C-O Bond Hydrogenolysis of Aqueous Mixtures of Sugar Polyols and Sugars over ReOx-Rh/ZrO2 Catalyst: Application to an Hemicelluloses Extracted Liquor

 and
and
Abstract
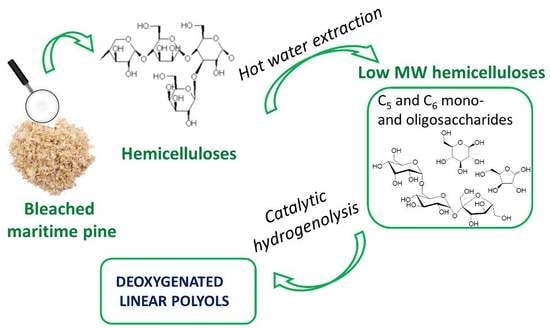
1. Introduction
2. Results and Discussions
2.1. Characterization of Catalysts
2.2. Hydrogenolysis of Polyols or Sugars Separately
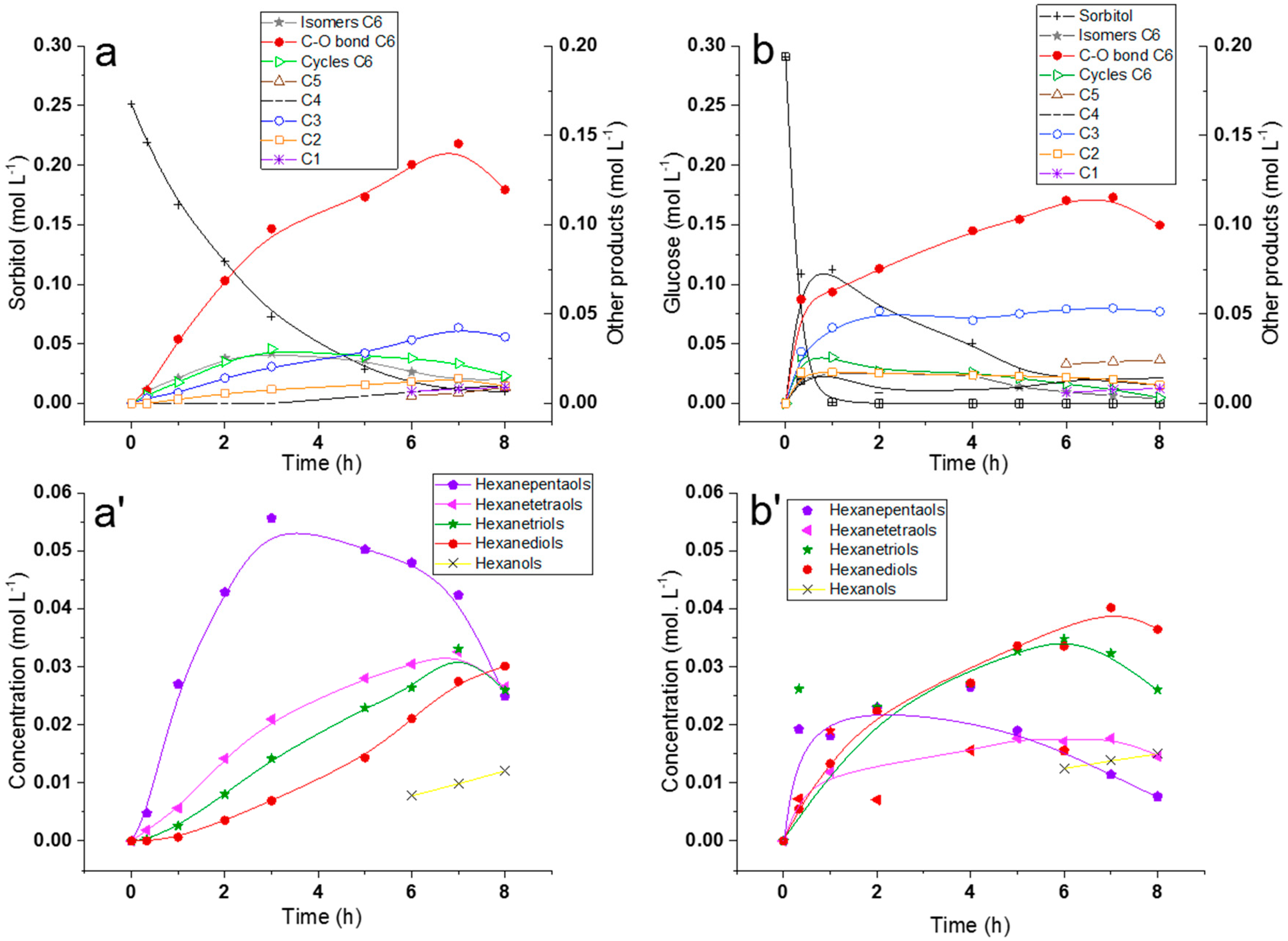
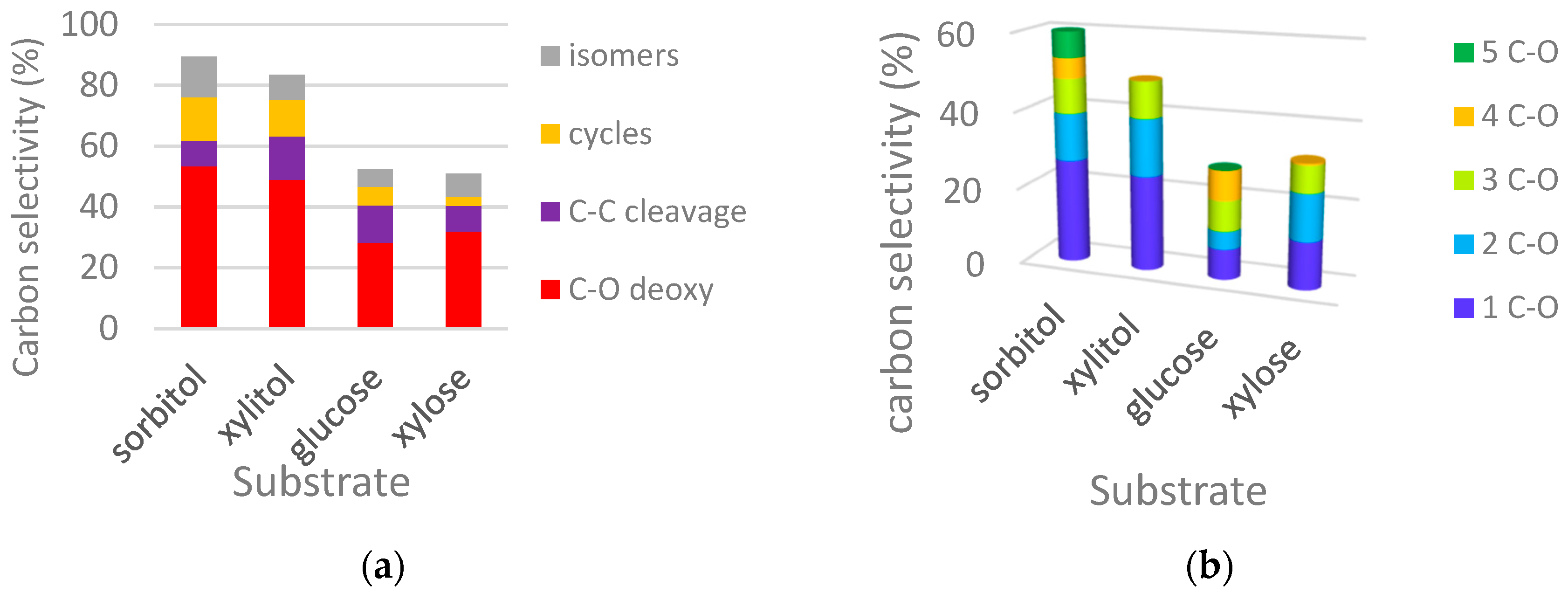
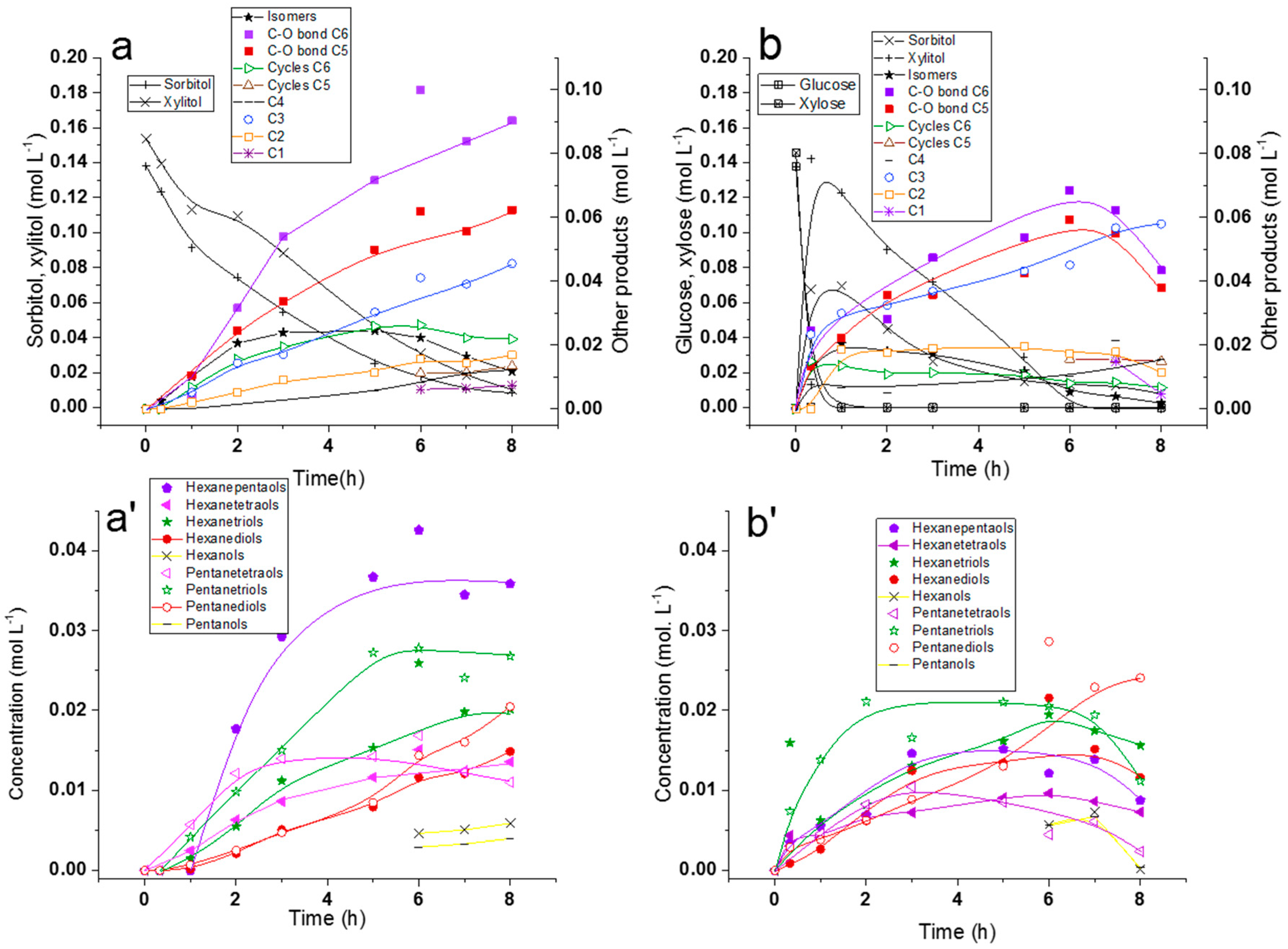
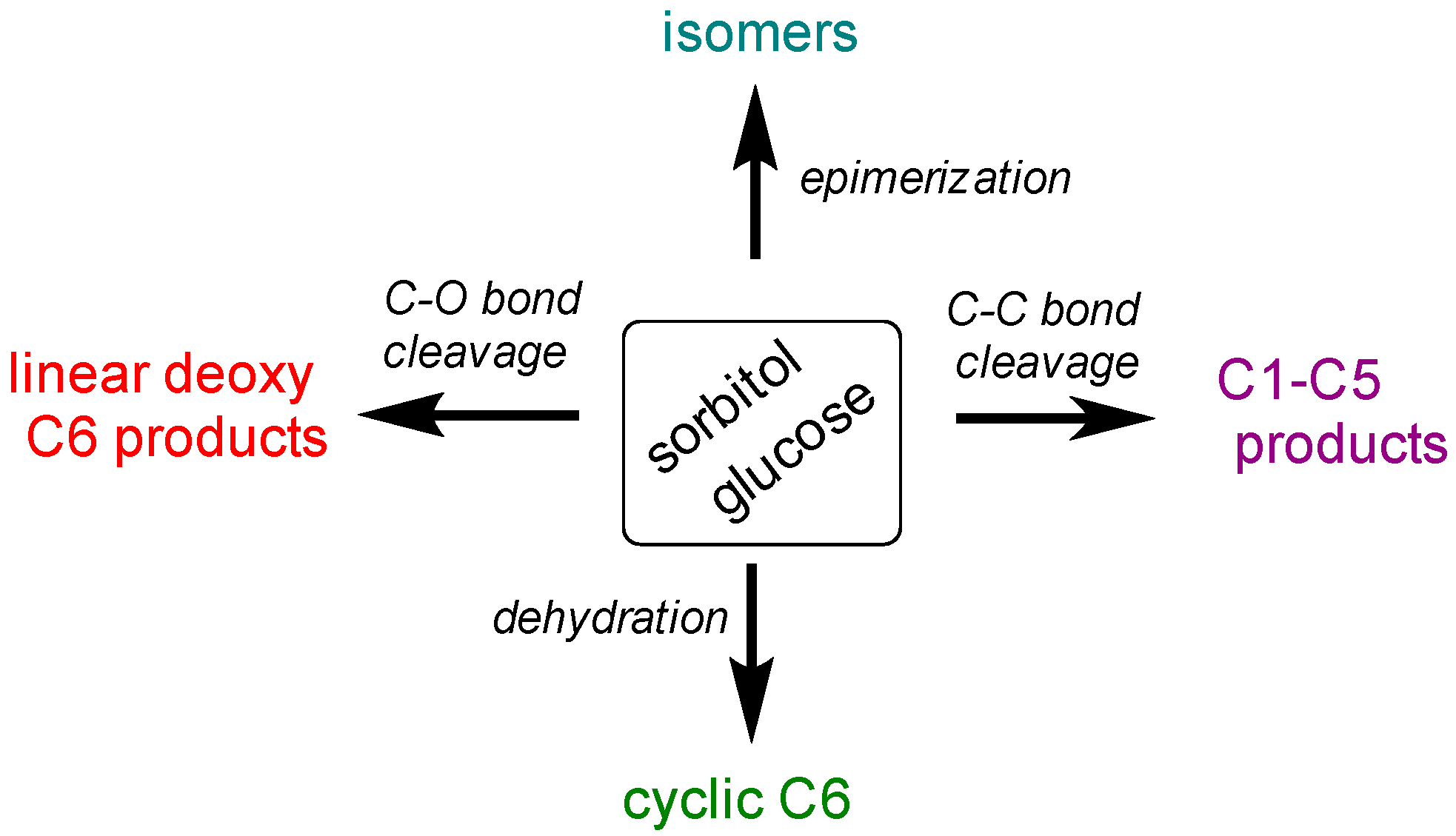
2.2.1. Comparison of Sorbitol and Glucose Hydrogenolysis
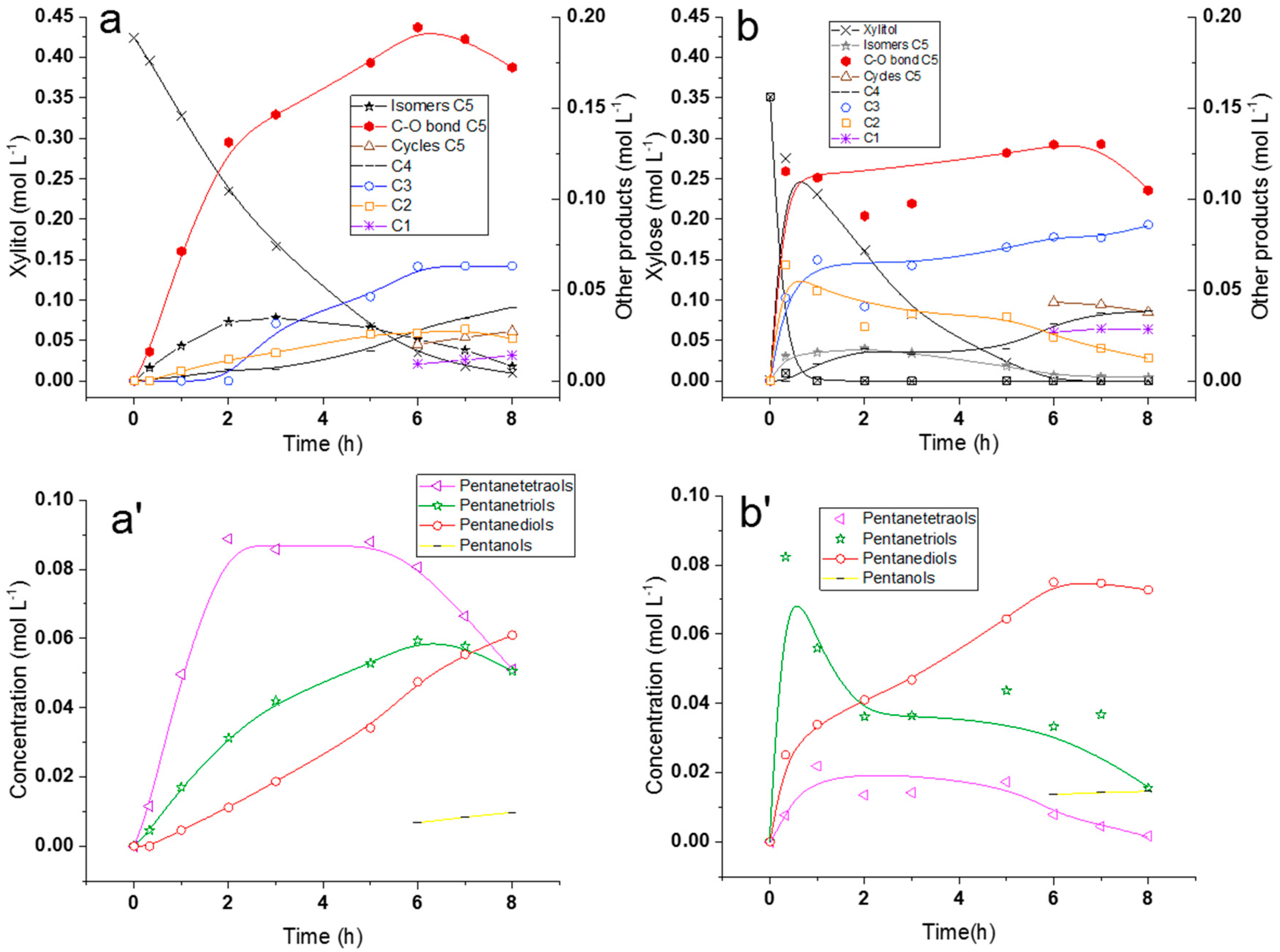
2.2.2. Comparison of Hydrogenolysis of Xylitol and Xylose
2.2.3. Discussion on Hydrogenolysis of Polyols vs. Hydrogenolysis of Sugars
2.3. Hydrogenolysis of Mixtures of Polyols and Sugars
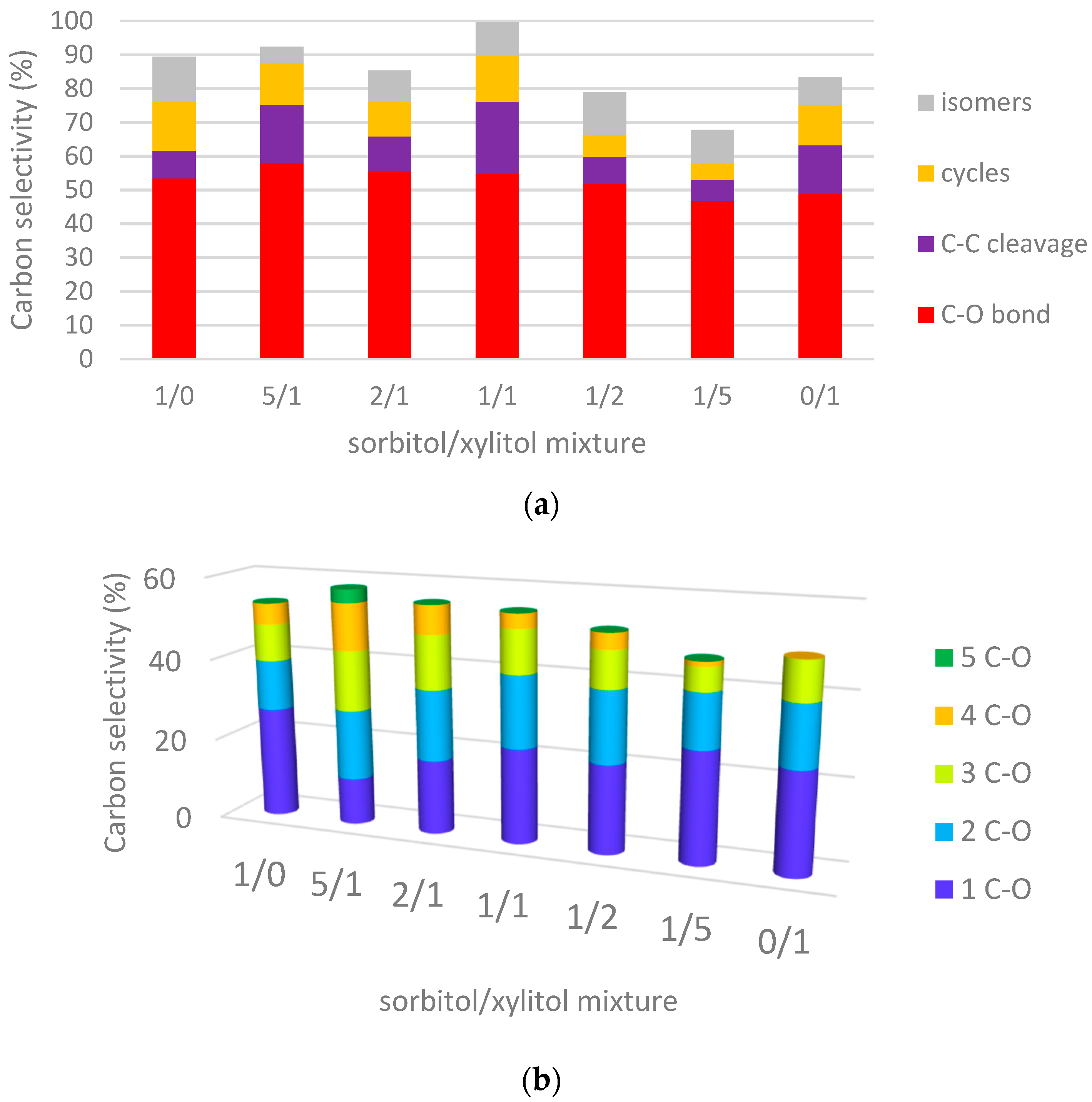
2.3.1. Hydrogenolysis of Sorbitol/Xylitol Mixtures
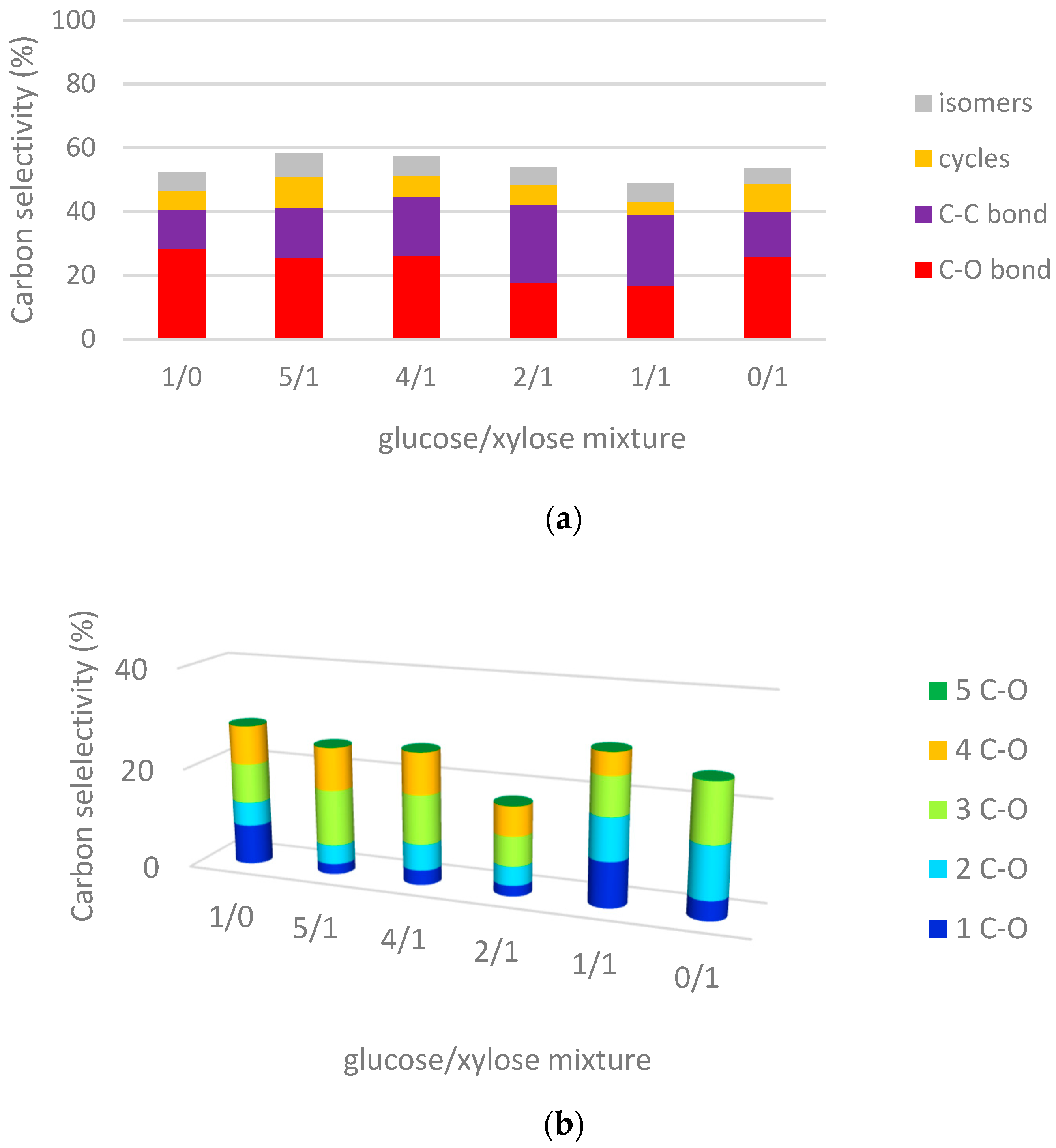
2.3.2. Hydrogenolysis of Mixtures of Glucose/Xylose
2.4. Hydrogenolysis of Sugar Isomers
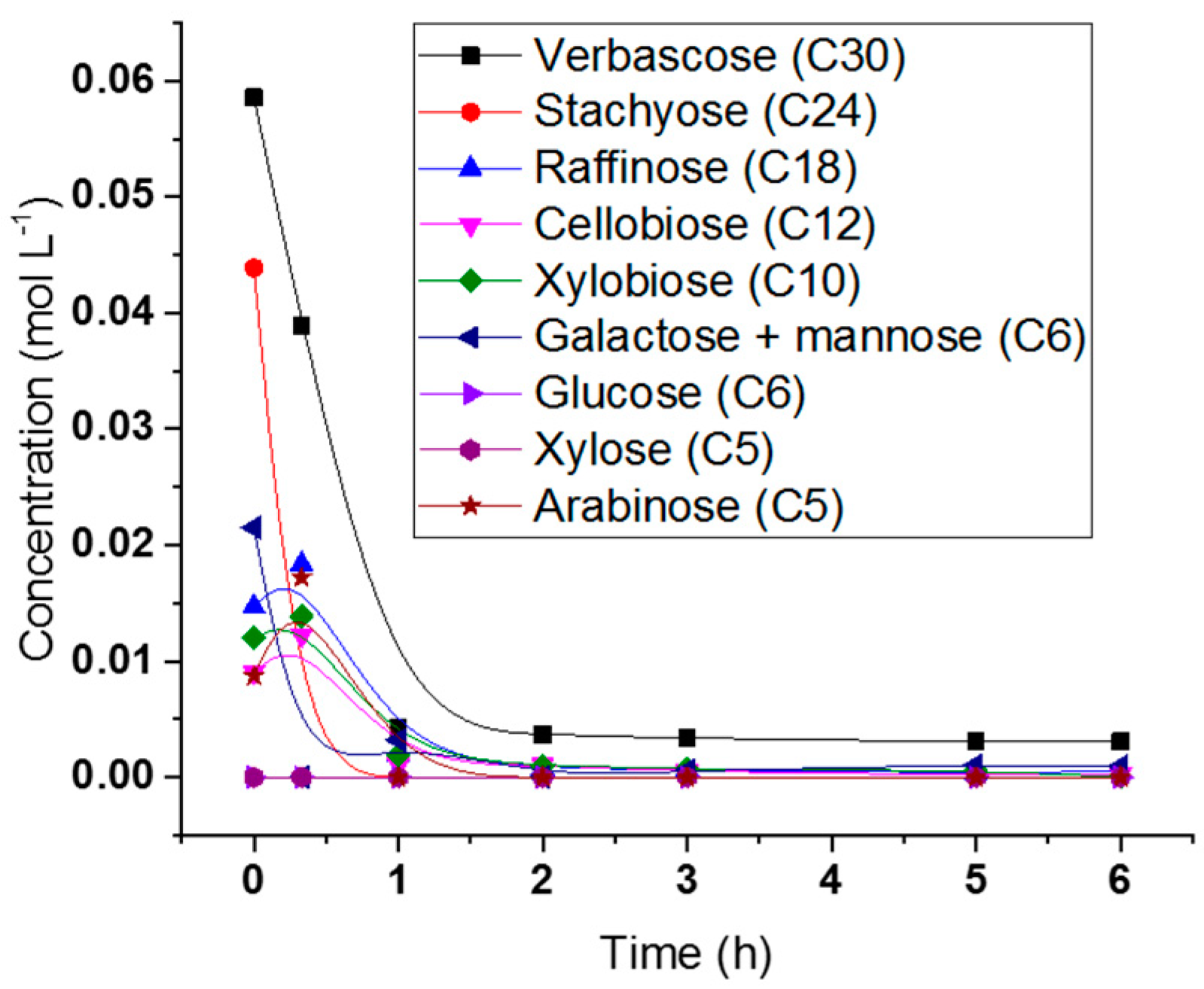
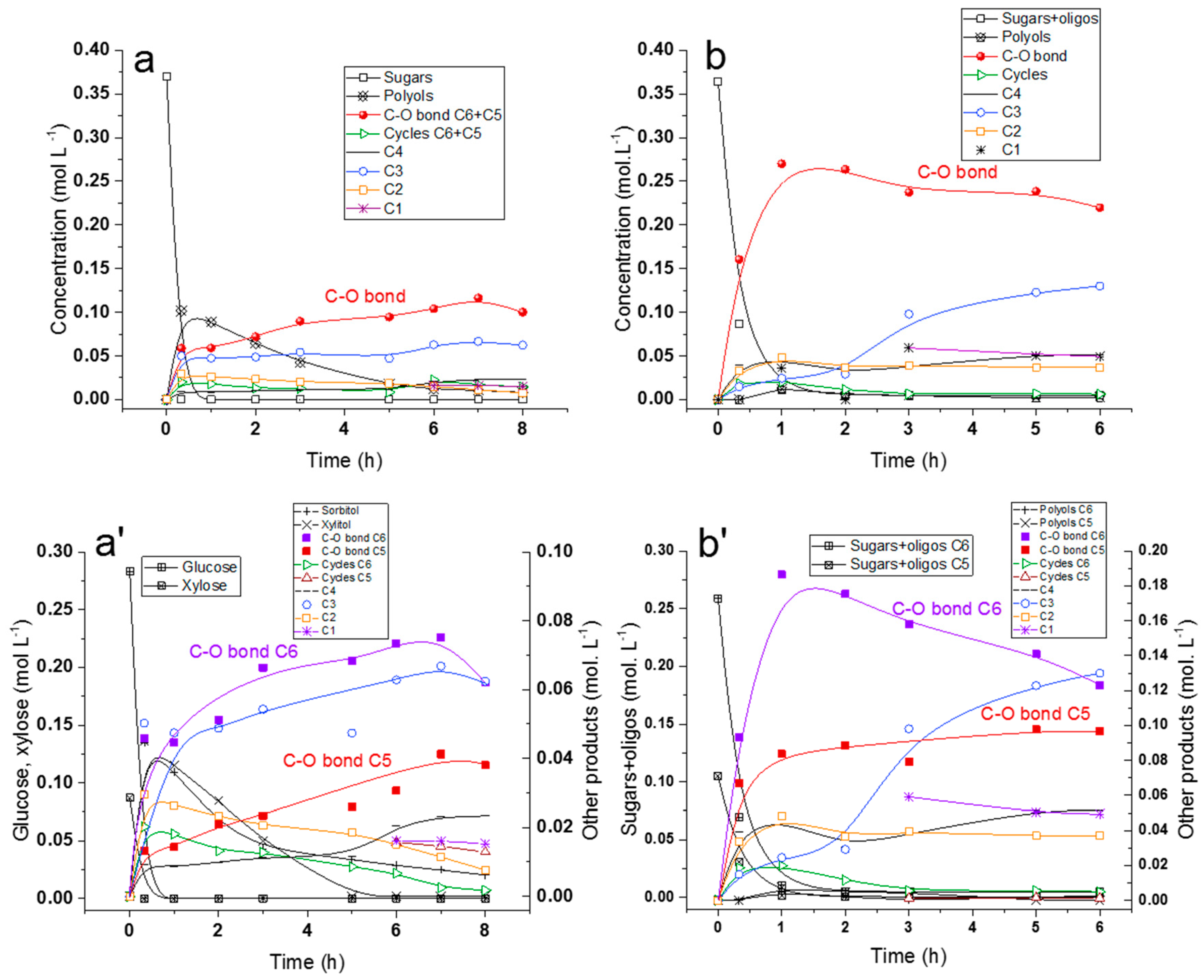
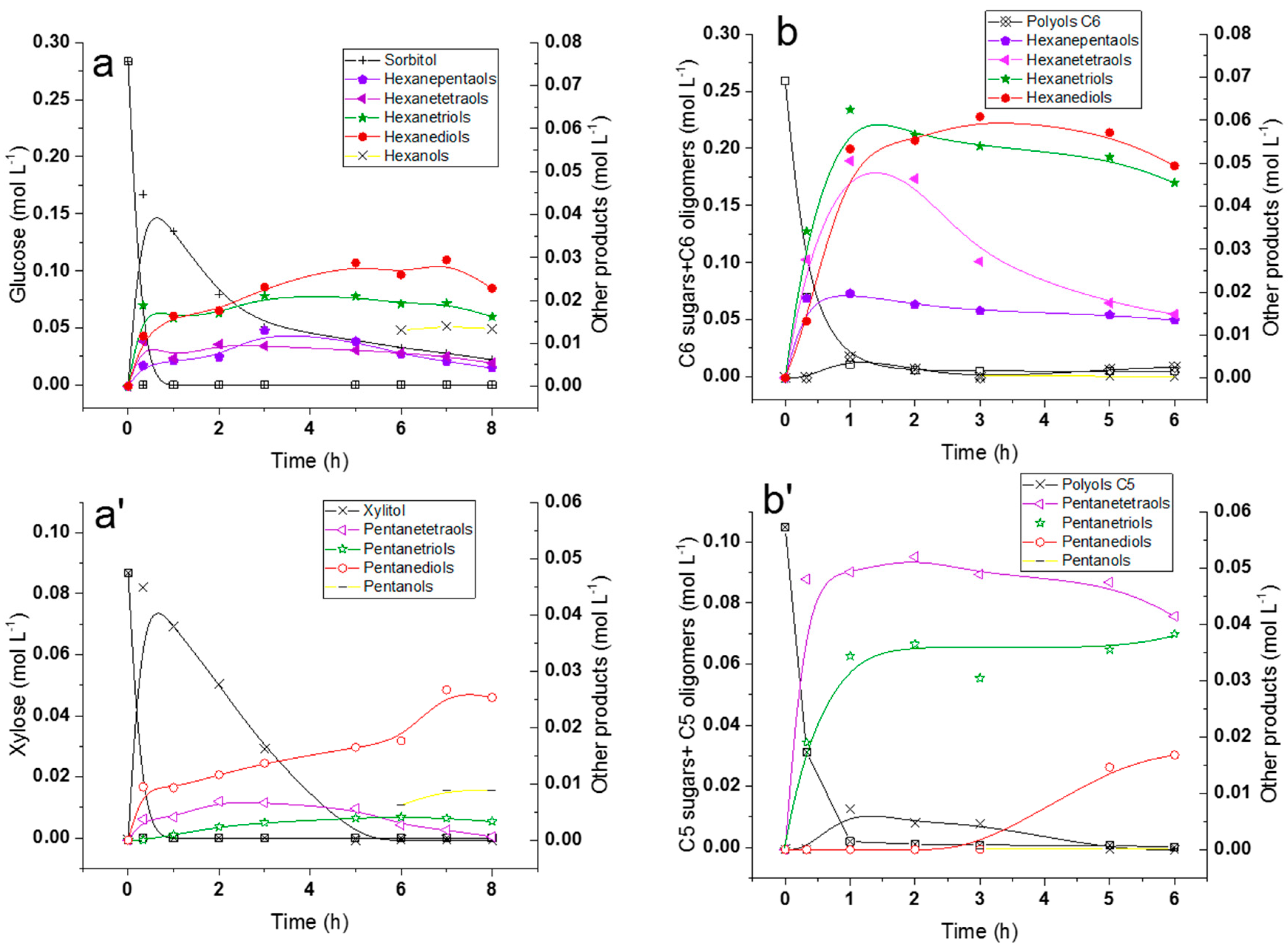
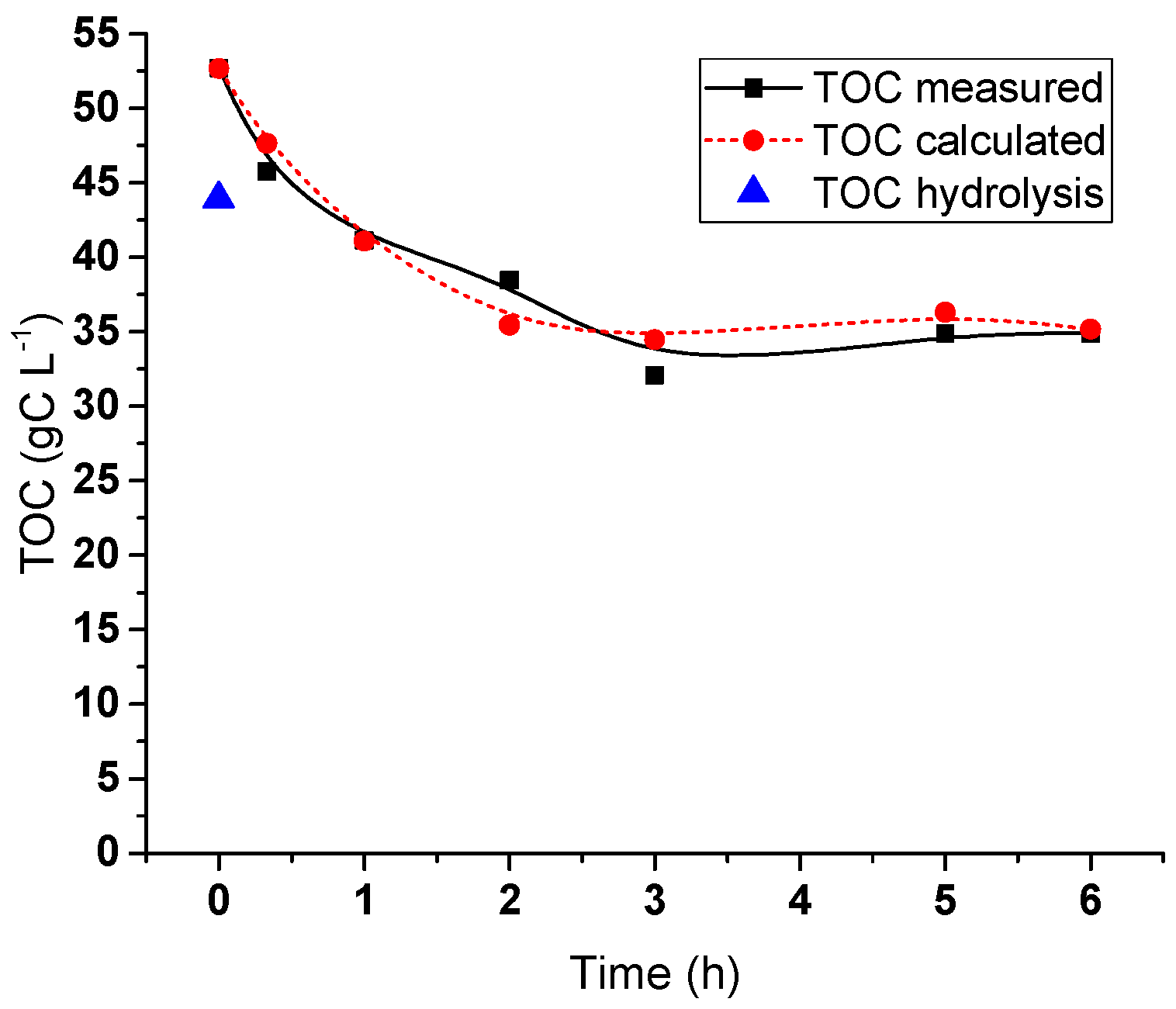
2.5. Hydrogenolysis of Hemicelluloses Liquor
3. Experimental
3.1. Catalyst Preparation
3.2. Maritime Pine Extraction and Hydrolysis
3.2.1. Extraction of Hemicelluloses
3.2.2. Analysis of Carbohydrates in Hemicellulose Extract
3.3. Catalyst Evaluation Experiments and Products Analysis
4. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- FitzPatrick, M.; Champagne, P.; Cunningham, M.F.; Whitney, R.A. A biorefinery prospective: Treatment of lignocellulosic materials for the production of value-added products. Bioresour. Technol. 2010, 101, 8915–8922. [Google Scholar] [CrossRef] [PubMed]
- Vennestrøm, P.N.R.; Osmundsen, C.M.; Christensen, C.H.; Taarning, E. Beyond petrochemicals: The renewable chemicals industry. Angew. Chem. Int. Ed. 2011, 50, 10502–10509. [Google Scholar] [CrossRef] [PubMed]
- Belgacem, M.N.; Gandini, A. Monomers, Polymers and Composites from Renewable Resources; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 978-0-08-045316-3. [Google Scholar]
- Corma, A.; Huber, G.W.; Sauvanaud, L.; O’Connor, P. Biomass to chemicals: Catalytic conversion of glycerol/water mixtures into acrolein, reaction network. J. Catal. 2008, 257, 163–171. [Google Scholar] [CrossRef]
- Lanfazame, P.; Barbera, K.; Papanikolaou, G.; Perathoner, S.; Centi, G.; Migliori, M.; Catizzone, E.; Giordano, G. Comparison of H+ and NH4+ forms of zeolites as acid catalysts for HMF etherification. Catal. Today 2018, 304, 97–102. [Google Scholar] [CrossRef]
