First Principles Density Functional Theory Prediction of the Crystal Structure and the Elastic Properties of Mo2ZrB2 and Mo2HfB2


Abstract
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
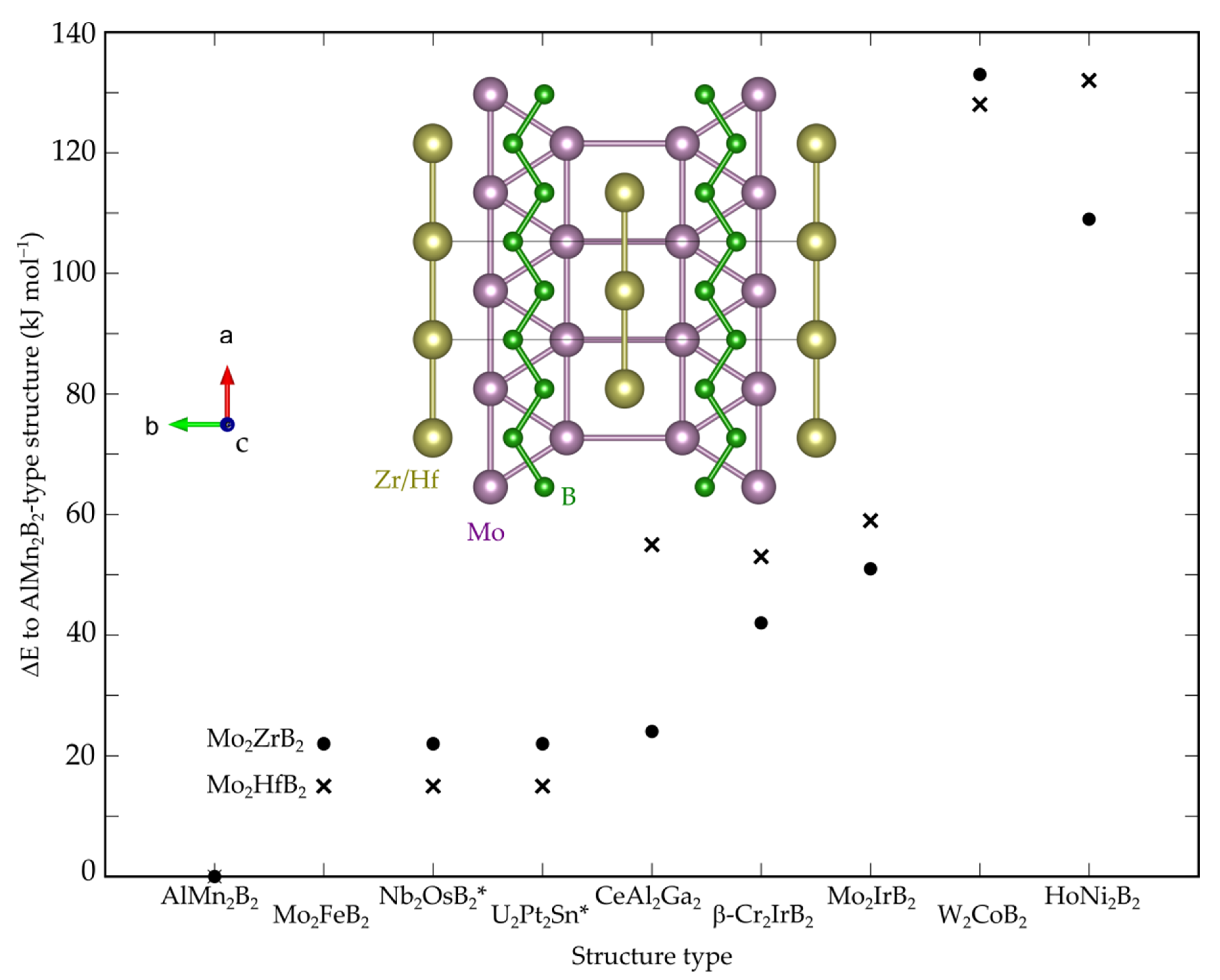
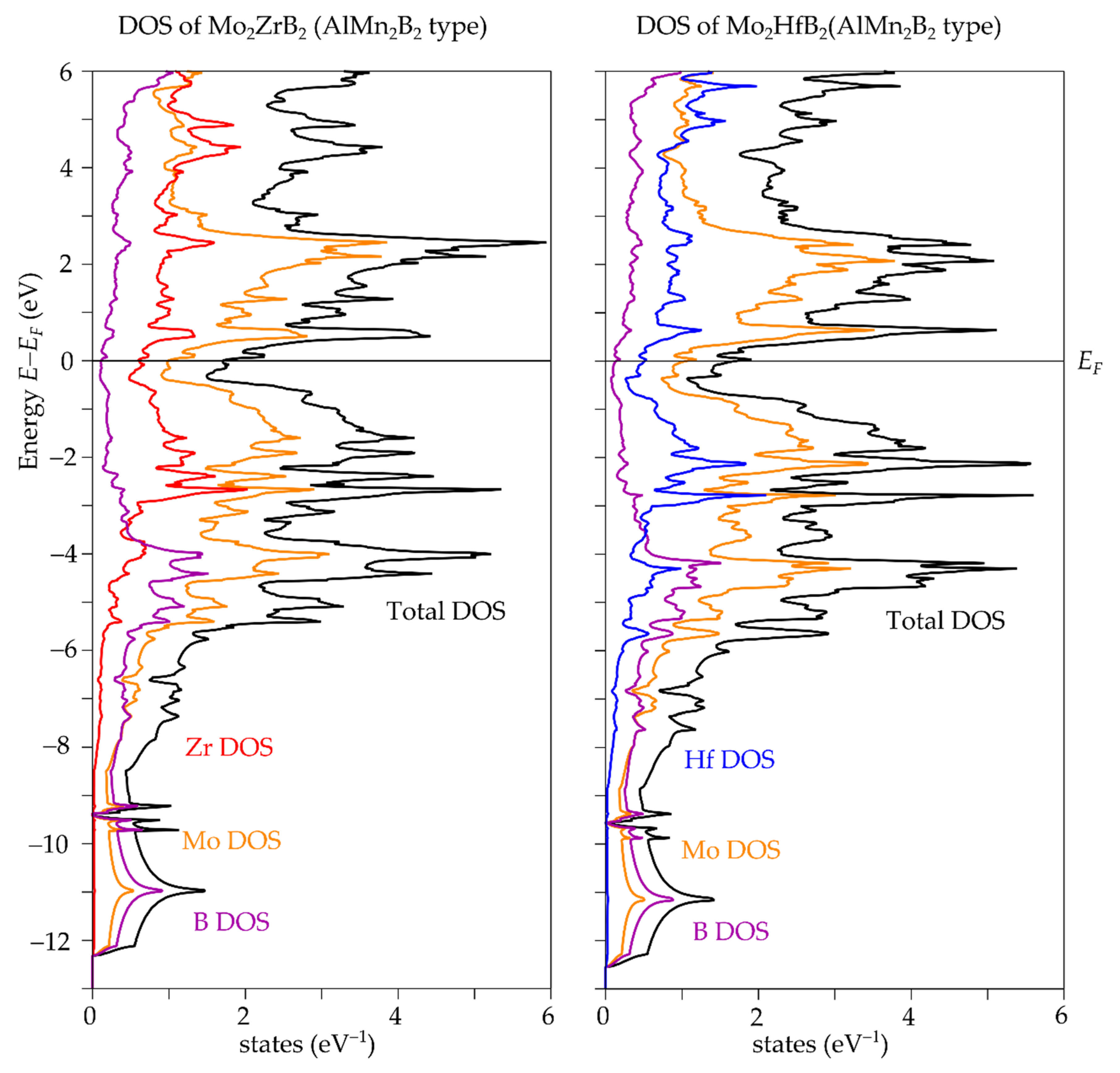
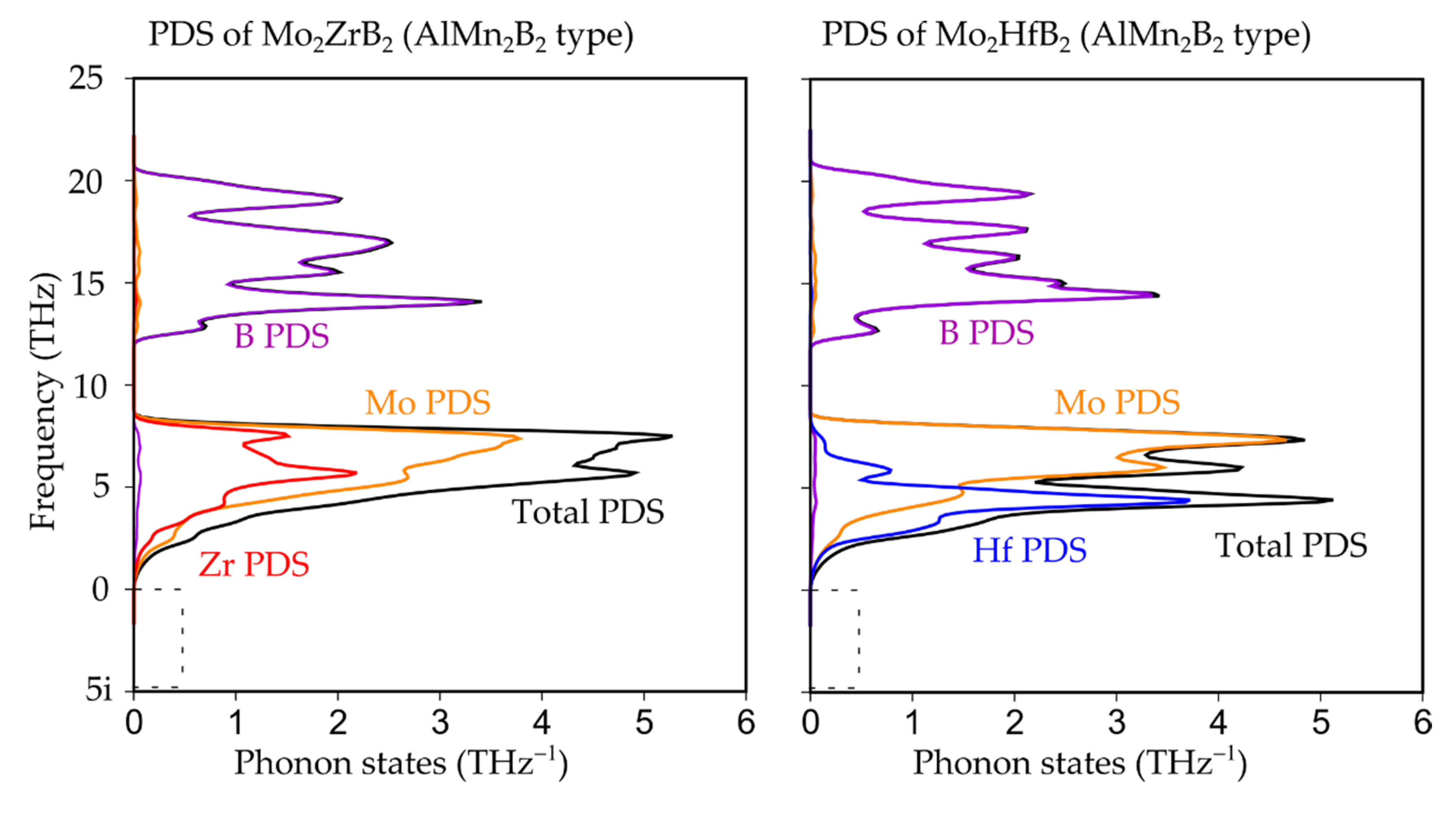
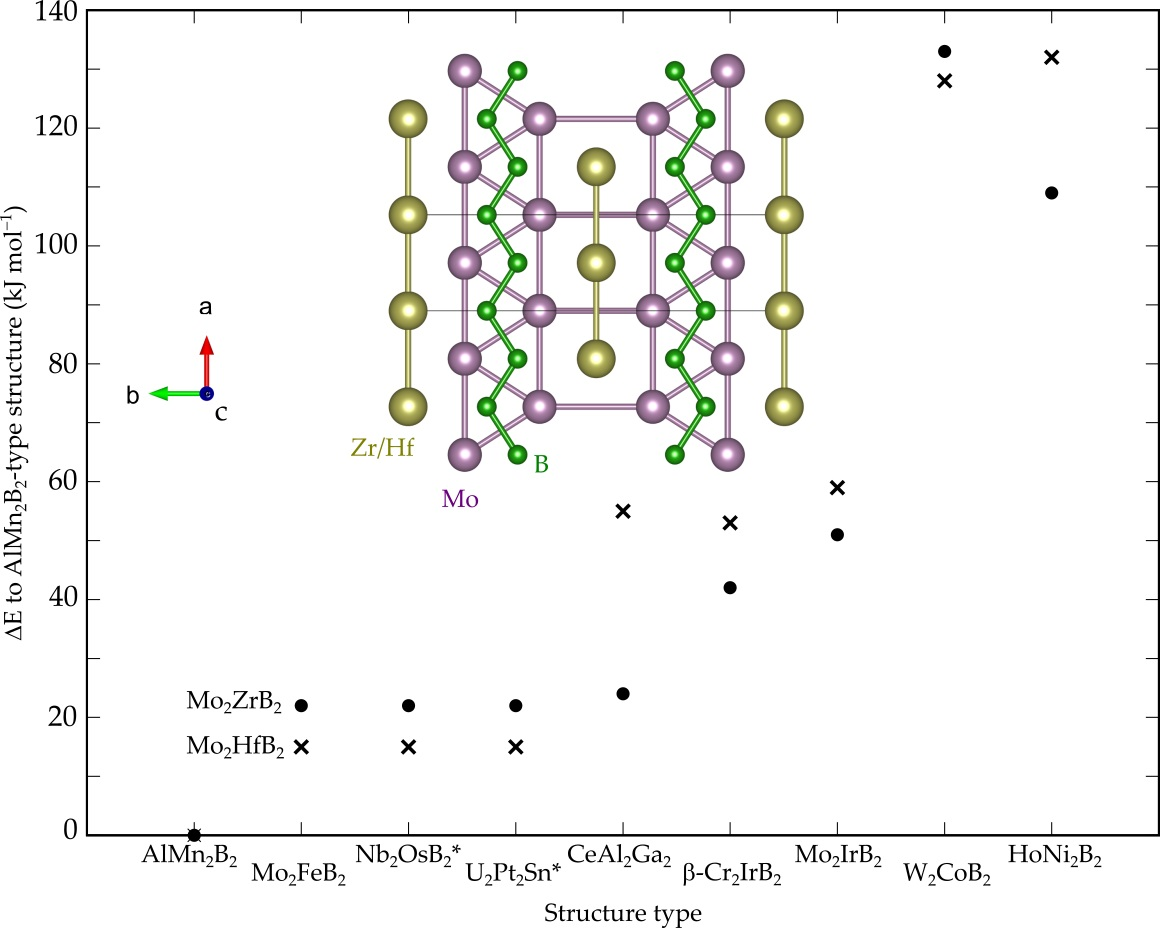
3.1. Crystal, Electronic and Phonon Structure
3.2. Elastic Properties
3.2.1. Isotropic Elastic Properties
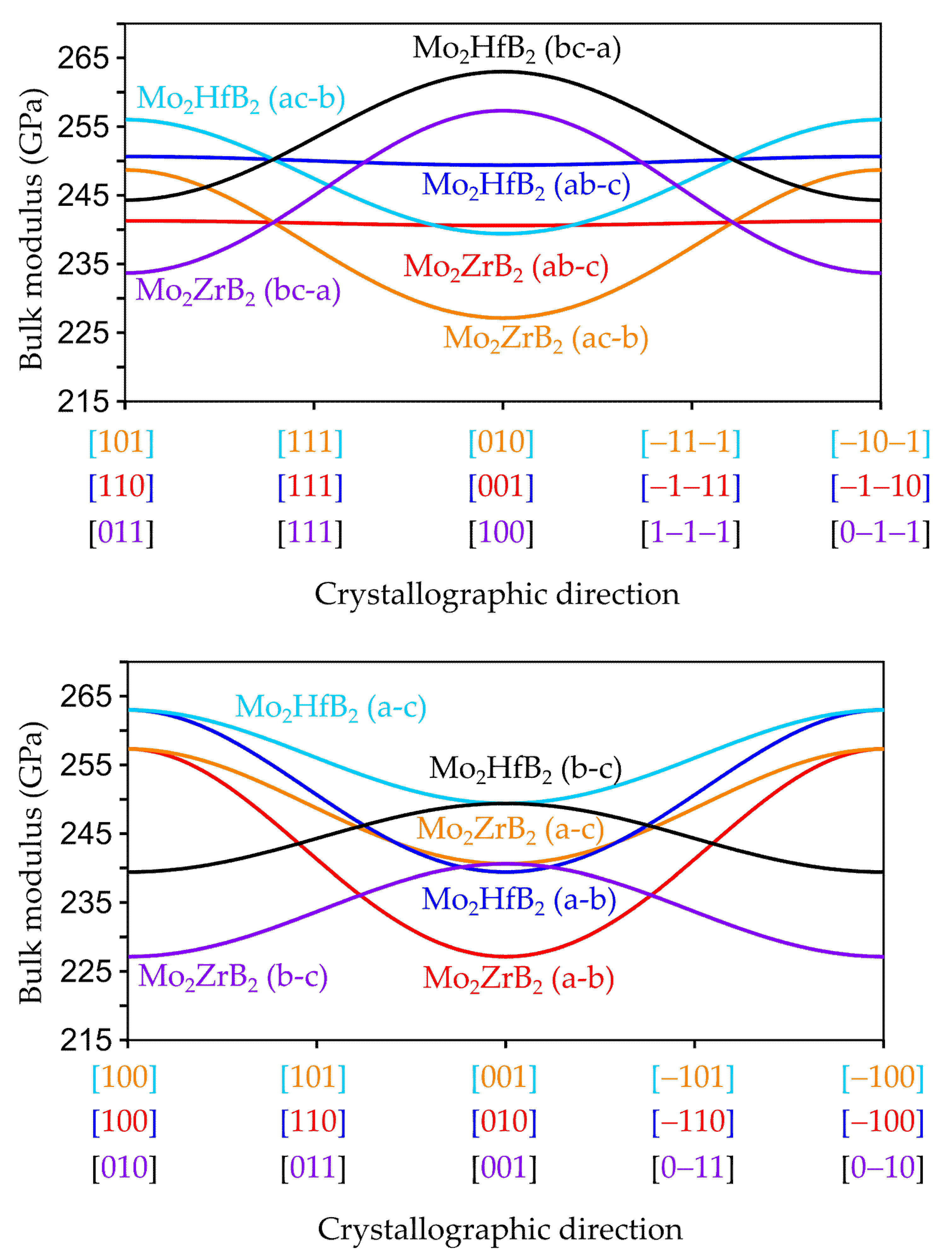
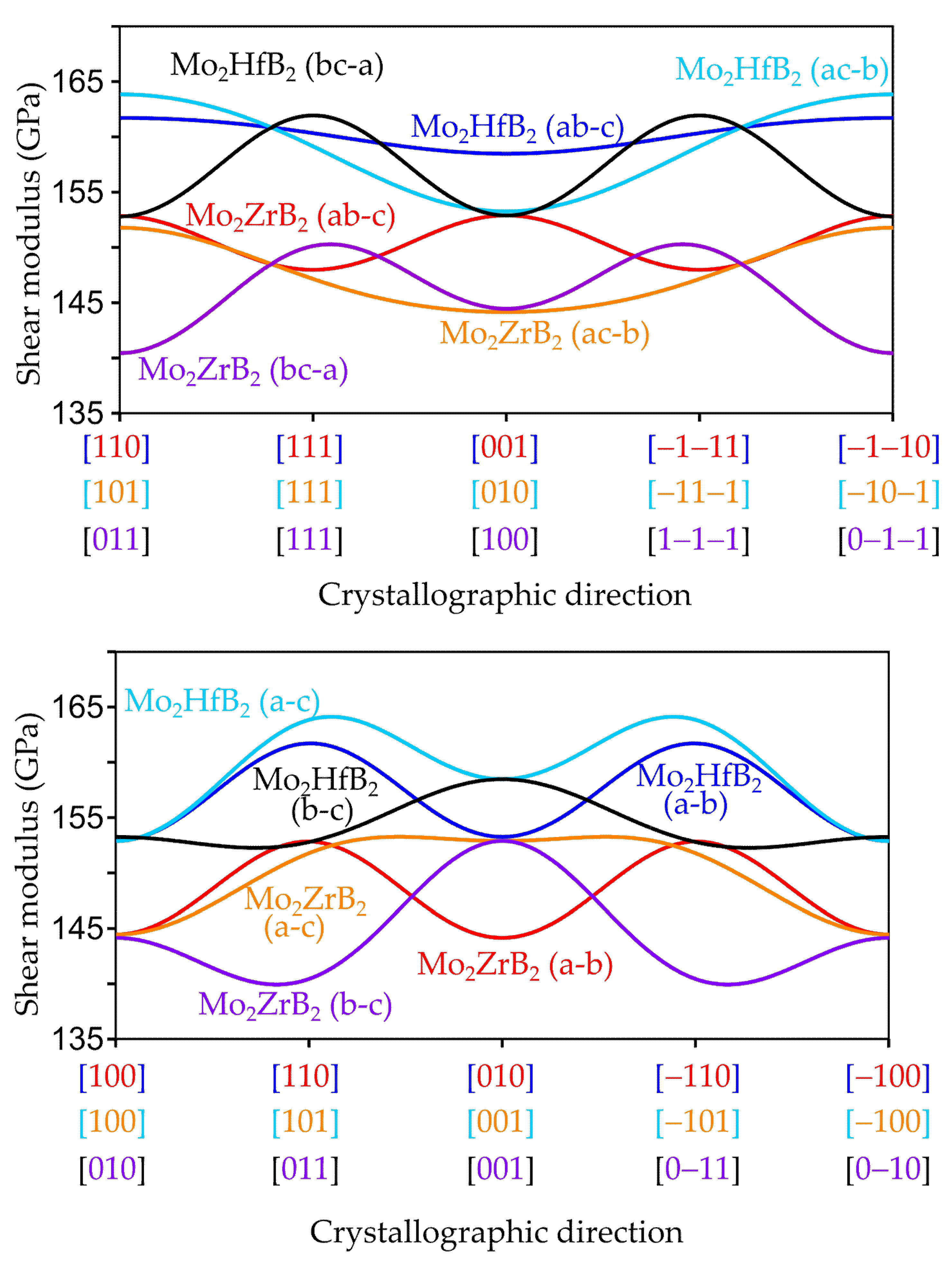
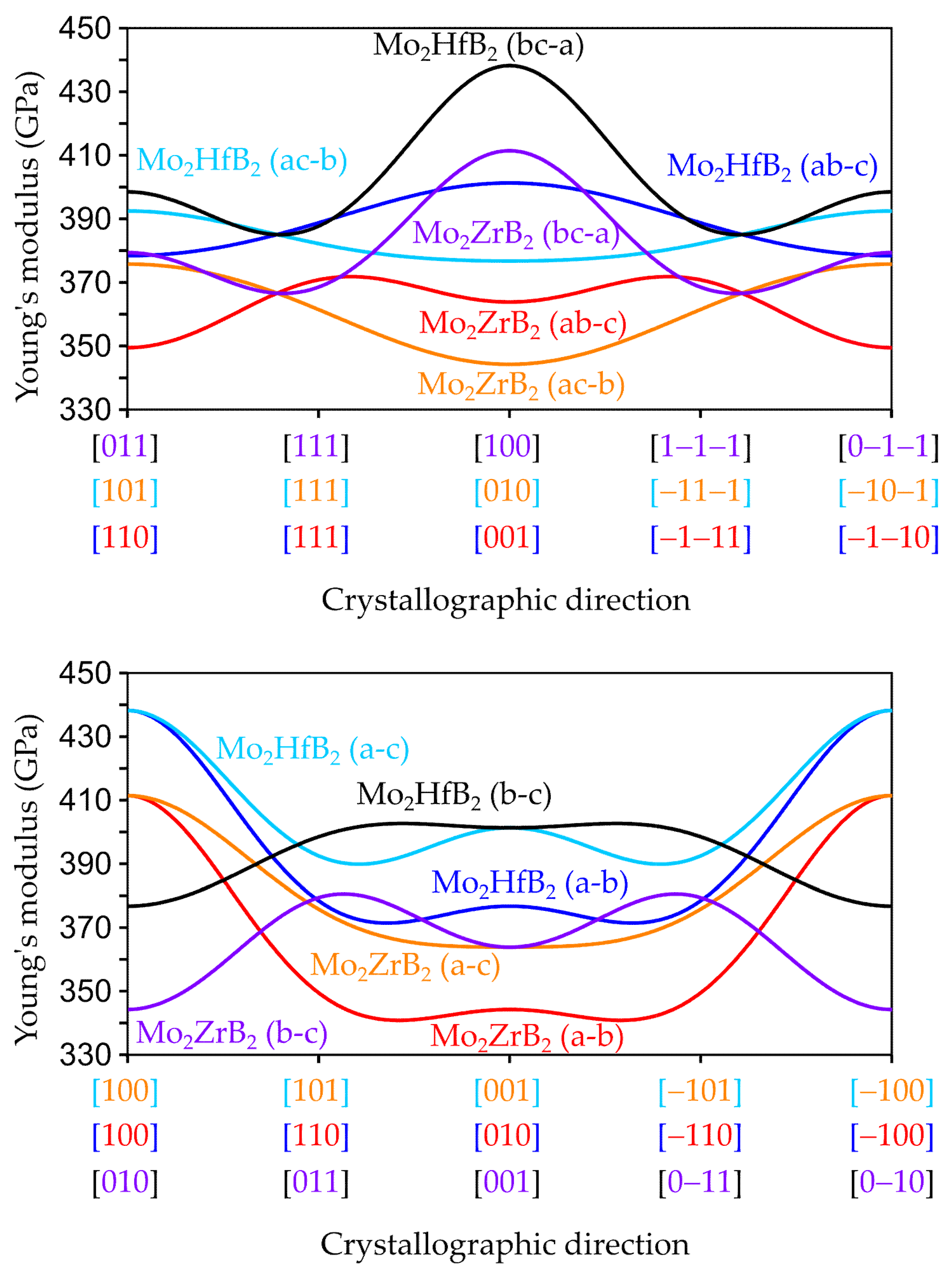
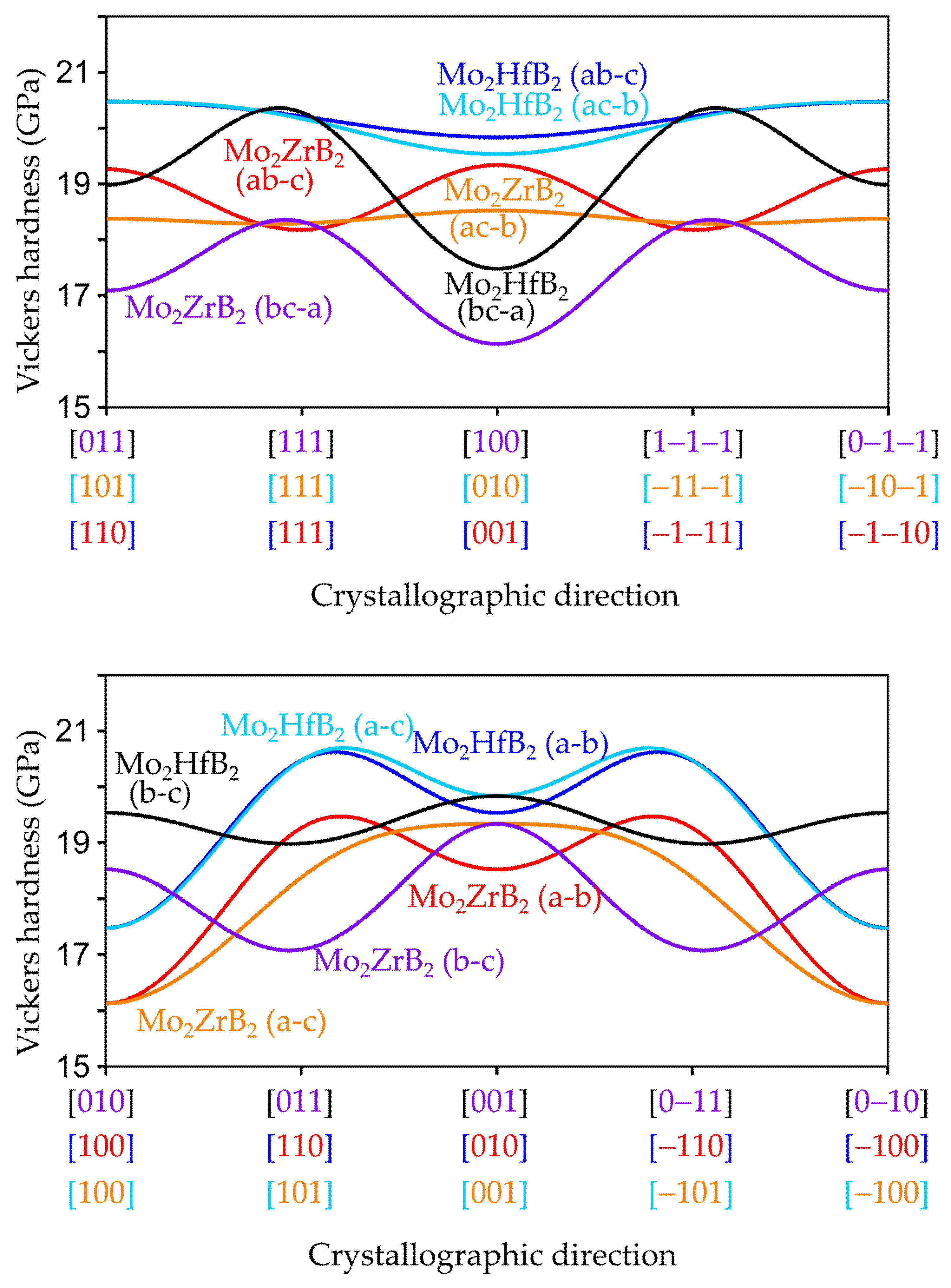
3.2.2. Anisotropic Elastic Properties
3.3. Influence of the Chemical Bonding on the Elastic Properties
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dimiduk, D.M.; Perepezko, J.H. Mo-Si-B Alloys: Developing a Revolutionary Turbine-Engine Material. MRS Bull. 2003, 28, 639–645. [Google Scholar] [CrossRef]
- Kruzic, J.J.; Schneibel, J.H.; Ritchie, R.O. Ambient- to elevated-temperature fracture and fatigue properties of Mo-Si-B alloys: Role of microstructure. Metall. Mater. Trans. A 2005, 36, 2393–2402. [Google Scholar] [CrossRef]
