Recent Advances in the Chemical Fixation of Carbon Dioxide: A Green Route to Carbonylated Heterocycle Synthesis





Abstract
1. Introduction
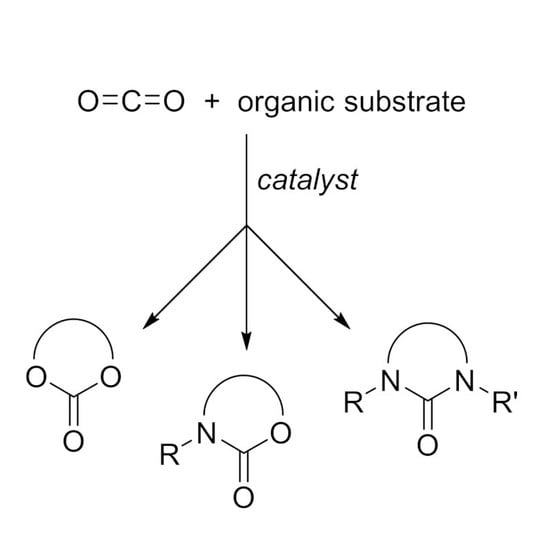
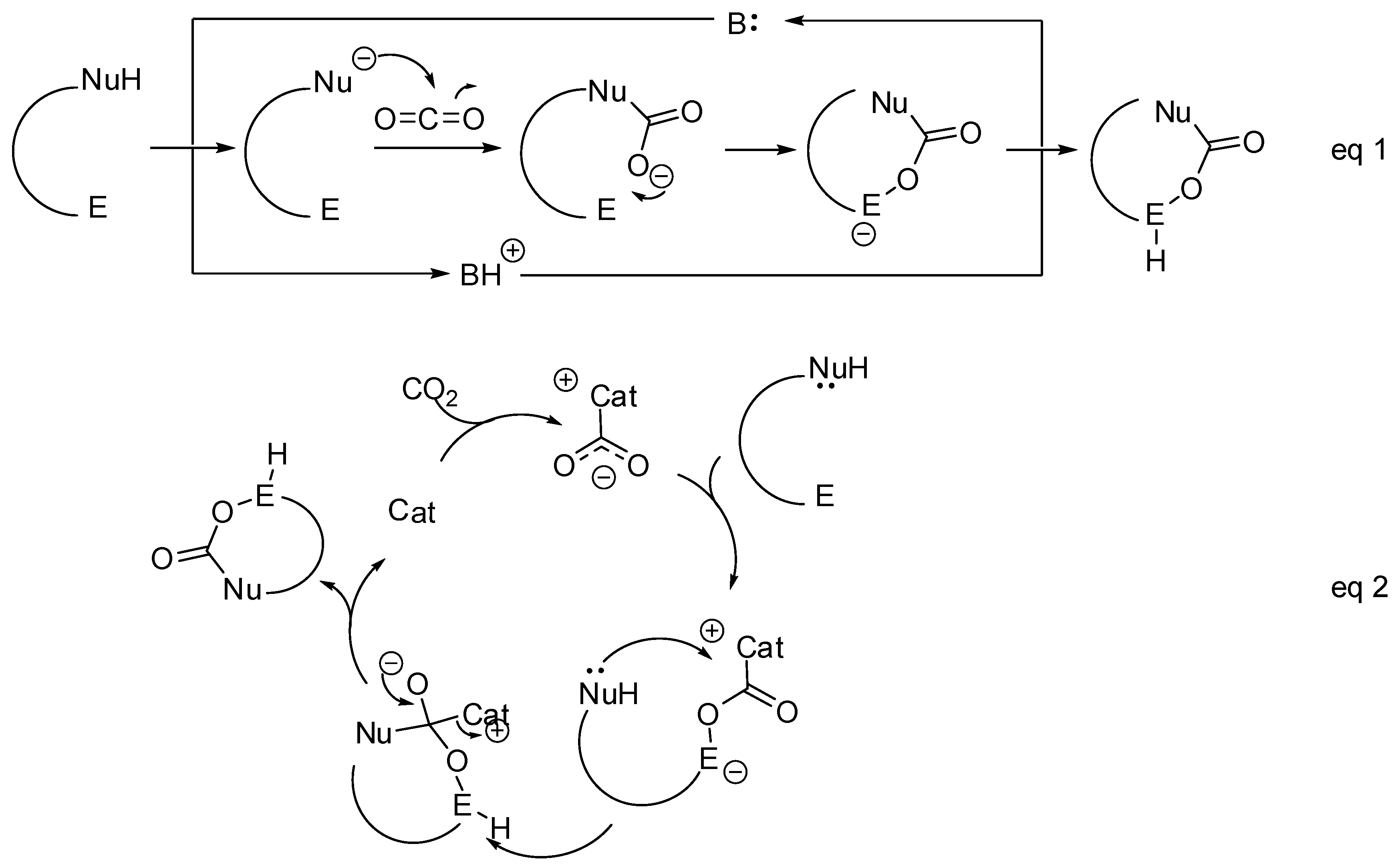
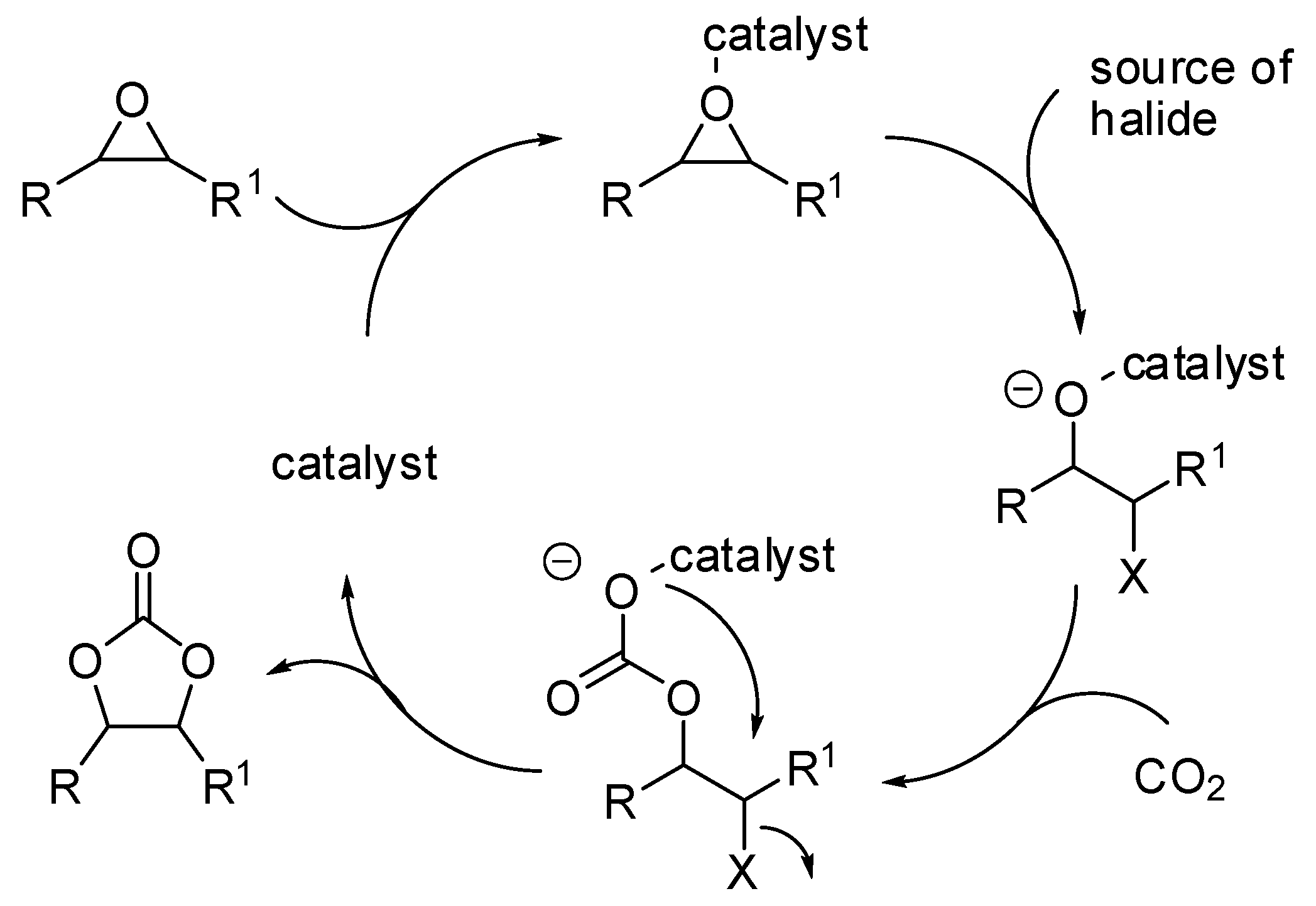
- The nucleophilic site (eventually enforced by a base) can add to the carbon atom, to form the corresponding carboxylate, which in turn adds at the electrophilic site to close the cycle (Scheme 1, Equation (1)).
- A catalyst is able to fix the carbon dioxide leading to a zwitterionic species. The carboxylate moiety of this activated species attacks an electrophilic site of the substrate molecule. Then the nucleophilic site closes the cycle, releasing the catalyst (Scheme 1, Equation (2)). N-heterocyclic carbenes (NHCs), N-heterocyclic olefins (NHOs), phosphorus ylides, polyoxometalates (POMs), ionic liquids (ILs), frustrated Lewis pairs (FLPs), metal-organic frameworks (MOFs) or superbases have been used for this scope.