- Sjöström, E. Wood Chemistry-Fundamentals and Applications, 2nd ed.; Academic Press: New York, NY, USA, 1993; ISBN 978-0-08-092589-9. [Google Scholar]
- Willför, S.; Sundberg, A.; Hemmings, J.; Holmbom, B. Polysaccharides in some industrially important softwood species. Wood Sci. Technol. 2005, 39, 245–258. [Google Scholar] [CrossRef]
- González-Muñoz, M.J.; Rivas, S.; Santos, V.; Parajó, J.C. Fractionation of extracted hemicellulosic saccharides from Pinus pinaster wood by multistep membrane processing. J. Membr. Sci. 2013, 428, 281–289. [Google Scholar] [CrossRef]
- González-Muñoz, M.J.; Rivas, S.; Santos, V.; Parajó, J.C. Aqueous processing of Pinus pinaster wood: Kinetics of polysaccharide breakdown. Chem. Eng. J. 2013, 231, 380–387. [Google Scholar] [CrossRef]
- Willför, S.; Sundberg, A.; Hemmings, J.; Holmbom, B. Polysaccharides in some industrially important hardwood species. Wood Sci. Technol. 2005, 39, 601–617. [Google Scholar] [CrossRef]
- Fradinho, D.M.; Pascola Neto, C.; Evtuguin, D.; Jorge, F.C.; Irle, M.A.; Gil, M.H.; Pedrosa de Jesus, J. Chemical characterisation of bark and of alkaline bark extracts from maritime pine grown in Portugal. Ind. Crops Prod. 2002, 16, 23–32. [Google Scholar] [CrossRef]
- Liu, S.; Lu, H.; Hu, R.; Shupe, A.; Lin, L.; Liang, B. A sustainable woody biomass biorefinery. Biotechnol. Adv. 2012, 30, 785–810. [Google Scholar] [CrossRef] [PubMed]
- Gürbüz, E.I.; Gallo, J.M.R.; Alonso, D.M.; Wettstein, S.G.; Lim, W.Y.; Dumesic, J.A. Conversion of Hemicellulose into Furfural Using Solid Acid Catalysts in γ-Valerolactone. Angew. Chem. Int. Ed. 2013, 52, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Whyman, C.E. Pretreatment: The key to unlocking low-cost cellulosic ethanol. Biofuel Bioprod. Bioref. 2008, 2, 26–40. [Google Scholar] [CrossRef]
- Timell, T.E. Recent progress in the chemistry of wood hemicelluloses. Wood Sci. Technol. 1967, 1, 45–70. [Google Scholar] [CrossRef]
- Song, T.; Pranovich, A.; Sumerskiy, I.; Holmbom, B. Extraction of galactoglucomannan from spruce wood with pressurised hot water. Holzforschung 2008, 62, 659–666. [Google Scholar] [CrossRef]
- Song, T.; Pranovich, A.; Holmbom, B. Characterisation of Norway spruce hemicelluloses extracted by pressurised hot-water extraction (ASE) in the presence of sodium bicarbonate. Holzforschung 2011, 65, 35–42. [Google Scholar] [CrossRef]