- Yu, X.J.; Kumar, K.S. The tensile response of Mo, Mo-Re and Mo-Si solid solutions. Int. J. Refract. Hard Met. 2013, 41, 329–338. [Google Scholar] [CrossRef]
- Perepezko, J.H. The hotter the engine, the better. Science 2009, 326, 1068–1069. [Google Scholar] [CrossRef]
- Rioult, F.A.; Imhoff, S.D.; Sakidja, R.; Perepezko, J.H. Transient oxidation of Mo-Si-B alloys: Effect of the microstructure size scale. Acta Mater. 2009, 57, 4600–4613. [Google Scholar] [CrossRef]
- Krüger, M.; Jain, P.; Kumar, K.S.; Heilmaier, M. Correlation between microstructure and properties of fine grained Mo-Mo3Si-Mo5SiB2 alloys. Intermetallics 2014, 48. [Google Scholar] [CrossRef]
- Schliephake, D.; Azim, M.; von Klinski-Wetzel, K.; Gorr, B.; Christ, H.J.; Bei, H.; George, E.P.; Heilmaier, M. High-temperature creep and oxidation behavior of Mo-Si-B alloys with high Ti contents. Metall. Mater. Trans. A Phys. Metall. Mater. Sci. 2014, 45, 1102–1111. [Google Scholar] [CrossRef]