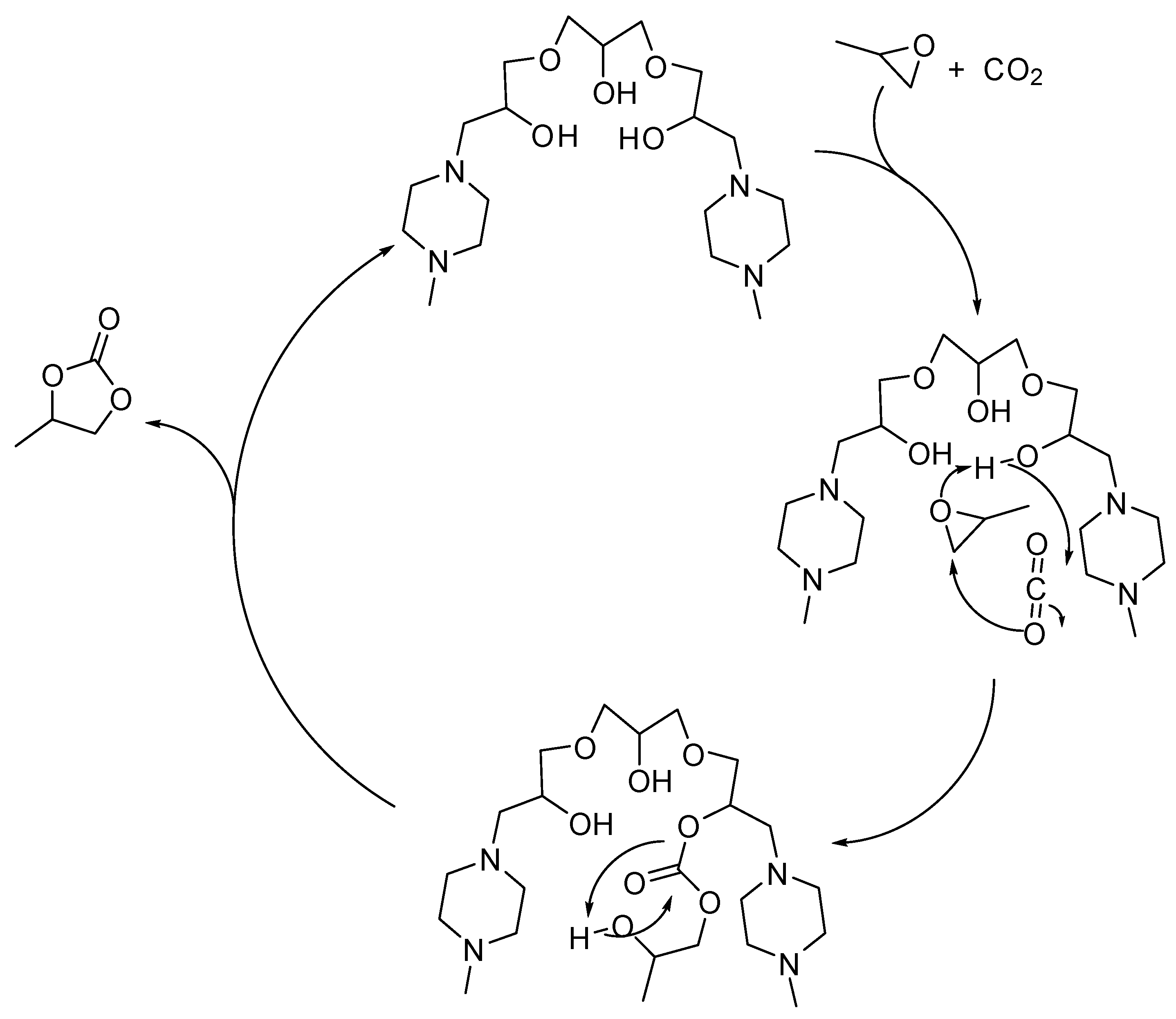
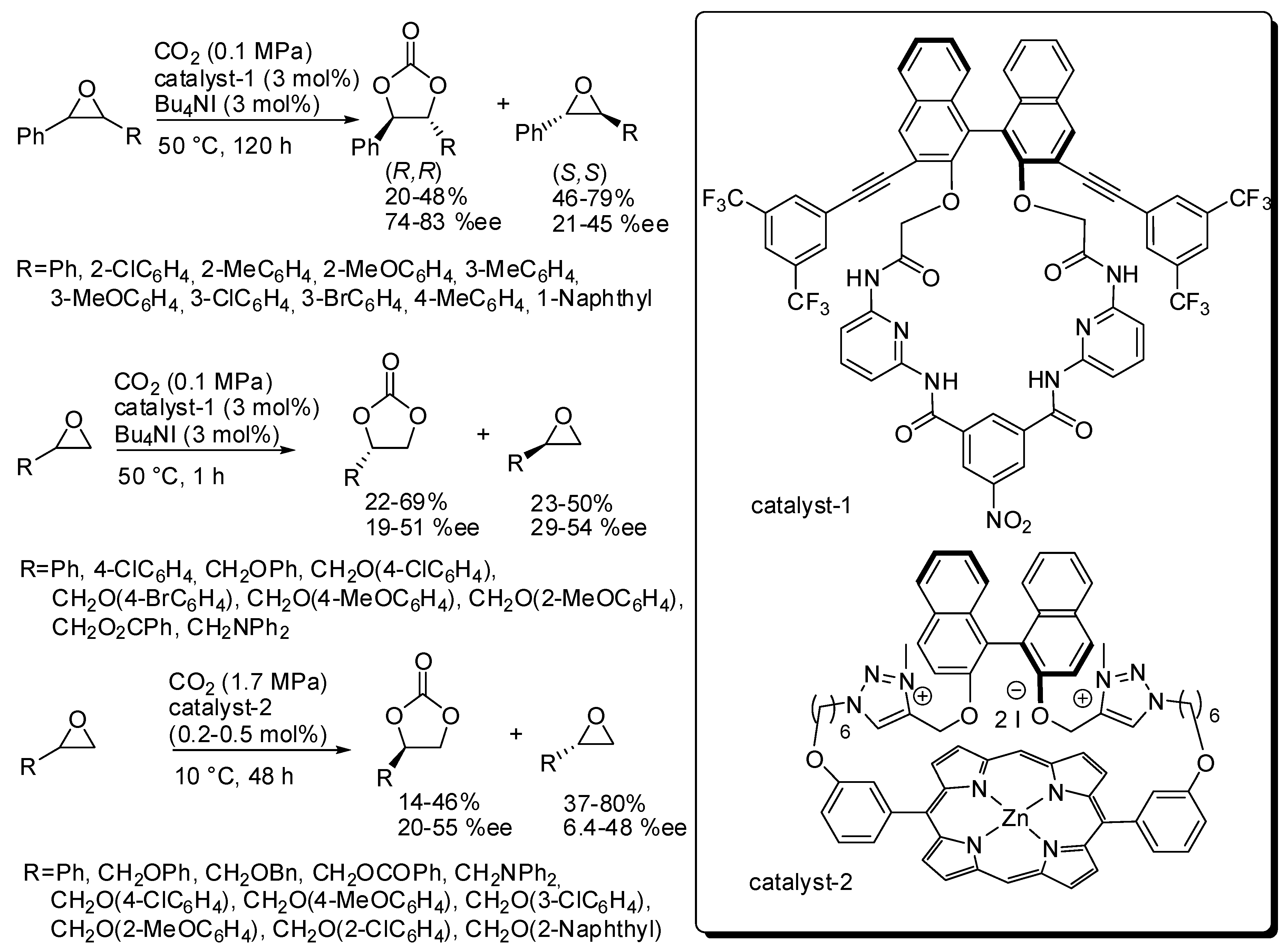
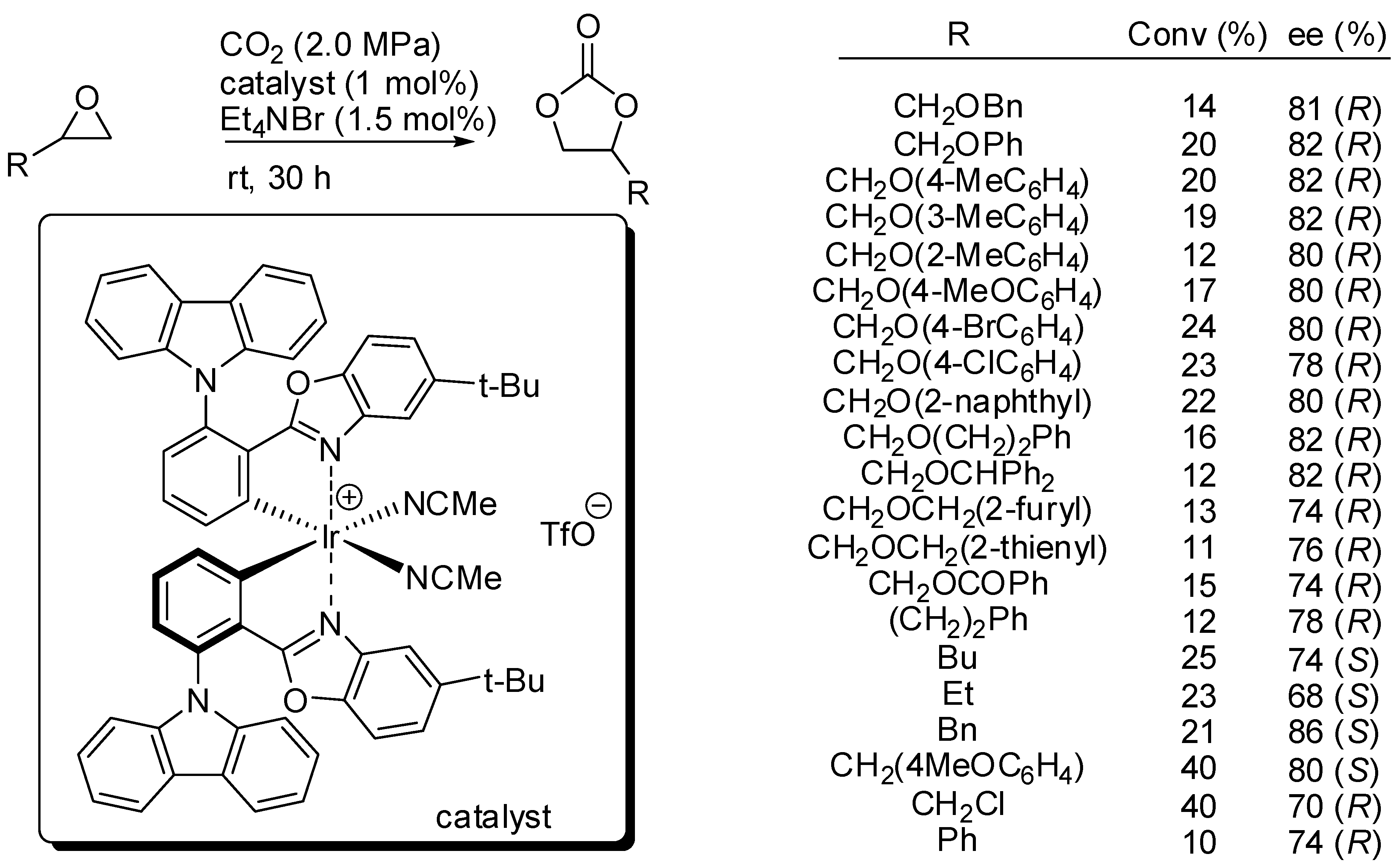
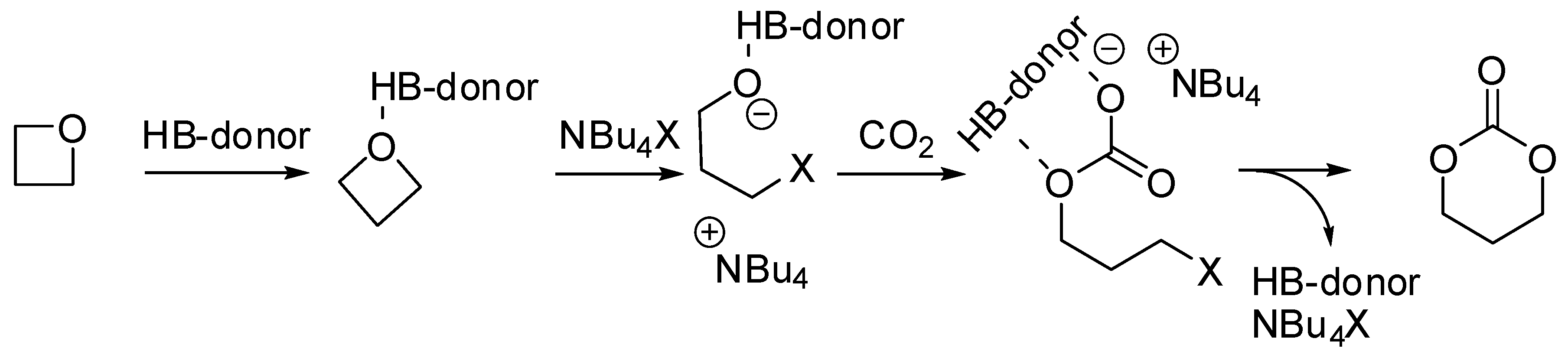
2. Cyclic Carbonates
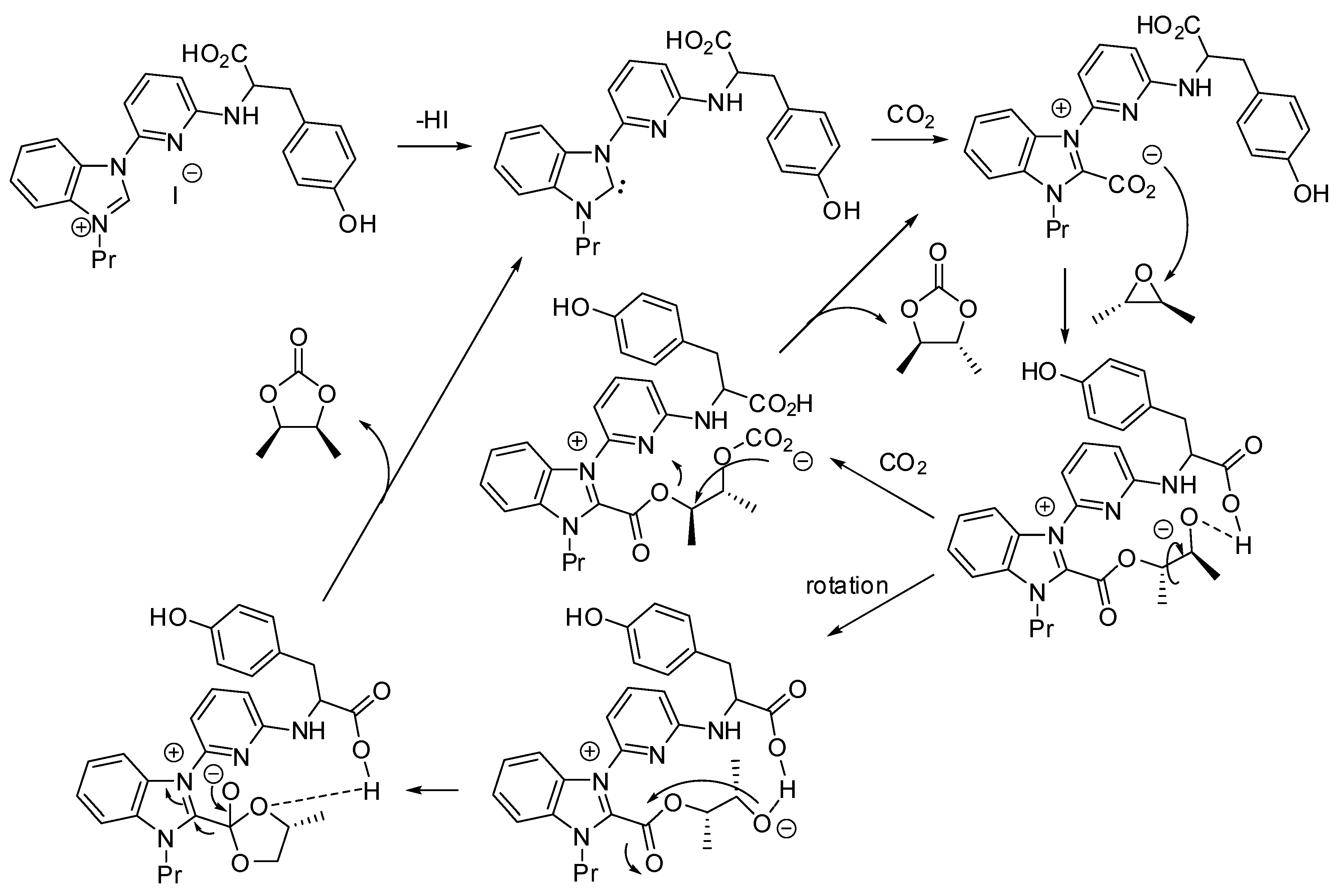
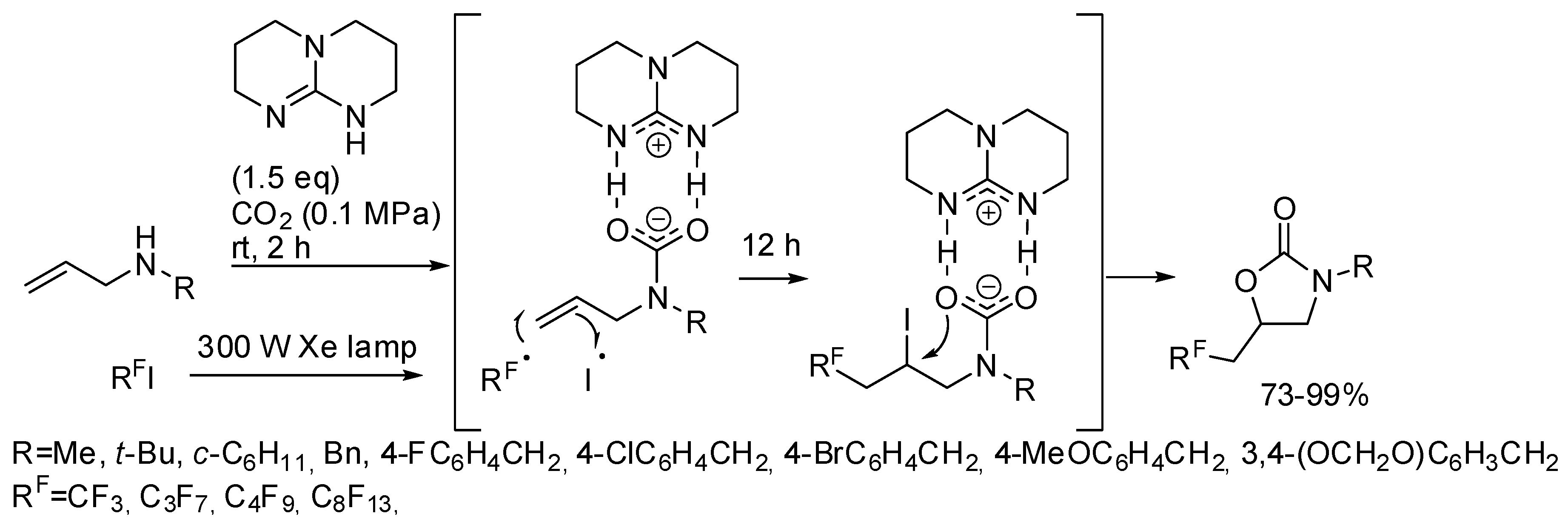