- Song, T.; Pranovich, A.; Holmbom, B. Effects of pH control with phthalate buffers on hot-water extraction of hemicelluloses from spruce wood. Bioresour. Technol. 2011, 102, 10518–10523. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qin, M.; Xu, C.; Chen, X. Hot water extraction of hemicelluloses from aspen wood chips of different sizes. BioResources 2013, 8, 5690–5700. [Google Scholar] [CrossRef]
- Bravo, C.; Garcés, D.; Faba, L.; Sastre, H.; Ordóñez, S. Selective arabinose extraction from Pinus sp. sawdust by two-step soft acid hydrolysis. Ind. Crops Prod. 2017, 104, 229–236. [Google Scholar] [CrossRef]
- Cebreiros, F.; Guigou, M.D.; Cabrera, M.N. Integrated forest biorefineries: Recovery of acetic acid as a by-product from eucalyptus wood hemicellulosic hydrolysates by solvent extraction. Ind. Crops Prod. 2017, 109, 101–108. [Google Scholar] [CrossRef]
- Martin-Sampedro, R.; Eugenio, M.E.; Moreno, J.A.; Revilla, E.; Villar, J.C. Integration of a kraft pulping mill into a forest biorefinery: Pre-extraction of hemicellulose by steam explosion versus steam treatment. Bioresour. Technol. 2014, 153, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Fišerová, M.; Opálená, E.; Illa, A. Hot water and oxalic acid pre-extraction of beech wood integrated with kraft pulping. Wood Res. 2013, 58, 381–394. [Google Scholar]
- Vena, P.F.; García-Aparicio, M.P.; Brienzo, M.; Görgens, J.F.; Rypstra, T. Effect of alkaline hemicellulose extraction on kraft pulp fibers from Eucalyptus Grandis. J. Wood Chem. Technol. 2013, 33, 157–173. [Google Scholar] [CrossRef]
- Sim, K.; Youn, H.J.; Cho, H.; Shin, H.; Lee, H.L. Improvements in pulp properties by alkali prextraction and subsequent kraft pulping with controlling H-factor and alkali charge. BioResources 2012, 7, 5864–5878. [Google Scholar] [CrossRef]
- Duarte, G.V.; Ramarao, B.V.; Amidon, T.E.; Ferreira, P.T. Effect of hot water extraction on hardwood kraft pulp fibers (Acer saccharum, sugar maple). Ind. Eng. Chem. Res. 2011, 50, 9949–9959. [Google Scholar] [CrossRef]
- Naidu, D.S.; Hlangothi, S.P.; John, M.J. Bio-based products from xylan: A review. Carbohydr. Polym. 2018, 179, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Belmokaddem, F.-Z.; Pinel, C.; Huber, P.; Petit-Conil, M.; da Silva Perez, D. Green Synthesis of xylan hemicellulose esters. Carbohydr. Res. 2011, 346, 2896–2904. [Google Scholar] [CrossRef]
- Bigand, V.; Pinel, C.; da Silva Perez, D.; Rataboul, F.; Huber, P.; Petit-Conil, M. Cationisation of galactomannan and xylan hemicelluloses. Carbohydr. Polym. 2011, 85, 138–149. [Google Scholar] [CrossRef]
- Bigand, V.; Pinel, C.; da Silva Perez, D.; Rataboul, F.; Petit-Conil, M.; Huber, P. Influence of liquid or solid phase preparation of cationic hemicelluloses on physical properties of paper. BioResources 2013, 8, 2118–2134. [Google Scholar] [CrossRef]