- Bolbut, V.; Seils, S.; Boll, T.; Chassaing, D.; Krüger, M. Controversial discussion on the existence of the Hf and Zr monoborides and experimental proof by atom probe tomography. Materialia 2019, 6, 100322. [Google Scholar] [CrossRef]
- Bolbut, V.; Bogomol, I.; Loboda, P.; Krüger, M. Microstructure and mechanical properties of a directionally solidified Mo-12Hf-24B alloy. J. Alloys Compd. 2018, 735, 2324–2330. [Google Scholar] [CrossRef]
- Bolbut, V.; Bogomol, I.; Bauer, C.; Krüger, M. Gerichtet erstarrte Mo-Zr-B-Legierungen/ Directionally solidified Mo-Zr-B alloys. Materialwissenschaft und Werkstofftechnik 2017, 48, 1113–1124. [Google Scholar] [CrossRef]
- Smokovych, I.; Bolbut, V.; Krüger, M.; Scheffler, M. Tailored Oxidation Barrier Coatings for Mo-Hf-B and Mo-Zr-B Alloys. Materials 2019, 12, 2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogl, B. Boron-Molybdenum-Zirconium. In Numerical Data and Functional Relationships in Science and Technology; Effenberg, G., Madelung, O., Eds.; Springer: Berlin, Germany, 2010; pp. 72–82. [Google Scholar]
- Rogl, P. Boron-Hafnium-Molybdenum. In Refractory Metal Systems; Martienssen, W., Effenberg, G., Ilyenko, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 695–707. [Google Scholar]