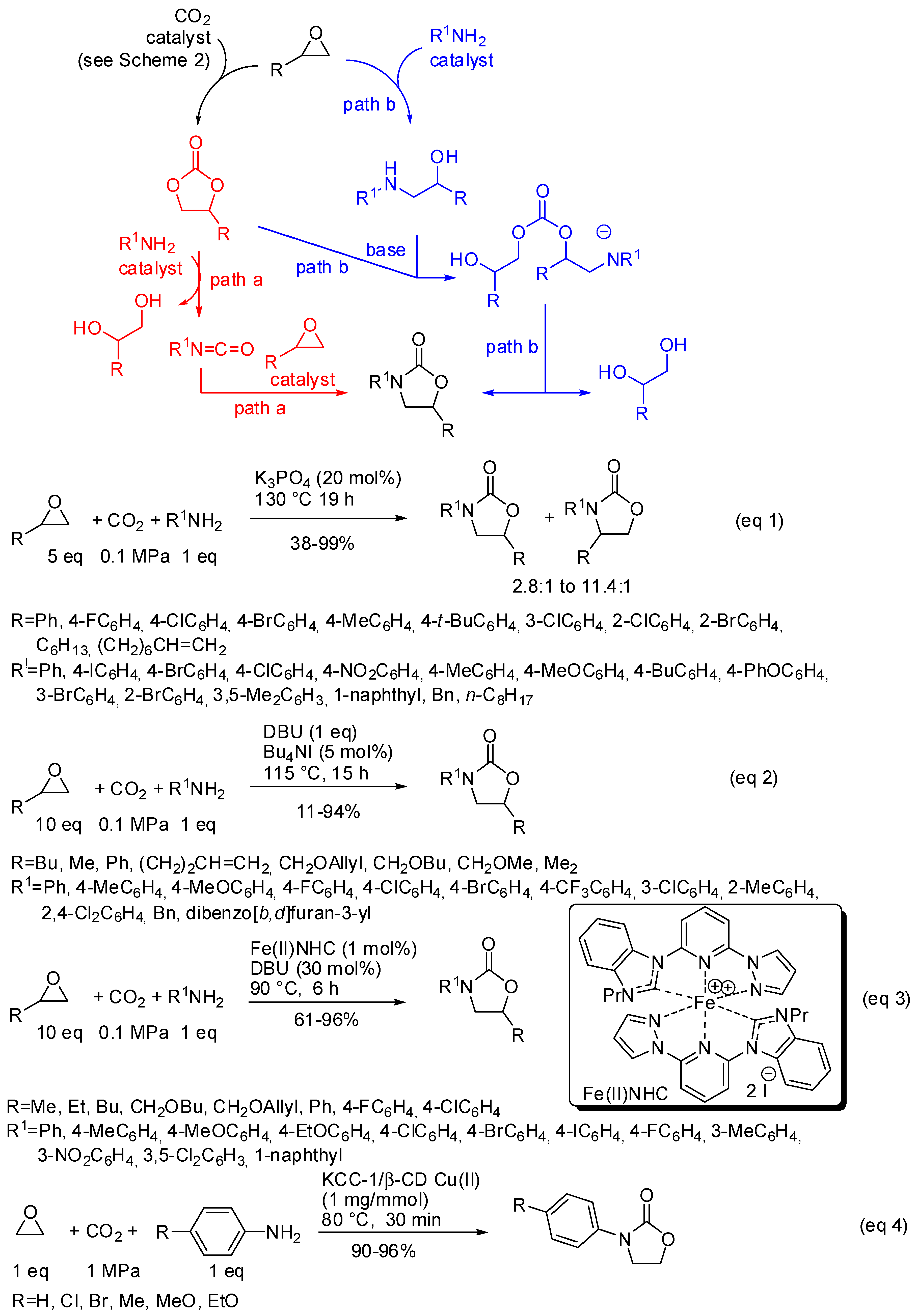
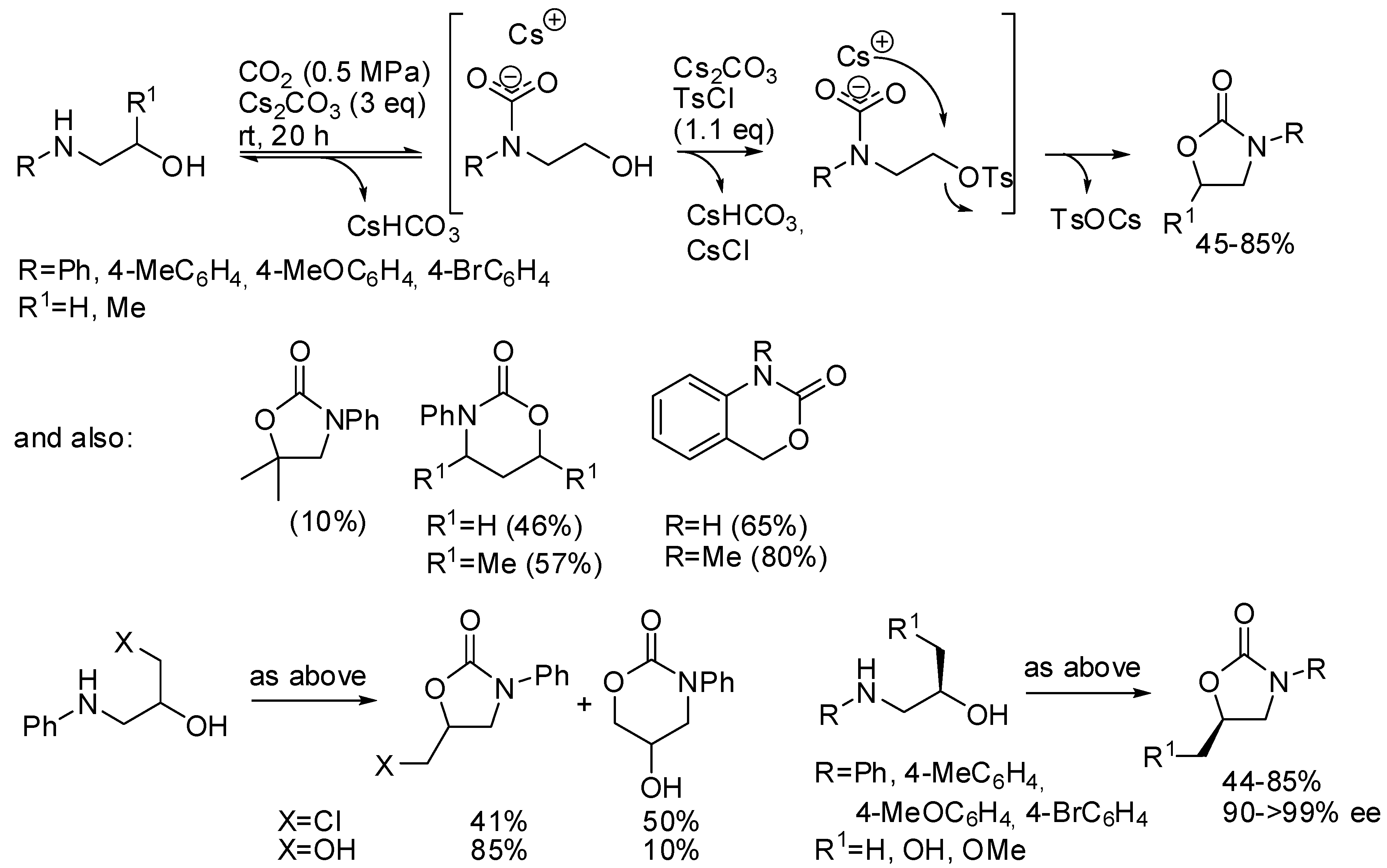
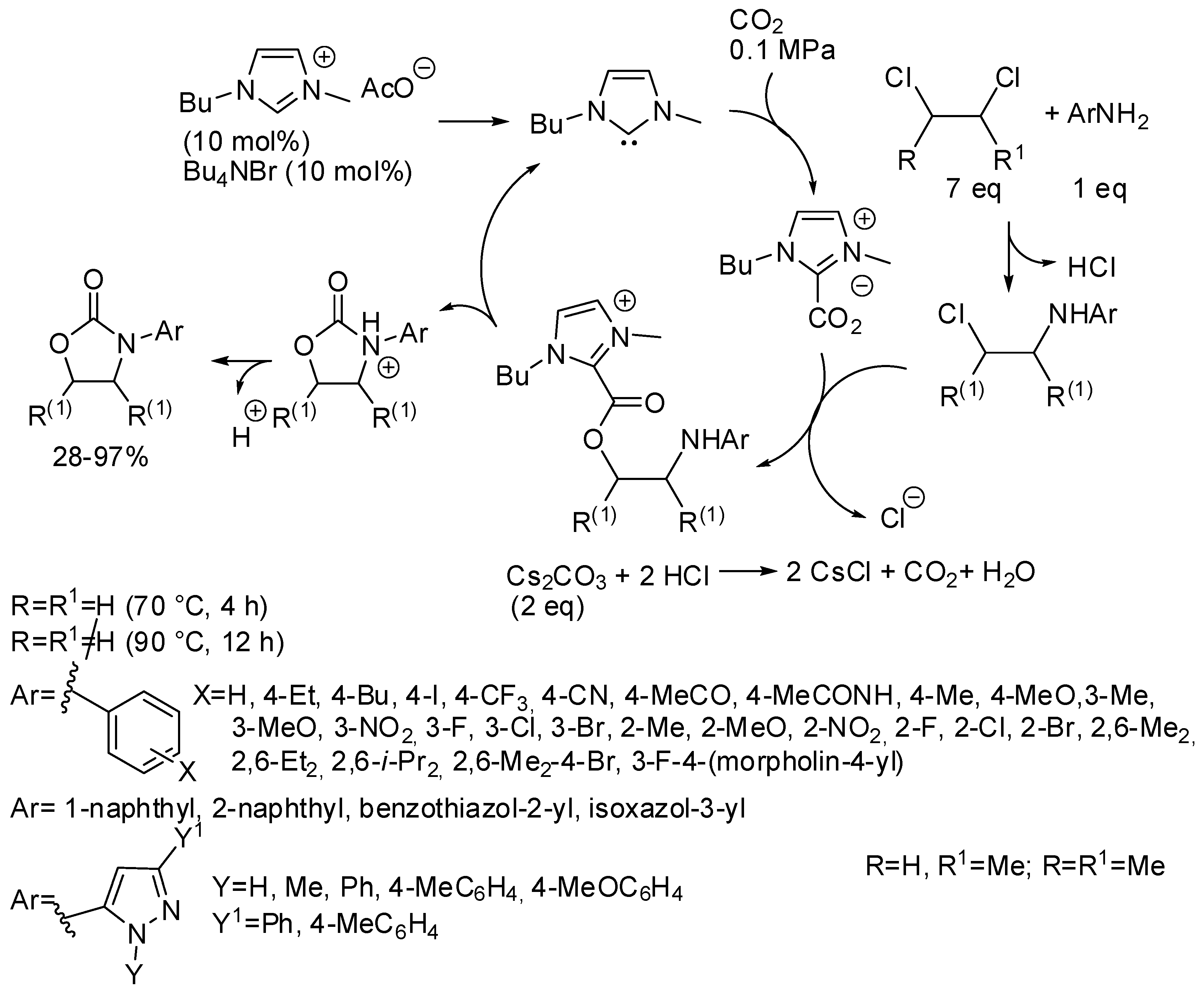
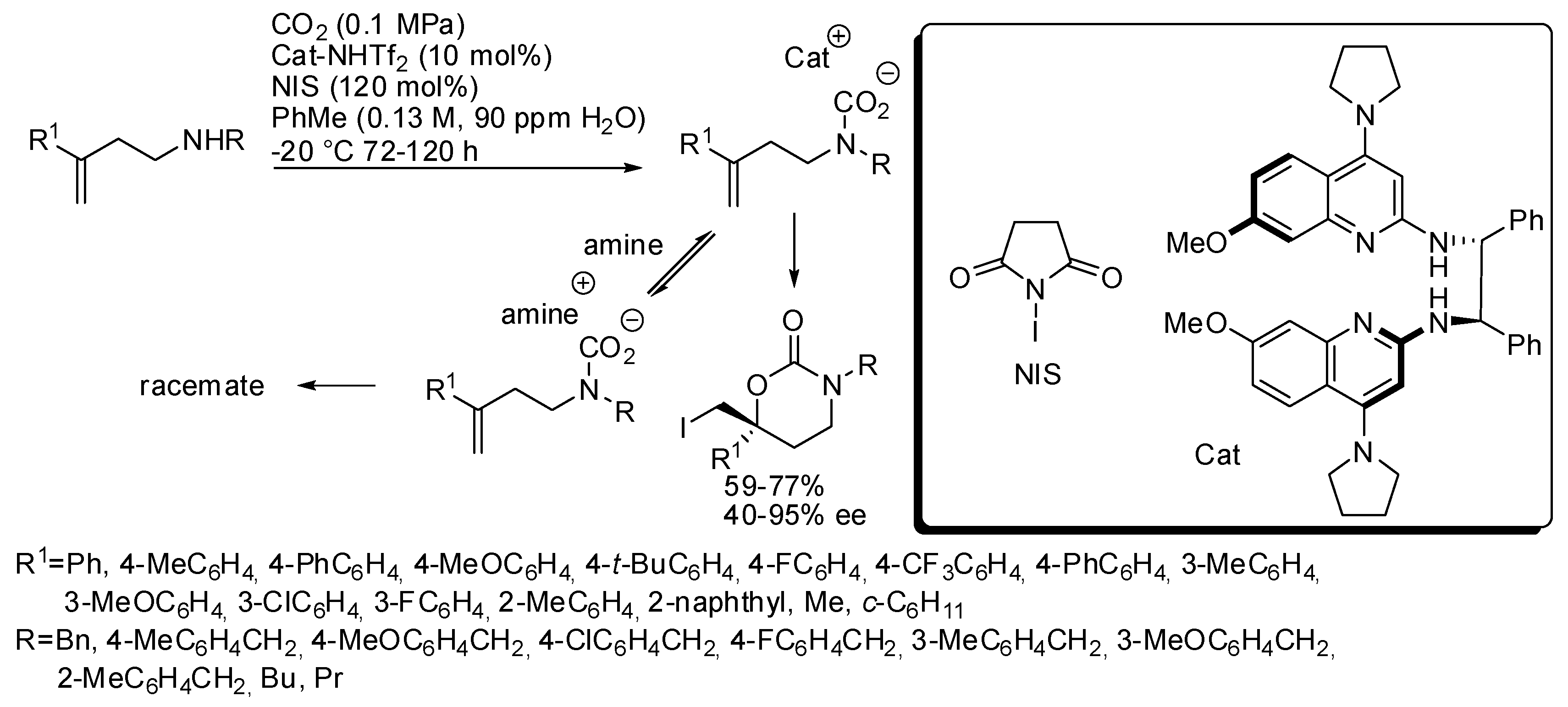
3. Cyclic Carbamates
- the first one involved the formation of cyclic carbonate by the classical mechanism (see Scheme 2), then amine opened the carbonate by releasing of diol and formation of a isocyanate, which in turn added another molecule of epoxide (path a);
- the second one involved the interaction between the cyclic carbonate and the amino-alcohol from nucleophilic opening of epoxide by amine(path b).