- Sedlmeyer, F.B. Xylan as by-product of biorefineries: Characteristics and potential use for food applications. Food Hydrocoll. 2011, 25, 1891–1898. [Google Scholar] [CrossRef]
- Willför, S.; Sundberg, K.; Tenkanen, M.; Holmbom, B. Spruce-derived mannans—A potential raw material for hydrocolloids and novel advanced natural materials. Carbohydr. Polym. 2008, 72, 197–210. [Google Scholar] [CrossRef]
- Dax, D.; Eklund, P.; Hemming, J.; Sarfraz, J.; Backman, P.; Xu, C.; Willför, S. Amphiphilic spruce galactoglucamannan derivatives based on naturally-occurring fatty acids. BioResources 2013, 8, 3771–3790. [Google Scholar] [CrossRef]
- Marinkovic, S.; Estrine, B. Direct conversion of wheat bran hemicelluloses into n-decyl-pentosides. Green Chem. 2010, 12, 1929–1932. [Google Scholar] [CrossRef]
- Sanglard, M.; Chirat, C.; Jarman, B.; Lachenal, D. Biorefinery in a pulp mill: Simultaneous production of cellulosic fibers from Eucalyptus globulus by soda-anthraquinone cooking and surface-active agents. Holzforschung 2013, 67, 481–488. [Google Scholar] [CrossRef]
- Hansen, N.M.L.; Plackett, D. Sustainable films and coatings from hemicelluloses: A review. Biomacromolecules 2008, 9, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Eckerman, C.; Smeds, A.; Reunanen, M.; Eklund, P.C.; Sjöholm, R.; Willför, S. Carboxymethylated spruce galactoglucomannas: Preparation, characterization, dispersion stability, water-in-oil emulsion stability, and sorption on cellulose surface. Nord. Pulp. Pap. Res. J. 2011, 26, 167–178. [Google Scholar] [CrossRef]
- Mikkonen, K.S.; Tenkanen, M. Sustainable food-packaging materials based on future biorefinery products: Xylans and mannans. Trends Food Sci. Technol. 2012, 28, 90–102. [Google Scholar] [CrossRef]
- Palm, M.; Zacchi, G. Extraction of hemicellulosic oligosaccharides from spruce using microwve oven or steam tretment. Biomacromolecules 2003, 4, 617–623. [Google Scholar] [CrossRef]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef]
- Morais, A.R.C.; Matuchaki, M.D.D.J.; Andreaus, J.; Bogel-Lukasik, R. A green and efficient approach to selective conversion of xylose and biomass hemicellulose into furfural in aqueous media using high-pressure CO2 as a sustainable catalyst. Green Chem. 2016, 18, 2985–2994. [Google Scholar] [CrossRef]
- Wei, L.; Shrestha, A.; Tu, M.; Adhikari, S. Effects of surfactant on biochemical and hydrothermal conversion of softwood hemicellulose to ethanol and furan derivatives. Process Biochem. 2011, 46, 1785–1792. [Google Scholar] [CrossRef]
