Scholar] [CrossRef]
- Opitz, A.K.; Nenning, A.; Rameshan, C.; Kubicek, M.; Götsch, T.; Blume, R.; Hävecker, M.; Knop-Gericke, A.; Rupprechter, G.; Klötzer, B.; et al. Surface Chemistry of Perovskite-Type Electrodes During High Temperature CO2 Electrolysis Investigated by Operando Photoelectron Spectroscopy. ACS Appl. Mater. Interfaces 2017, 9, 35847–35860. [Google Scholar] [CrossRef]
- Opitz, A.K.; Rameshan, C.; Kubicek, M.; Rupp, G.M.; Nenning, A.; Götsch, T.; Blume, R.; Hävecker, M.; Knop-Gericke, A.; Rupprechter, G.; et al. The Chemical Evolution of the La0.6Sr0.4CoO3−δ Surface Under SOFC Operating Conditions and Its Implications for Electrochemical Oxygen Exchange Activity. Top. Catal. 2018, 61, 2129–2141. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Hong, W.T.; Crumlin, E.J.; Bluhm, H.; Shao-Horn, Y. Insights into Electrochemical Reactions from Ambient Pressure Photoelectron Spectroscopy. Accounts Chem. Res. 2015, 48, 2976–2983. [Google Scholar] [CrossRef]
- Nurk, G.; Kooser, K.; Korjus, O.; Kanarbik, R.; Urpelainen, S.; Käämbre, T.; Joost, U.; Kook, M.; Kodu, M.; Möller, P.; et al. Operando NAP-HT-XPS and Impedance Spectroscopy Study of Pulsed Laser Deposited Ni-Ce0.9Gd0.1O2-δ Solid Oxide Fuel Cell Electrode. ECS Trans. 2019, 91, 555–561. [Google Scholar] [CrossRef]
- Kooser, K.; Käämbre, T.; Vestli, M.; Joost, U.; Urpelainen, S.; Kook, M.; Bournel, F.; Gallet, J.-J.; Lust, E.; Kukk, E.; et al. Operando high-temperature near-ambient pressure X-ray photoelectron spectroscopy and impedance spectroscopy study of Ni−Ce0.9Gd0.1O2−δ solid oxide fuel cell anode. Int. J. Hydrogen Energy 2020, 45, 25286–25298. [Google Scholar] [CrossRef]
- Adler, S.B. Reference Electrode Placement in Thin Solid Electrolytes. J. Electrochem. Soc. 2002, 149, E166–E172. [Google Scholar] [CrossRef]
- Bluhm, H. Photoelectron spectroscopy of surfaces under humid conditions. J. Electron Spectrosc. Relat. Phenom. 2010, 177, 71–84. [Google Scholar] [CrossRef]
- Ahamer, C.; Opitz, A.K.; Rupp, G.M.; Fleig, J. Revisiting the Temperature Dependent Ionic Conductivity of Yttria Stabilized Zirconia (YSZ). J. Electrochem. Soc. 2017, 164, F790–F803. [Google Scholar] [CrossRef]
- Schlesinger, R. Modeling and Evaluation of Three-Terminal Impedance Measurement Configurations for Solid Oxide Fuel Cell Electrodes; TU Wien: Vienna, Austria, 2018. [Google Scholar]
- Lorenz, H.; Zhao, Q.; Turner, S.; Lebedev, O.I.; Van Tendeloo, G.; Klötzer, B.; Rameshan, C.; Penner, S. Preparation and structural characterization of SnO2 and GeO2 methanol steam reforming thin film model catalysts by (HR)TEM. Mater. Chem. Phys. 2010, 122, 623–629. [Google Scholar] [CrossRef]
- Lorenz, H.; Zhao, Q.; Turner, S.; Lebedev, O.I.; Van Tendeloo, G.; Klötzer, B.; Rameshan, C.; Pfaller, K.; Konzett, J.; Penner, S. Origin of different deactivation of Pd/SnO2 and Pd/GeO2 catalysts in methanol dehydrogenation and reforming: A comparative study. Appl. Catal. A Gen. 2010, 381, 242–252. [Google Scholar] [CrossRef]
- Lindenthal, L.; Rameshan, R.; Summerer, H.; Ruh, T.; Popovic, J.; Nenning, A.; Löffler, S.; Opitz, A.K.; Blaha, P.; Rameshan, C. Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution. Catalysts 2020, 10, 268. [Google Scholar] [CrossRef]
- Schmid, A.; Rupp, G.M.; Fleig, J. Voltage and partial pressure dependent defect chemistry in (La,Sr)FeO3−δ thin films investigated by chemical capacitance measurements. Phys. Chem. Chem. Phys. 2018, 20, 12016–12026. [Google Scholar] [CrossRef]
- Kuhn, M.; Hashimoto, S.; Sato, K.; Yashiro, K.; Mizusaki, J. Oxygen nonstoichiometry, thermo-chemical stability and lattice expansion of La0.6Sr0.4FeO3−δ. Solid State Ionics 2011, 195, 7–15. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.; Gerson, A.R.; Smart, R.S. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Crumlin, E.J.; Mutoro, E.; Hong, W.T.; Biegalski, M.D.; Christen, H.M.; Liu, Z.; Bluhm, H.; Shao-Horn, Y. In Situ Ambient Pressure X-ray Photoelectron Spectroscopy of Cobalt Perovskite Surfaces under Cathodic Polarization at High Temperatures. J. Phys. Chem. C 2013, 117, 16087–16094. [Google Scholar] [CrossRef]