- Bolbut, V. Development of Mo-Hf-B and Mo-Zr-B Alloys for High Temperature Application. Ph.D. Thesis, Otto-von-Guericke University Magdeburg, Magdeburg, Germany, 2018. [Google Scholar]
- Bolbut, V.; Seils, S.; Chassaing, D.; Boll, T.; Krüger, M. Assessment of ternary borides in the Mo-Hf-B and Mo-Zr-B systems and new description on phase equilibria. Unpublished work.
- Schmidt, M.U.; Englert, U. Prediction of crystal structures. J. Chem. Soc. Dalton Trans. 1996, 10, 2077–2082. [Google Scholar] [CrossRef]
- Kirkpatrick, S.; Gelatt, C.D., Jr.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becher, H.J.; Krogmann, K.; Peisker, E. Über das ternäre Borid Mn2AlB2. Z. Anorg. Allg. Chem. 1966, 344, 140–147. (In German) [Google Scholar] [CrossRef]
- Rieger, W.; Nowotny, H.; Benesovsky, F. Die Kristallstruktur von Mo2FeB2. Monatsh. Chem. 1964, 95, 1502–1503. (In German) [Google Scholar] [CrossRef]
- Mbarki, M.; Touzani, R.S.; Fokwa, B.P.T. Nb2OsB2, with a new twofold superstructure of the U3Si2 type: Synthesis, crystal chemistry and chemical bonding. J. Sol. Sta. Chem. 2013, 203, 304–309. [Google Scholar] [CrossRef]