- Boucher, J.; Chirat, C.; Lachenal, D. Extraction of hemicelluloses from wood in a pulp biorefinery, and subsequent fermentation into ethanol. Energy Convers. Manag. 2014, 88, 1120–1126. [Google Scholar] [CrossRef]
- Yi, G.; Zhang, Y. One-Pot Selective Conversion of hemicellulose (Xylan) to xylitol under mild Conditions. ChemSusChem 2012, 5, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Murzin, D.Y.; Kusema, B.; Murzina, E.V.; Aho, A.; Tokarev, A.; Boymirzaev, A.S.; Wärna, J.; Dapsens, P.Y.; Mondelli, C.; Perez-Ramirez, J.; et al. Hemicellulose arabinogalactan hydrolytic hydrogenation over Ru-modified H-USY zeolites. J. Catal. 2015, 330, 93–105. [Google Scholar] [CrossRef]
- Tathod, A.P.; Dhepe, P.L. Towards efficient synthesis of sugar alcohols from mono-and poly-saccharides: Role of metals, supports & promoters. Green Chem. 2014, 16, 4944–4954. [Google Scholar] [CrossRef]
- Derrien, E.; Ahmar, M.; Martin-Sisteron, E.; Raffin, G.; Queneau, Y.; Marion, P.; Beyerle, M.; Pinel, C.; Besson, M. Oxidation of aldoses contained in softwood hemicellulose acid hydrolysates into aldaric acids under alkaline or non-controlled pH conditions. Ind. Eng. Chem. Res. 2018, 57, 4543–4552. [Google Scholar] [CrossRef]
- Sanborn, A.; Binder, T. Synthesis of R-Glucosides, Sugar Alcohols, Reduced Sugar Alcohols, and Furan Derivatives of Reduced Sugar Alcohols. U.S. Patent WO2015/156806, 9 July 2015. [Google Scholar]
- Cao, Y.; Niu, W.; Guo, J.; Xian, M.; Liu, H. Biotechnological production of 1,2,4-butanetriol: An efficient process to synthesize energetic material precursor from renewable biomass. Sci. Rep. 2015, 5, 18149–18159. [Google Scholar] [CrossRef]
- Mueller, H.; Mesch, W.; Broellos, K. Preparation of 1,2,4-Butanetriol. U.S. Patent US4973769, 27 November 1990. [Google Scholar]
- Hasegawa, R.; Hayashi, K. Polyester Containing Impure 1,2-Butanediol. U.S. Patent US 4596886, 24 June 1986. [Google Scholar]
- Espro, C.; Gumina, B.; Szumelda, T.; Paone, E.; Mauriello, F. Catalytic transfer hydrogenolysis as an effective tool for the reductive upgrading of cellulose, hemicellulose, lignin, and their derived molecules. Catalysts 2018, 8, 313. [Google Scholar] [CrossRef]
- Furikado, I.; Miyazawa, T.; Koso, S.; Shimao, A.; Kunimori, K.; Tomishige, K. Catalytic performance of Rh/SiO2 in glycerol reaction under hydrogen. Green Chem. 2007, 9, 582–588. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Weinberg, K.; Palkovits, R. Hydrogenolysis goes bio: From carbohydrates and sugar alcohols to platform chemicals. Angew. Chem. Int. Ed. 2012, 51, 2564–2601. [Google Scholar] [CrossRef]