4. Cyclic Ureas
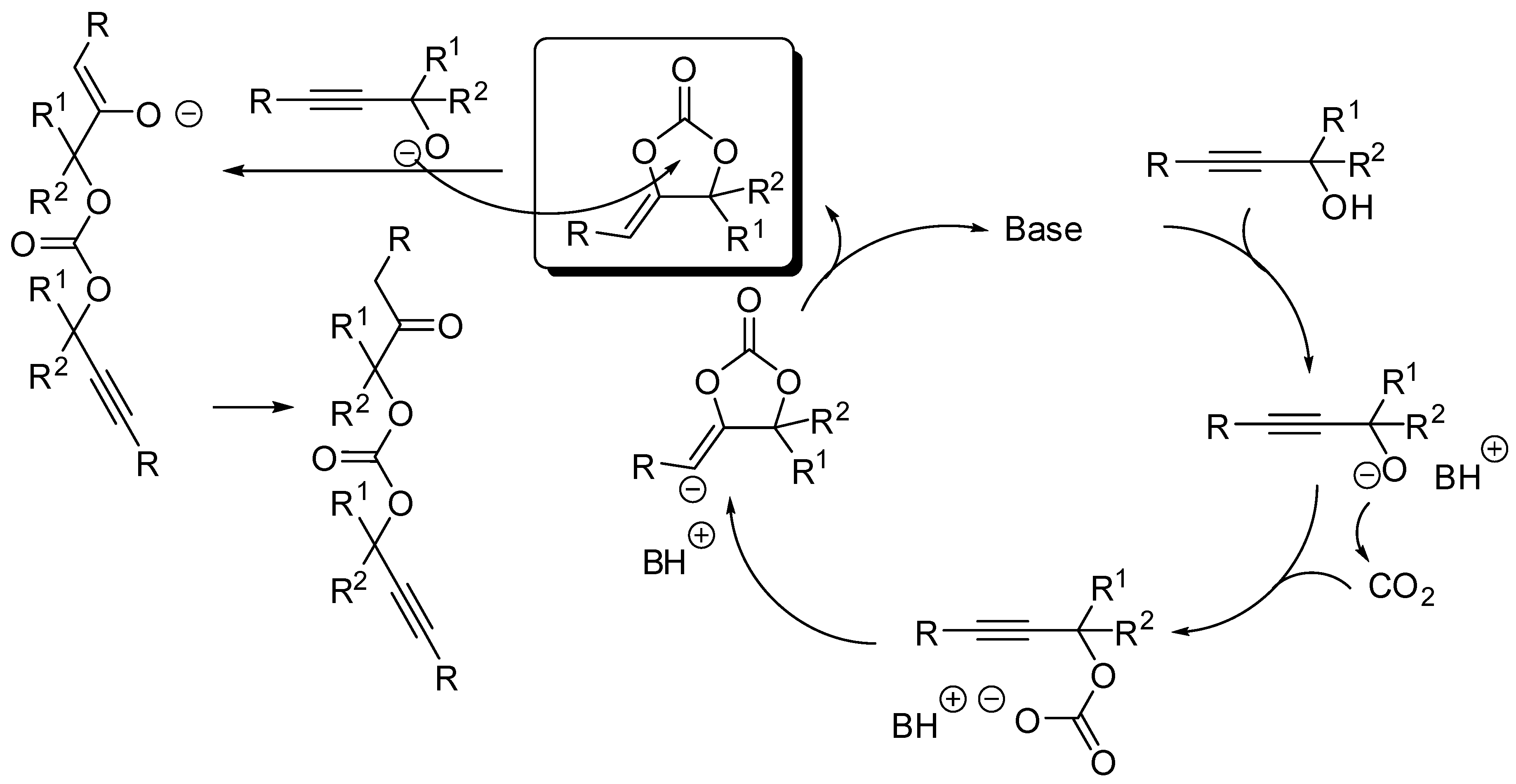
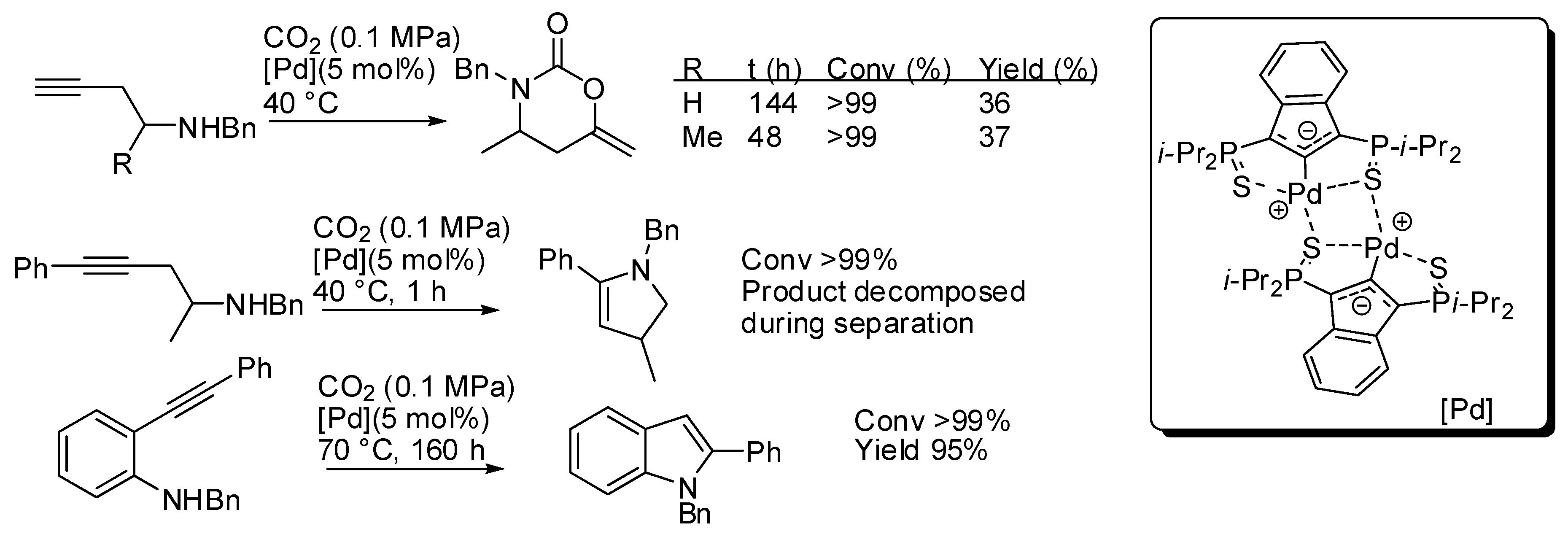
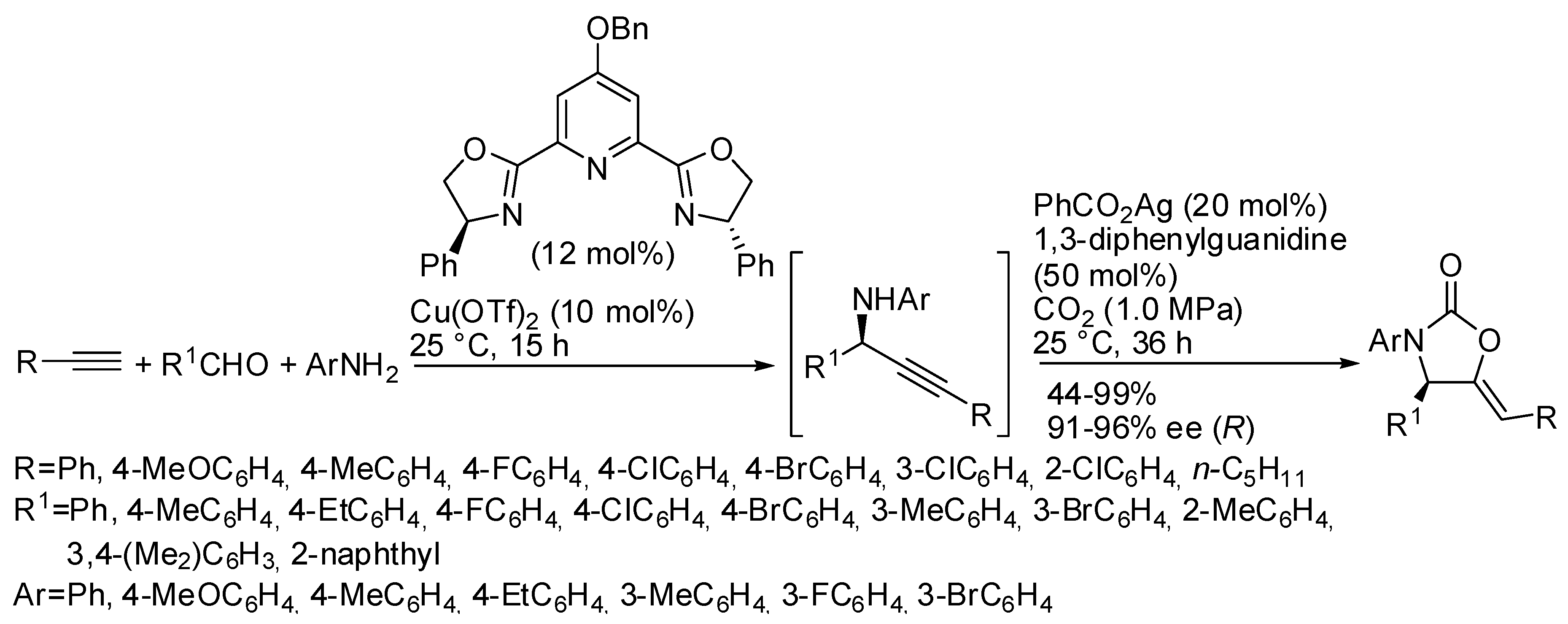
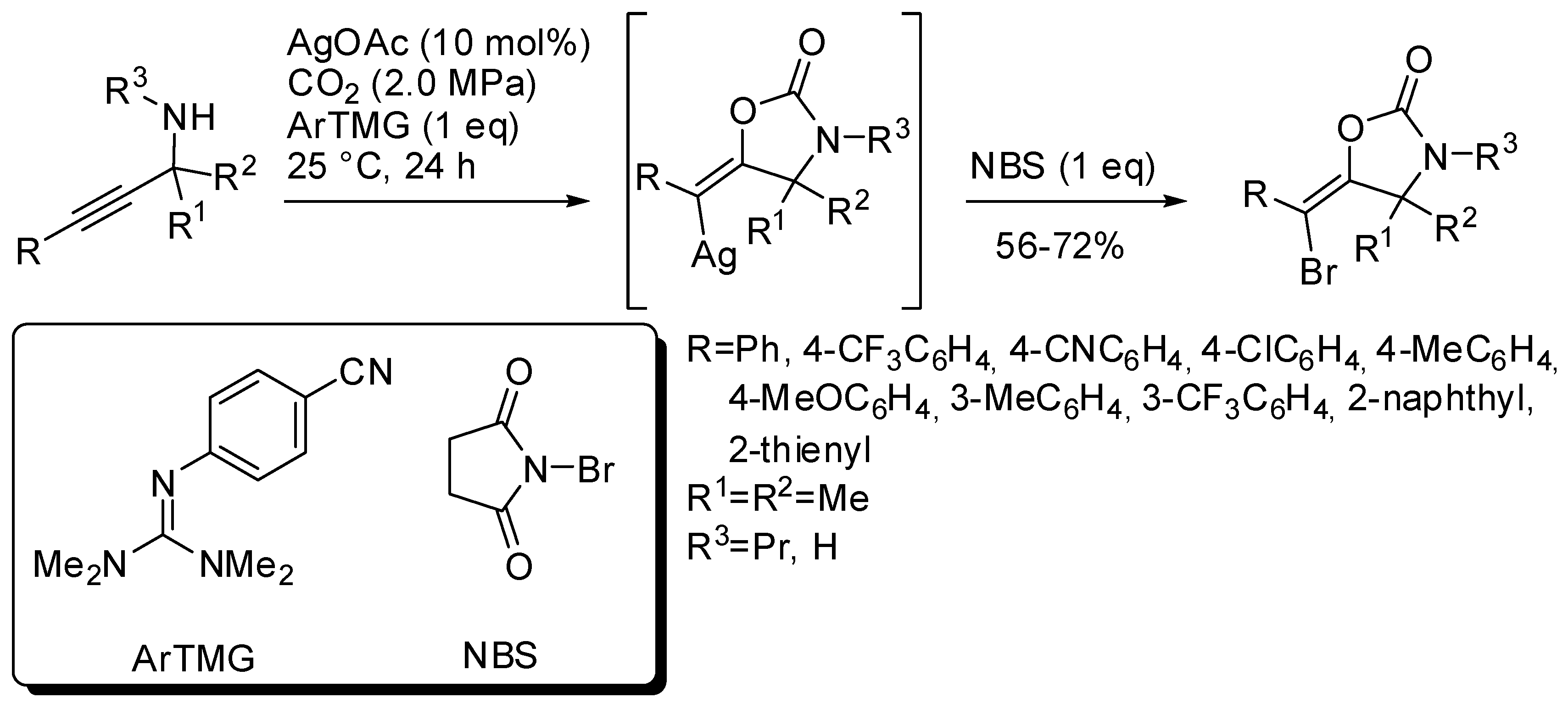
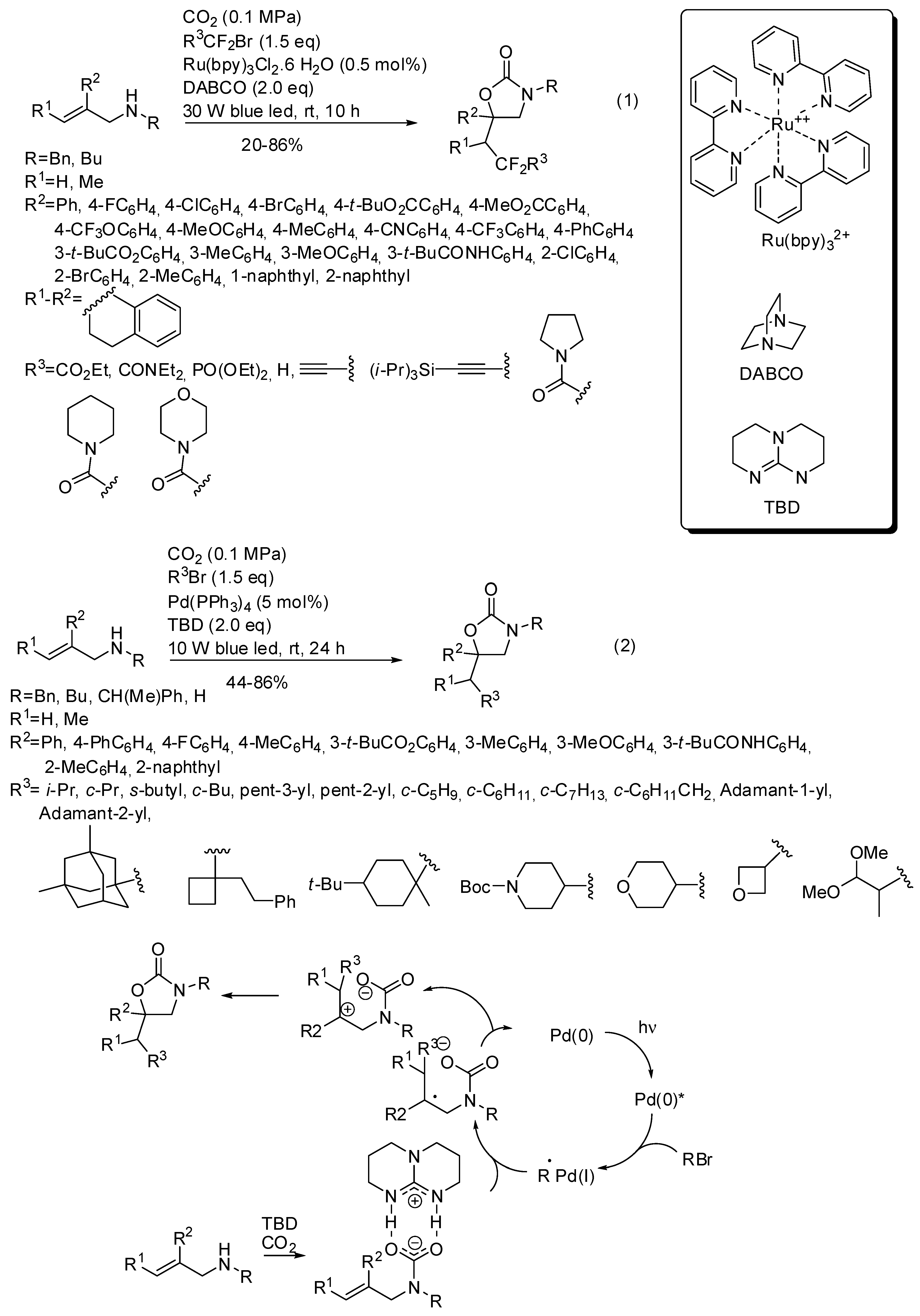
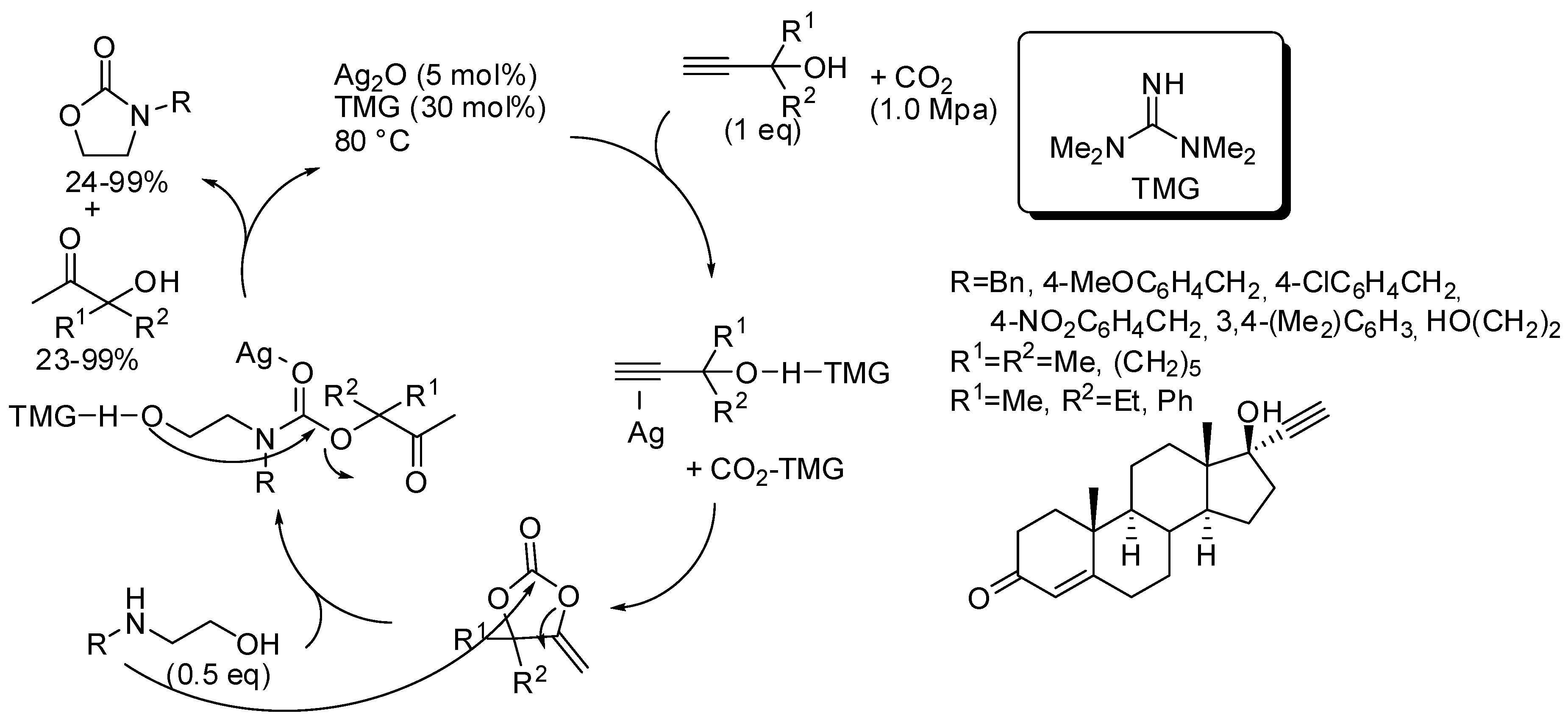
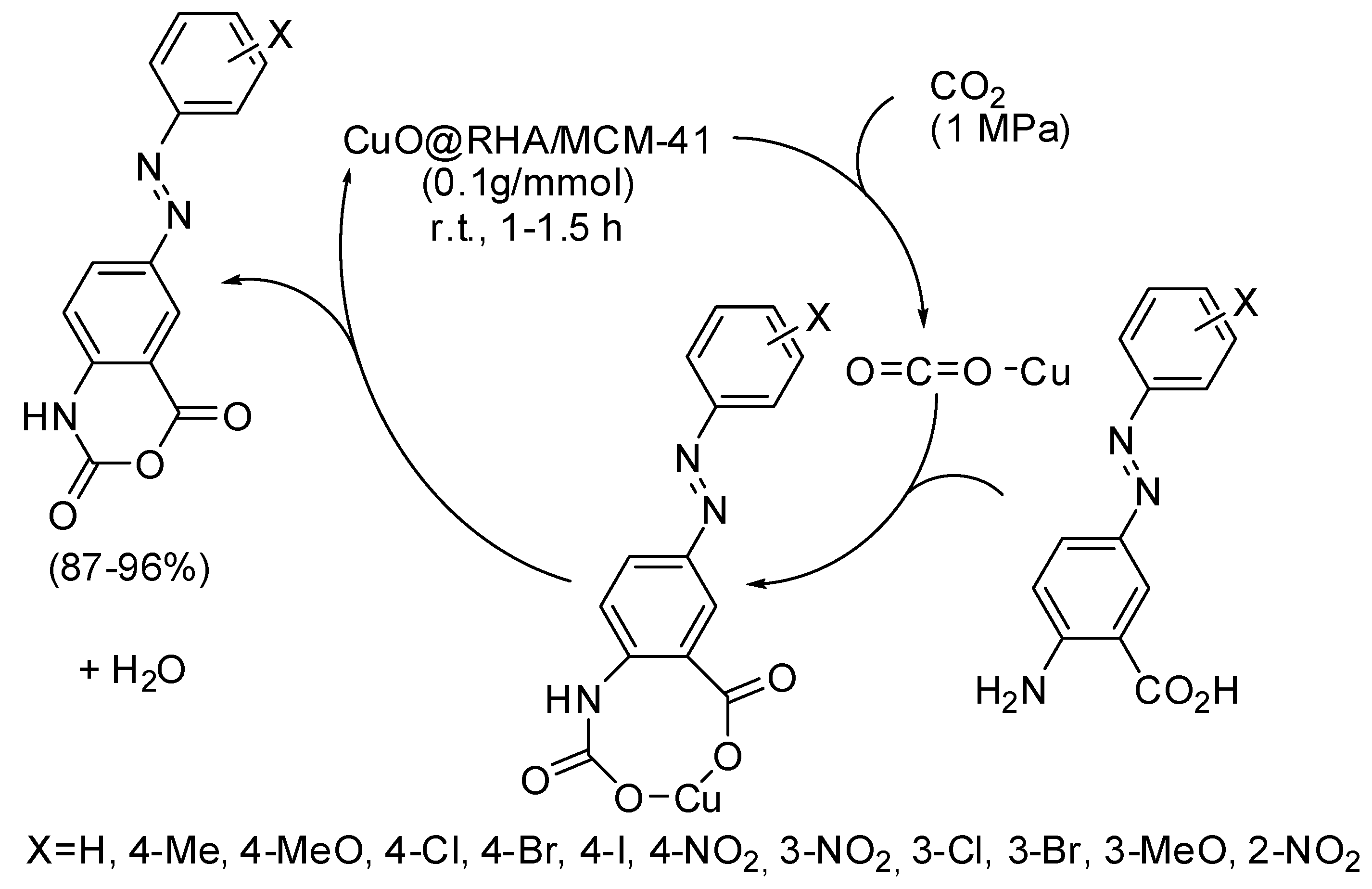
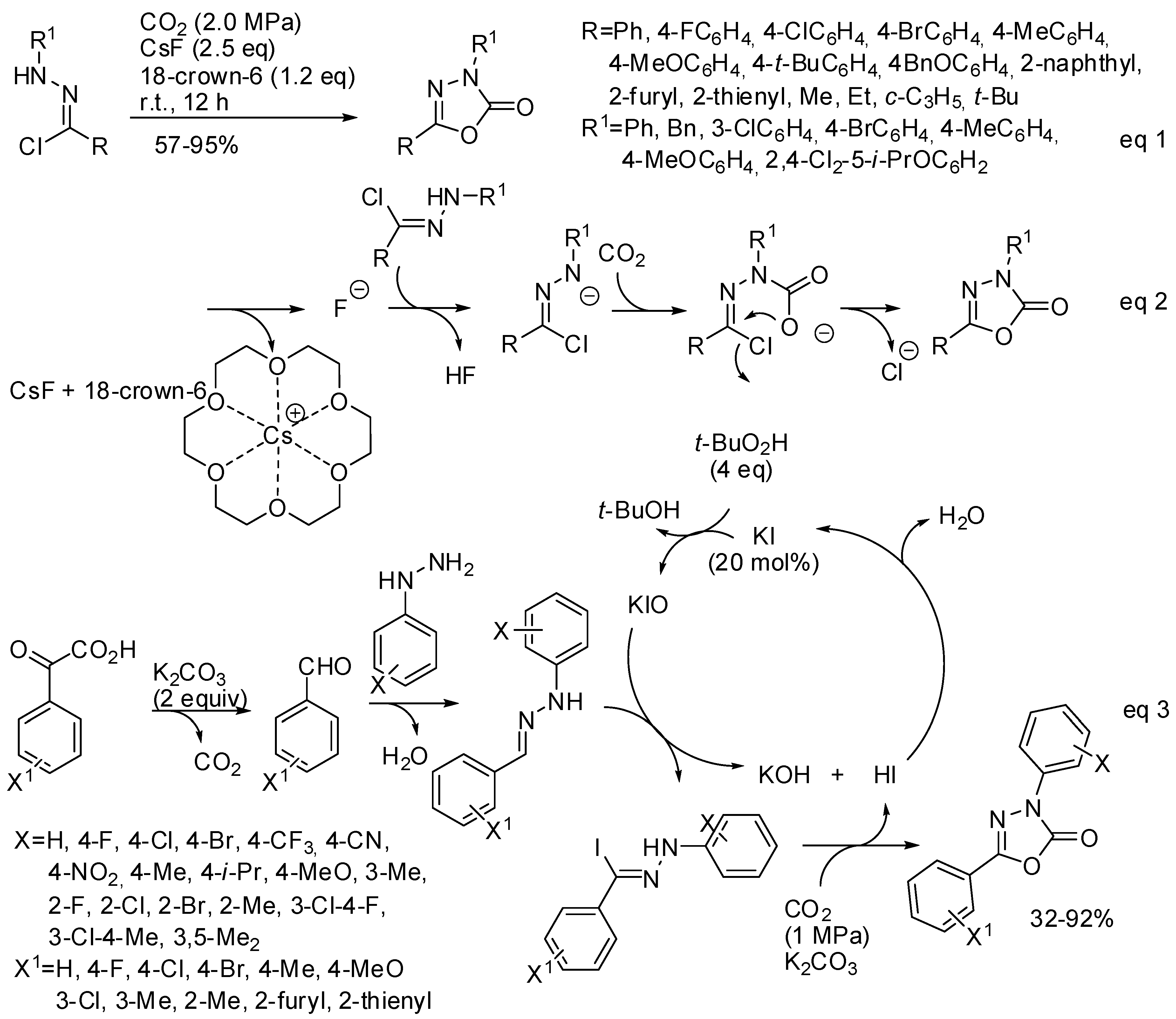
5. Other Heterocycles
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Solomon, S.; Plattner, G.-K.; Knutti, R.; Friedlingstein, P. Irreversible climate change due to carbon dioxide emissions. Proc. Natl. Acad. Sci. USA 2009, 106, 1704–1709. [Google Scholar] [CrossRef]
- Zhou, Z.; Xia, S.; He, L. Green Catalysis for Three-Component Reaction of Carbon Dioxide, Propargylic Alcohols and Nucleophiles. Acta Phys. Chim. Sin. 2018, 34, 838–844. [Google Scholar] [CrossRef]
- Cantat, T.; He, L.-N. (Eds.) CO2 Capture and Chemistry 2017. Curr. Opin. Green Sustain. Chem. 2017, 3, 1–66. Available online: https://www.sciencedirect.com/journal/current-opinion-in-green-and-sustainable-chemistry/vol/3/suppl/C (accessed on 5 June 2019).
- Song, Q.; Zhou, Z.; He, L. Efficient: Selective and sustainable catalysis of carbon dioxide. Green Chem. 2017, 19, 3707–3728. [Google Scholar] [CrossRef]
- ChemSusChem: Hot Topic: Carbon Dioxide First published: 1 April 2017, Last updated: 23 April 2018. Available online: https://onlinelibrary.wiley.com/doi/toc/10.1002/(ISSN)1864-564X.hottopic-co2 (accessed on 5 June 2019).
- Lu, X.B. Carbon Dioxide and Organometallics. In Topics in Organometallic Chemistry; Springer: Cham, Switzerland, 2015; Volume 53. [Google Scholar]
- Fiorani, G.; Guo, W.; Kleij, A.W. Sustainable conversion of carbon dioxide: The advent of organocatalysis. Green Chem. 2015, 17, 1375–1389. [Google Scholar] [CrossRef]
- Zhou, H.; Lu, X. Lewis base-CO2 adducts as organocatalysts for CO2 transformation. Sci. China Chem. 2017, 60, 904–911. [Google Scholar] [CrossRef]
- Tortajada, A.; Juliá-Hernández, F.; Börjesson, M.; Moragas, T.; Martin, R. Transition-Metal-Catalyzed Carboxylation Reactions with Carbon Dioxide. Angew. Chem. Int. Ed. 2018, 57, 15948–15982. [Google Scholar] [CrossRef]
- Poliakoff, M.; Leitner, W.; Streng, E.S. The Twelve Principles of CO2 Chemistry. Faraday Discuss. 2015, 183, 9–17. [Google Scholar] [CrossRef]
- Kumar, M.; Sundaram, S.; Gnansounou, E.; Larroche, C.; Thakura, I.S. Carbon dioxide capture, storage and production of biofuel and biomaterials by bacteria: A review. Bioresour. Technol. 2018, 247, 1059–1068. [Google Scholar] [CrossRef]
- Muthuramalingam, S.; Velusamy, M.; Mayilmurugan, R. Fixation and sequestration of carbon dioxide by copper(II) complexes. J. Chem. Sci. 2018, 130, 78. [Google Scholar] [CrossRef]
- Speight, J.G. Carbonylation. In Environmental Organic Chemistry for Engineers; Elsevier: Oxford, UK, 2017. [Google Scholar]
- Qiao, C.; Cao, Y.; He, L.-N. Transition Metal-Catalyzed Carboxylation of Terminal Alkynes with CO2. Mini-Rev. Org. Chem. 2018, 15, 283–290. [Google Scholar] [CrossRef]
- Zou, B.; Hu, C. Halogen-free processes for organic carbonate synthesis from CO2. Curr. Opin. Green Sustain. Chem. 2017, 3, 11–16. [Google Scholar] [CrossRef]
- Vessally, E.; Hosseinian, A.; Babazadeh, M.; Edjlali, L.; Hosseinzadeh-Khanmiri, R. Metal Catalyzed Carboxylative Coupling of Terminal Alkynes, Organohalides and Carbon Dioxide: A Novel and Promising Synthetic Strategy Toward 2-Alkynoates (A Review). Curr. Org. Chem. 2018, 22, 315–322. [Google Scholar] [CrossRef]