- Gravereau, P.; Mirambet, F.; Chevalier, B.; Weill, F.; Fournès, L.; Laffargue, D.; Bourée, F.; Etourneau, J. Crystal structure of U2Pt2Sn: A new derivative of the tetragonal U3Si2-type structure. J. Mater. Chem. 1994, 4, 1893–1895. [Google Scholar] [CrossRef]
- Rogl, P. Über SE-Metall-Kobaltboride. Monatsh. Chem. 1973, 104, 1623–1631. (In German) [Google Scholar] [CrossRef]
- Kotzott, D.; Ade, M.; Hillebrecht, H. Synthesis and Crystal Structures of alpha- and beta-Modifications of Cr2IrB2 containing B4-Chains, τ-Borides Cr23−xIrxB6 and Cr2B. Sol. Sta. Sci. 2008, 10, 291–302. [Google Scholar] [CrossRef]
- Rogl, P.; Beneshovsky, F.; Nowotny, H. Über einige Komplexboride mit Platinmetallen. Monatsh. Chem. 1972, 103, 965–989. (In German) [Google Scholar] [CrossRef]
- Bruskov, V.A.; Gubich, I.B.; Kuz’ma, Y.B. Crystal structure of a new boride HoNi2B2. Kristallografiya 1991, 36, 1123–1125. [Google Scholar]
- Rieger, W.; Nowotny, H.; Benesovsky, F. Die Kristallstruktur von W2CoB2 und isotypen Phasen. Monatsh. Chem. 1966, 97, 378–382. (In German) [Google Scholar] [CrossRef]