- Blanc, B.; Bourrel, A.; Gallezot, P.; Haas, T.; Taylor, P. Starch-derived polyols for polymer technologies: Preparation by hydrogenolysis on metal catalysts. Green Chem. 2000, 2, 89–91. [Google Scholar] [CrossRef]
- Kühne, B.; Vogel, H.; Meusinger, R.; Kunz, S.; Kunz, M. Mechanistic study on –C–O–and –C–C– hydrogenolysis over Cu catalysts: Identification of reaction pathways and key intermediates. Catal. Sci. Technol. 2018, 8, 755–767. [Google Scholar] [CrossRef]
- Tomishige, K.; Nakagawa, Y.; Tamura, M. Selective hydrogenolysis and hydrogenation using metal catalysts directly modified with metal oxide species. Green Chem. 2017, 19, 2876–2924. [Google Scholar] [CrossRef]
- Chia, M.; Pagan-Torres, H.J.; Hibbits, D.; Davis, R.J.; Dumesic, J.A. Selective hydrogenolysis of polyols and cyclic ethers over bifunctional surface sites on rhodium-rhenium catalysts. J. Am. Chem. Soc. 2011, 133, 12675–12689. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Mori, K.; Watanabe, H.; Tomishige, K. C-O bond hydrogenolysis of cyclic ethers with OH groups over rhenium-modified supported iridium catalysts. J. Catal. 2012, 294, 171–183. [Google Scholar] [CrossRef]
- Behr, A.; Eilting, J.; Irawadi, K.; Lescinski, J.; Lindner, F. Improved utilisation of renewable resources: New important derivatives of glycerol. Green Chem. 2008, 10, 13–20. [Google Scholar] [CrossRef]
- Zhou, C.-H.; Beltrami, J.N.; Fan, Y.-X.; Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Ten Dam, J.; Hanefeld, U. Renewable chemicals: Dehydroxylation of glycerol and polyols. ChemSusChem 2011, 4, 1017–1034. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Tomishige, K. Heterogeneous catalysis of the glycerol hydrogenolysis. Catal. Sci. Technol. 2011, 1, 179–190. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Tamura, M.; Tomishige, K. Catalytic materials for the hydrogenolysis of glycerol to 1,3-propanediol. J. Mater. Chem. A 2014, 2, 6688–6702. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of biomass into chemicals over metal catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Amada, Y.; Watanabe, H.; Hirai, Y.; Kajikawa, Y.; Nakagawa, Y.; Tomishige, K. Production of butanediols by the hydrogenolysis of erythritol. ChemSusChem 2012, 5, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Tamura, M.; Yuan, Z.; Nakagawa, Y.; Tomishige, K. One-pot conversion of sugar and sugar polyols to n-alkanes without C-C dissociation over the Ir-ReOx/SiO2 catalyst combined with H-ZSM65. ChemSusChem 2013, 6, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Okuyama, Y.; Tamura, M.; Nakagawa, Y.; Imai, A.; Tomishige, K. Selective transformation of hemicellulose (xylan) into n-pentane, pentanols or xylitol over a rhenium-modified iridium catalyst combined with acids. Green Chem. 2016, 16, 165–175. [Google Scholar] [CrossRef]