- Buttner, H.; Longwitz, L.; Steinbauer, J.; Wulf, C.; Werner, T. Recent Developments in the Synthesis of Cyclic Carbonates from Epoxides and CO2. Top. Curr. Chem. 2017, 375, 50. [Google Scholar] [CrossRef]
- Francke, R.; Schille, B.; Roemelt, M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide—Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118, 4631–4701. [Google Scholar] [CrossRef]
- Cotton, C.A.R.; Edlich-Muth, C.; Bar-Even, A. Reinforcing carbon fixation: CO2 reduction replacing and supporting carboxylation. Curr. Opin. Biotechnol. 2018, 49, 49–56. [Google Scholar] [CrossRef]
- Pander, J.E., III; Ren, D.; Huang, Y.; Loo, N.W.X.; Hong, S.H.L.; Yeo, B.S. Understanding the Heterogeneous Electrocatalytic Reduction of Carbon Dioxide on Oxide-Derived Catalysts. ChemElectroChem 2018, 5, 219–237. [Google Scholar] [CrossRef]
- Voiry, D.; Shin, H.S.; Loh, K.P.; Chhowalla, M. Low-dimensional catalysts for hydrogen evolution and CO2 reduction. Nat. Rev. Chem. 2018, 2, 0105. [Google Scholar] [CrossRef]
- Pagliaro, M.; Fidalgo, A.; Palmisano, L.; Ilharco, L.M.; Parrino, F.; Ciriminna, R. Polymers of limonene oxide and carbon dioxide: Polycarbonates of the solar economy. Preprints 2018, 2018, 040067. [Google Scholar] [CrossRef]
- Muthuraj, R.; Mekonnen, T. Recent progress in carbon dioxide (CO2) as feedstock for sustainable materials development: Co-polymers and polymer blends. Polymer 2018, 145, 348–373. [Google Scholar] [CrossRef]
- Kamphuis, A.J.; Picchioni, F.; Pescarmona, P.P. CO2-fixation into cyclic and polymeric carbonates: Principles and applications. Green Chem. 2019, 21, 406–448. [Google Scholar] [CrossRef]
- Tomishige, K.; Tamura, M.; Nakagawa, Y. CO2 Conversion with Alcohols and Amines into Carbonates, Ureas, and Carbamates over CeO2 Catalyst in the Presence and Absence of 2-Cyanopyridine. Chem. Rec. 2018, 18, 1–26. [Google Scholar] [CrossRef]
- Liang, J.; Huang, J.-B.; Cao, R. Metal-organic frameworks and porous organic polymers for sustainable fixation of carbon dioxide into cyclic carbonates. Coord. Chem. Rev. 2019, 378, 32–65. [Google Scholar] [CrossRef]
- Li, J.-Y.; Song, Q.-W.; Zhang, K.; Liu, P. Catalytic Conversion of Carbon Dioxide through C-N Bond Formation. Molecules 2019, 24, 182. [Google Scholar] [CrossRef]
- Farshbaf, S.; Fekri, L.Z.; Nikpassand, M.; Mohammadi, R.; Vessally, E. Dehydrative condensation of β-aminoalcohols with CO2: An environmentally benign access to 2-oxazolidinone derivatives. J. CO2 Util. 2018, 25, 194–204. [Google Scholar] [CrossRef]
- Didehbana, K.; Vessally, E.; Salary, M.; Edjlalic, L.; Babazadeh, M. Synthesis of a variety of key medicinal heterocyclic compounds via chemical fixation of CO2 onto o-alkynylaniline derivatives. J. CO2 Util. 2018, 23, 42–50. [Google Scholar] [CrossRef]
- Vessally, E.; Babazadeh, M.; Hosseinian, A.; Arshadi, S.; Edjlali, L. Nanocatalysts for chemical transformation of carbon dioxide. J. CO2 Util. 2017, 21, 491–502. [Google Scholar] [CrossRef]
- Vessally, E.; Didehban, K.; Babazadeh, M.; Hosseinian, A.; Edjlali, L. Chemical fixation of CO2 with aniline derivatives: A new avenue to the synthesis of functionalized azole compounds (A review). J. CO2 Util. 2017, 21, 480–490. [Google Scholar] [CrossRef]
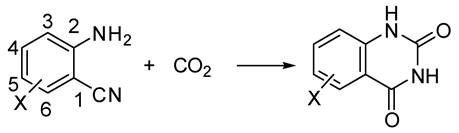