- Vande Vondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Commun. Comput. Phys. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- Hutter, J.; Ianuzzi, M.; Schiffmann, F.; Vande Vondele, J. CP2k: Atomistic Simulations of Condensed Matter System. WIRES Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Lippert, G.; Hutter, J.; Parrinello, M. A hybrid Gaussian and plane wave density functional scheme. Mol. Phys. 1997, 92, 477–488. [Google Scholar] [CrossRef]
- Vande Vondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [Green Version]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [Green Version]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef] [Green Version]
- Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 2005, 114, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scripta Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Touzani, R.S.; Mbarki, M.; Chen, X.; Fokwa, B.P.T. Peierls-Distorted Ru-Chains and Boron Dumbbells in Nb2RuB2 and Ta2RuB2 from First-Principles Calculations and Experiments. Eur. J. Inorg. Chem. 2016, 25, 4104–4110. [Google Scholar] [CrossRef]
- Andersen, O.K.; Skriver, H.L.; Nohl, H.; Johansson, B. Electronic structure of transition metal compounds; ground-state properties of the 3d-monoxides in the atomic sphere approximation. Pure Appl. Chem. 1980, 52, 93–118. [Google Scholar] [CrossRef]
- Andersen, O.K.; Jepsen, O. Explicit, First-Principles Tight-Binding Theory. Phys. Rev. Lett. 1984, 53, 2571–2574. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blöchl, P. Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Dal Corso, A. Pseudopotentials periodic table: From H to Pu. Comput. Mat. Sci. 2014, 95, 337–350. [Google Scholar] [CrossRef]
- Marzari, N.; Vanderbilt, D.; De Vita, A.; Payne, M.C. Thermal Contraction and Disordering of the Al(110) Surface. Phys. Rev. Lett. 1999, 82, 3296–3299. [Google Scholar] [CrossRef] [Green Version]
- Thermo_pw is an Extension of the Quantum ESPRESSO (QE) Package Which Provides an Alternative Organization of the QE Work-flow for the Most Common Tasks. Available online: https://dalcorso.github.io/thermo_pw/ (accessed on 17 August 2020).