- Said, A.; Da Silva Perez, D.; Perret, N.; Pinel, C.; Besson, M. Selective C−O Hydrogenolysis of erythritol over Supported Rh-ReOx Catalysts in the Aqueous Phase. ChemCatChem 2017, 9, 2768–2783. [Google Scholar] [CrossRef]
- Sadier, A.; Perret, N.; Da Silva Perez, D.; Besson, M.; Pinel, C. Effect of carbon chain length on catalytic C-O bond cleavage of polyols over Rh-ReOx/ZrO2 in aqueous phase. Appl. Catal. A Gen. 2019. [Google Scholar] [CrossRef]
- Rivière, M.; Perret, N.; Cabiac, A.; Delcroix, D.; Pinel, C.; Besson, M. Xylitol hydrogenolysis over ruthenium-based catalysts: Effect of alkaline promoters and basic oxide-modified catalysts. ChemCatChem 2017, 9, 2145–2159. [Google Scholar] [CrossRef]
- Kanie, K.; Akiyama, K.; Iwamoto, M. Reaction pathways of glucose and fructose on Pt nanoparticles in subcritial water under a hydroge a hydrogen atmosphere. Catal. Today 2011, 178, 58–63. [Google Scholar] [CrossRef]
- Lazaridis, P.A.; Karakoulia, S.A.; Teodorescu, C.; Apostol, N.; Macovei, D.; Panteli, A.; Delimitis, A.; Coman, S.M.; Parvulecu, V.I.; Triantafyllidis, K.S. High hexitols selectivity in cellulose hydrolytic hydrogenation over platinum (Pt) vs. ruthenium (Ru) catalysts supported on micro/mesoporous carbon. Appl. Catal. B Environ. 2017, 214, 1–14. [Google Scholar] [CrossRef]
- Cerning-Béroard, J.; Filiatre-Verel, A. Characterization and distribution of soluble and insoluble carbohydrates in lupin seeds. Z. Lebensm. Unters. Forchung 1980, 171, 281–285. [Google Scholar] [CrossRef]
- Rivas, S.; Raspolli-Galletti, A.M.; Antonetti, C.; Santos, V.; Parajo, J.C. Sustainable conversion of Pinus pinaster wood into biofuel precursors: A biorefinery approach. Fuel 2016, 164, 51–58. [Google Scholar] [CrossRef]
- Ahlgren, P.A.; Goring, D.A.I. Removal of wood components during chlorite delignification of black spruce. Can. J. Chem. 1971, 49, 1272–1275. [Google Scholar] [CrossRef]
- Buranov, A.U.; Mazza, G. Extraction and characterization of hemicelluloses from flax shives by different methods. Carbohydr. Polym. 2010, 79, 17–25. [Google Scholar] [CrossRef]
- Sifontes Herrera, V.A.; Saleem, F.; Kusema, B.; Eränen, K.; Salmi, T. Hydrogenation of L-arabinose and D-galactose mixtures over a heterogeneous Ru/C catalyst. Top. Catal. 2012, 55, 550–555. [Google Scholar] [CrossRef]