- Vessally, E.; Soleimani-Amiri, S.; Hosseinian, A.; Edjlali, L.; Babazadeh, M. Chemical fixation of CO2 to 2-aminobenzonitriles: A straightforward route to quinazoline-2,4(1H,3H)-diones with green and sustainable chemistryperspectives. J. CO2 Util. 2017, 21, 342–352. [Google Scholar] [CrossRef]
- Arshadi, S.; Vessally, E.; Sobati, M.; Hosseinian, A.; Bekhradnia, A. Chemical fixation of CO2 to N-propargylamines: A straightforward route to 2-oxazolidinones. J. CO2 Util. 2017, 19, 120–129. [Google Scholar] [CrossRef]
- Hosseinian, A.; Ahmadi, S.; Mohammadi, R.; Monfared, A.; Rahmani, Z. Three-component reaction of amines, epoxides, and carbon dioxide: A straightforward route to organic carbamates. J. CO2 Util. 2018, 27, 381–389. [Google Scholar] [CrossRef]
- Liu, X.-F.; Wang, M.-Y.; He, L.-N. Heterogeneous Catalysis for Oxazolidinone Synthesis from Aziridines and CO2. Curr. Org. Chem. 2017, 21, 698–707. [Google Scholar] [CrossRef]
- Arshadi, S.; Vessally, E.; Hosseinian, A.; Soleimani-Amiri, S.; Edjlali, L. Three component coupling of CO2, propargyl alcohols, and amines: An environmentally benign access to cyclic and acyclic carbamates (A Review). J. CO2 Util. 2017, 21, 108–118. [Google Scholar] [CrossRef]
- Yu, B.; He, L.-N. Upgrading Carbon Dioxide by Incorporation into Heterocycles. ChemSusChem 2015, 8, 52–62. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Making Plastics from Carbon Dioxide: Salen Metal Complexes as Catalysts for the Production of Polycarbonates from Epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef]
- Poland, S.J.; Darensbourg, D.J. A quest for polycarbonates provided via sustainable epoxide/CO2 copolymerization processes. Green Chem. 2017, 19, 4990–5011. [Google Scholar] [CrossRef]
- Zhang, S.S. A review on electrolyte additives for lithium-ion batteries. J. Power Sources 2006, 162, 1379–1394. [Google Scholar] [CrossRef]
- He, Q.; O’Brien, J.W.; Kitselman, K.A.; Tompkins, L.E.; Curtis, G.C.T.; Kerton, F.M. Synthesis of cyclic carbonates from CO2 and epoxides using ionic liquids and related catalysts including choline chloride–metal halide mixtures. Catal. Sci. Technol. 2014, 4, 1513–1528. [Google Scholar] [CrossRef]
- Lan, D.-H.; Fan, N.; Wang, Y.; Gao, X.; Zhang, P.; Chen, L.; Au, C.-T.; Yin, S.-F. Recent advances in metal-free catalysts for the synthesis of cyclic carbonates from CO2 and epoxides. Chin. J. Catal. 2016, 37, 826–845. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Dyson, P.J. Synthesis of carbonates and related compounds incorporating CO2 using ionic liquid-type catalysts: State-of-the-art and beyond. J. Catal. 2016, 343, 52–61. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Mereau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Organocatalyzed coupling of carbon dioxide with epoxides for the synthesis of cyclic carbonates: Catalyst design and mechanistic studies. Catal. Sci. Technol. 2017, 7, 2651–2684. [Google Scholar] [CrossRef]
- Shaikh, R.R.; Pornpraprom, S.; D’Elia, V. Catalytic Strategies for the Cycloaddition of Pure, Diluted, and Waste CO2 to Epoxides under Ambient Conditions. ACS Catal. 2018, 8, 419–450. [Google Scholar] [CrossRef]
- Kim, H.-U.; Babu, R.; Roshan, R.; Park, D.-W. Catalytic performance of metal azolate frameworks in the solventless synthesis of cyclic carbonates from CO2 and epoxides. Appl. Catal. A Gen. 2017, 538, 59–65. [Google Scholar] [CrossRef]