- Voigt, W. Lehrbuch der Kristallphysik; Springer: Berlin/Leipzig, Germany, 1928. (In German) [Google Scholar]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. Z. Angew Math. Mech. 1929, 9, 49–58. (In German) [Google Scholar] [CrossRef]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Met. Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals: Their Representation by Tensors and Matrices; Oxford University Press: New York, NY, USA, 1985. [Google Scholar]
- Wu, D.-H.; Wang, H.-C.; Wei, L.-T.; Pan, R.-K.; Tang, B.-Y. First-principles study of structural stability and elastic properties of MgPd3 and its hydride. J. Magnes. Alloy 2014, 2, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal Elastic Anisotropy Index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [Green Version]
- Touzani, R.S. Quantenchemische Untersuchung der Elastischen und Magnetischen Eigenschaften Einiger Übergangsmetallboride. Ph.D. Thesis, RWTH Aachen University, Aachen, Germany, 2016. (In German). [Google Scholar]
- Slater, J.C. Atomic Radii in Crystals. J. Chem. Phys. 1964, 41, 3199–3204. [Google Scholar] [CrossRef]
- Touzani, R.S.; Rehorn, C.W.G.; Fokwa, B.P.T. Influence of chemical bonding and mahnetism on elastic properties of the A2MB2 borides (A = Nb, Ta; M = Fe, Ru, Os) from first-principles calculations. Comput. Mater. Sci. 2015, 104, 52–59. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | a (Å) | b (Å) | c (Å) | V (Å3) |
|---|---|---|---|---|
| Mo2ZrB2 | 3.145 (3.146) | 12.59 (12.60) | 3.208 (3.208) | 127.1 (127.2) |
| Mo2HfB2 | 3.143 (3.141) | 12.33 (12.35) | 3.213 (3.212) | 124.6 (124.6) |
| Compound | B (GPa) | G (GPa) | Y (GPa) | HV (GPa) | AU ( ) |
|---|---|---|---|---|---|
| Mo2ZrB2 | 241.19 | 148.34 | 369.30 | 18.24 | 0.03 |
| Mo2HfB2 | 250.31 | 158.73 | 393.11 | 19.81 | 0.02 |
| Compound | B Anisotropy Index ( ) | G Anisotropy Index ( ) | Y Anisotropy Index ( ) | HV Anisotropy Index ( ) | B and G Averaged Anisotropy Index ( ) |
|---|---|---|---|---|---|
| Mo2ZrB2 | 0.13 | 0.10 | 0.21 | 0.21 | 0.12 |
| Mo2HfB2 | 0.10 | 0.08 | 0.18 | 0.18 | 0.09 |
| Bonds (Number) | Direction Along Axis | ICOHP of the Bonds in Mo2ZrB2 (eV) | ICOHP of the Bonds in Mo2HfB2 (eV) |
|---|---|---|---|
| B-B (2×) | ab | −6.56 | −6.67 |
| M-B (2×) | b | −2.51 | −2.79 |
| Mo-B, short (4×) | ac | −9.09 | −8.92 |
| Mo-B, long (2×) | bc | −3.80 | −3.74 |
| M-M, short (2×) | a | −1.54 | −1.66 |
| M-M, long (2×) | c | −1.51 | −1.58 |
| Mo-M (4×) | ab-c | −5.24 | −5.62 |
| Mo-Mo, short (2×) | ab | −2.23 | −2.13 |
| Mo-Mo, medium (2×) | a | −1.65 | −1.63 |
| Mo-Mo, long (2×) | c | −1.29 | −1.24 |
| Mo-Mo, longest (1×) | b | −0.26 | −0.36 |
| Sum | −35.68 | −36.33 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Touzani, R.S.; Krüger, M. First Principles Density Functional Theory Prediction of the Crystal Structure and the Elastic Properties of Mo2ZrB2 and Mo2HfB2. Crystals 2020, 10, 865. https://doi.org/10.3390/cryst10100865
Touzani RS, Krüger M. First Principles Density Functional Theory Prediction of the Crystal Structure and the Elastic Properties of Mo2ZrB2 and Mo2HfB2. Crystals. 2020; 10(10):865. https://doi.org/10.3390/cryst10100865
Chicago/Turabian StyleTouzani, Rachid Stefan, and Manja Krüger. 2020. "First Principles Density Functional Theory Prediction of the Crystal Structure and the Elastic Properties of Mo2ZrB2 and Mo2HfB2" Crystals 10, no. 10: 865. https://doi.org/10.3390/cryst10100865
APA StyleTouzani, R. S., & Krüger, M. (2020). First Principles Density Functional Theory Prediction of the Crystal Structure and the Elastic Properties of Mo2ZrB2 and Mo2HfB2. Crystals, 10(10), 865. https://doi.org/10.3390/cryst10100865