- Kusema, B.T.; Xu, C.; Mäki-Arvela, P.; Willför, S.; Holmbom, B.; Salmi, T.; Murzin, D.Y. Kinetics of acid hydrolysis of arabinogalactans. Int. J. Chem. Reactor Eng. 2010, 8, 1–16. [Google Scholar] [CrossRef]
- Grénman, H.; Eränen, K.; Krogell, J.; Willför, S.; Salmi, T.; Murzin, D.Y. Kinetics of aqueous extraction of hemicelluloses from spruce in an intensified reactor system. Ind. Eng. Chem. Res. 2011, 50, 3818–3828. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon Selectivity to Products (%) | Glucose | Mannose | Galactose | Xylose | Arabinose |
|---|---|---|---|---|---|
| C-O bond scission | 26.3 | 34.3 | 47.4 | 31.9 | 32.9 |
| C-C products | 9.1 | 23.5 | 10.1 | 15.2 | 18.9 |
| Cycles | 6.8 | 11.3 | 5.3 | 2.9 | 1.4 |
| Isomers | 7.8 | 1.2 | 6.3 | 7.6 | 3.5 |
| 1 C-O breaking | 10 | 13.4 | 17.1 | 12.3 | 12.3 |
| 2 C-O breakings | 5.0 | 3.7 | 11.7 | 12.1 | 12.6 |
| 3 C-O breakings | 5.5 | 8.5 | 10.1 | 7.0 | 7.5 |
| 4 C-O breakings | 5.8 | 8.7 | 8.5 | 0.5 | 0.5 |
| 5 C-O breakings | - | - | - | - | - |
| Glucose (mg L−1) | Xylose (mg L−1) | Mannose (mg L−1) | Galactose (mg L−1) | Arabinose (mg L−1) | |
|---|---|---|---|---|---|
| Non-hydrolyzed liquor | 35.3 | 235.7 | 113.6 | 95.0 | 818.8 |
| Post-hydrolysis liquor | 1229.9 | 2195.2 | 5575.5 | 437.4 | 1088.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mounguengui-Diallo, M.; Sadier, A.; Noly, E.; Da Silva Perez, D.; Pinel, C.; Perret, N.; Besson, M. C-O Bond Hydrogenolysis of Aqueous Mixtures of Sugar Polyols and Sugars over ReOx-Rh/ZrO2 Catalyst: Application to an Hemicelluloses Extracted Liquor. Catalysts 2019, 9, 740. https://doi.org/10.3390/catal9090740
Mounguengui-Diallo M, Sadier A, Noly E, Da Silva Perez D, Pinel C, Perret N, Besson M. C-O Bond Hydrogenolysis of Aqueous Mixtures of Sugar Polyols and Sugars over ReOx-Rh/ZrO2 Catalyst: Application to an Hemicelluloses Extracted Liquor. Catalysts. 2019; 9(9):740. https://doi.org/10.3390/catal9090740
Chicago/Turabian StyleMounguengui-Diallo, Modibo, Achraf Sadier, Eddi Noly, Denilson Da Silva Perez, Catherine Pinel, Noémie Perret, and Michèle Besson. 2019. "C-O Bond Hydrogenolysis of Aqueous Mixtures of Sugar Polyols and Sugars over ReOx-Rh/ZrO2 Catalyst: Application to an Hemicelluloses Extracted Liquor" Catalysts 9, no. 9: 740. https://doi.org/10.3390/catal9090740
APA StyleMounguengui-Diallo, M., Sadier, A., Noly, E., Da Silva Perez, D., Pinel, C., Perret, N., & Besson, M. (2019). C-O Bond Hydrogenolysis of Aqueous Mixtures of Sugar Polyols and Sugars over ReOx-Rh/ZrO2 Catalyst: Application to an Hemicelluloses Extracted Liquor. Catalysts, 9(9), 740. https://doi.org/10.3390/catal9090740