- Pawlak, D.A.; Ito, M.; Oku, M.; Shimamura, K.; Fukuda, T. Interpretation of XPS O (1s) in Mixed Oxides Proved on Mixed Perovskite Crystals. J. Phys. Chem. B 2002, 106, 504–507. [Google Scholar] [CrossRef]
- Merino, N.A.; Barbero, B.P.; Eloy, P.; Cadús, L.E. La1−xCaxCoO3 perovskite-type oxides: Identification of the surface oxygen species by XPS. Appl. Surf. Sci. 2006, 253, 1489–1493. [Google Scholar] [CrossRef]
- Jung, W.; Tuller, H.L. A New Model Describing Solid Oxide Fuel Cell Cathode Kinetics: Model Thin Film SrTi1-xFexO3-δ Mixed Conducting Oxides-a Case Study. Adv. Energy Mater. 2011, 1, 1184–1191. [Google Scholar] [CrossRef]
- Kogler, S.; Nenning, A.; Rupp, G.M.; Opitz, A.K.; Fleig, J.; Fleig, J. Comparison of Electrochemical Properties of La0.6Sr0.4FeO3-δ Thin Film Electrodes: Oxidizing vs. Reducing Conditions. J. Electrochem. Soc. 2015, 162, F317–F326. [Google Scholar] [CrossRef]
- Kubicek, M.; Huber, T.M.; Welzl, A.; Penn, A.; Rupp, G.M.; Bernardi, J.; Stöger-Pollach, M.; Hutter, H.; Fleig, J. Electrochemical properties of La0.6Sr0.4CoO3−δ thin films investigated by complementary impedance spectroscopy and isotope exchange depth profiling. Solid State Ionics 2014, 256, 38–44. [Google Scholar] [CrossRef]
- Nenning, A.; Navickas, E.; Hutter, H.; Fleig, J. Water-Induced Decoupling of Tracer and Electrochemical Oxygen Exchange Kinetics on Mixed Conducting Electrodes. J. Phys. Chem. Lett. 2016, 7, 2826–2831. [Google Scholar] [CrossRef]
- Fleig, J.; Schmid, A.; Rupp, G.M.; Slouka, C.; Navickas, E.; Andrejs, L.; Hutter, H.; Volgger, L.; Nenning, A.; Juergen, F. The Chemical Capacitance as a Fingerprint of Defect Chemistry in Mixed Conducting Oxides. Acta Chim. Slov. 2016, 63, 509–518. [Google Scholar] [CrossRef]
- Chueh, W.C.; McDaniel, A.H.; Grass, M.E.; Hao, Y.; Jabeen, N.; Liu, Z.; Haile, S.M.; Mccarty, K.F.; Bluhm, H.; El Gabaly, F. Highly Enhanced Concentration and Stability of Reactive Ce3+ on Doped CeO2 Surface Revealed In Operando. Chem. Mater. 2012, 24, 1876–1882. [Google Scholar] [CrossRef]
- Gopal, C.B.; El Gabaly, F.; McDaniel, A.H.; Chueh, W.C. Origin and Tunability of Unusually Large Surface Capacitance in Doped Cerium Oxide Studied by Ambient-Pressure X-Ray Photoelectron Spectroscopy. Adv. Mater. 2016, 28, 4692–4697. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ratio Co0/Fe0 | |
|---|---|
| 0.7 mbar H2 + H2O, 500 °C | 0.6 |
| 0.7 mbar H2 + H2O, 600 °C | 2.5 |
| 0.7 mbar H2 + H2O, 600 °C, −250 mV bias | 0.5 |
| Conditions | Rpol (Ω) | (mol cm−2 s−1) | Fe2+ + Co2+ (EIS) | Fe0 (XPS) | Co0 (XPS) |
|---|---|---|---|---|---|
| O2 | - | - | - | 0% | 0% |
| H2 + H2O 500 °C | 147,000 | 0.4% | <3% | 7% | |
| H2 + H2O 600 °C | 36,000 | 0.36% | <4% | 42% | |
| H2 + H2O 600 °C, −250 mV bias | 3600 | (net reaction) | 2.2% | 17% | 49% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rameshan, R.; Nenning, A.; Raschhofer, J.; Lindenthal, L.; Ruh, T.; Summerer, H.; Opitz, A.K.; Martin Huber, T.; Rameshan, C. Novel Sample-Stage for Combined Near Ambient Pressure X-ray Photoelectron Spectroscopy, Catalytic Characterization and Electrochemical Impedance Spectroscopy. Crystals 2020, 10, 947. https://doi.org/10.3390/cryst10100947
Rameshan R, Nenning A, Raschhofer J, Lindenthal L, Ruh T, Summerer H, Opitz AK, Martin Huber T, Rameshan C. Novel Sample-Stage for Combined Near Ambient Pressure X-ray Photoelectron Spectroscopy, Catalytic Characterization and Electrochemical Impedance Spectroscopy. Crystals. 2020; 10(10):947. https://doi.org/10.3390/cryst10100947
Chicago/Turabian StyleRameshan, Raffael, Andreas Nenning, Johannes Raschhofer, Lorenz Lindenthal, Thomas Ruh, Harald Summerer, Alexander Karl Opitz, Tobias Martin Huber, and Christoph Rameshan. 2020. "Novel Sample-Stage for Combined Near Ambient Pressure X-ray Photoelectron Spectroscopy, Catalytic Characterization and Electrochemical Impedance Spectroscopy" Crystals 10, no. 10: 947. https://doi.org/10.3390/cryst10100947
APA StyleRameshan, R., Nenning, A., Raschhofer, J., Lindenthal, L., Ruh, T., Summerer, H., Opitz, A. K., Martin Huber, T., & Rameshan, C. (2020). Novel Sample-Stage for Combined Near Ambient Pressure X-ray Photoelectron Spectroscopy, Catalytic Characterization and Electrochemical Impedance Spectroscopy. Crystals, 10(10), 947. https://doi.org/10.3390/cryst10100947