- Elkurtehi, A.I.; Kerton, F.M. Coupling Reactions of Carbon Dioxide with Epoxides Catalyzed by Vanadium Aminophenolate Complexes. ChemSusChem 2017, 10, 1249–1254. [Google Scholar] [CrossRef]
- Chen, F.; Liu, N.; Dai, B. Iron(II) Bis-CNN Pincer Complex-Catalyzed Cyclic Carbonate Synthesis at Room Temperature. ACS Sustain. Chem. Eng. 2017, 5, 9065–9075. [Google Scholar] [CrossRef]
- Wu, X.; North, M. A Bimetallic Aluminium(Salphen) Complex for the Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide. ChemSusChem 2017, 10, 74–78. [Google Scholar] [CrossRef]
- Luo, R.; Yang, Z.; Zhang, W.; Zhou, X.; Ji, H. Recyclable bifunctional aluminum salen catalyst for CO2 fixation: The efficient formation of five-membered heterocyclic compounds. Sci. China Chem. 2017, 60, 979–989. [Google Scholar] [CrossRef]
- Zhou, F.; Xie, S.-L.; Gao, X.-T.; Zhang, R.; Wang, C.-H.; Yin, G.-Q.; Zhou, J. Activation of (salen)CoI complex by phosphorane for carbon dioxide transformation at ambient temperature and pressure. Green Chem. 2017, 19, 3908–3915. [Google Scholar] [CrossRef]
- Kaneko, S.; Shirakawa, S. Potassium Iodide−Tetraethylene Glycol Complex as a Practical Catalyst for CO2 Fixation Reactions with Epoxides under Mild Conditions. ACS Sustain. Chem. Eng. 2017, 5, 2836–2840. [Google Scholar] [CrossRef]
- Steinbauer, J.; Werner, T. Poly(ethylene glycol)s as Ligands in Calcium-Catalyzed Cyclic Carbonate Synthesis. ChemSusChem 2017, 10, 3025–3029. [Google Scholar] [CrossRef]
- Steinbauer, J.; Spannenberg, A.; Werner, T. An in situ formed Ca2+–crown ether complex and its use in CO2-fixation reactions with terminal and internal epoxides. Green Chem. 2017, 19, 3769–3779. [Google Scholar] [CrossRef]
- Longwitz, L.; Steinbauer, J.; Spannenberg, A.; Werner, T. Calcium-Based Catalytic System for the Synthesis of Bio-Derived Cyclic Carbonates under Mild Conditions. ACS Catal. 2018, 8, 665–672. [Google Scholar] [CrossRef]
- Zhao, T.-X.; Zhang, Y.-Y.; Liang, J.; Li, P.; Hu, X.-B.; Wu, Y.-T. Multisite activation of epoxides by recyclable CaI2/N-methyldiethanolamine catalyst for CO2 fixation: A facile access to cyclic carbonates under mild conditions. Mol. Catal. 2018, 450, 87–94. [Google Scholar] [CrossRef]
- Chowdhury, A.H.; Bhanja, P.; Salam, N.; Bhaumik, A.; Islam, S.M. Magnesium oxide as an efficient catalyst for CO2 fixation and N-formylation reactions under ambient conditions. Mol. Catal. 2018, 450, 46–54. [Google Scholar] [CrossRef]
- Kilic, A.; Durgun, M.; Aytar, E.; Yavuz, R. Synthesis and characterization of novel positively charged organocobaloximes as catalysts for the fixation of CO2 to cyclic carbonates. J. Organomet. Chem. 2018, 858, 78–88. [Google Scholar] [CrossRef]
- Della Monica, F.; Buonerba, A.; Paradiso, V.; Milione, S.; Grassi, A.; Capacchione, C. [OSSO]-Type Fe(III) Metallate as Single-Component Catalyst for the CO2 Cycloaddition to Epoxides. Adv. Synth. Catal. 2018, 361, 283–288. [Google Scholar] [CrossRef]
- Kuznetsova, S.A.; Rulev, Y.A.; Larionov, V.A.; Smol’yakov, A.F.; Zubavichus, Y.V.; Maleev, V.I.; Li, H.; North, M.; Saghyan, A.S.; Belokon, Y.N. Self-Assembled Ionic Composites of Negatively Charged Zn(salen) Complexes and Triphenylmethane Derived Polycations as Recyclable Catalysts for the Addition of Carbon Dioxide to Epoxides. ChemCatChem 2019, 11, 511–519. [Google Scholar] [CrossRef]
- Milani, J.L.S.; Oliveira, I.S.; Dos Santos, P.A.; Valdo, A.K.S.M.; Martins, F.T.; Cangussu, D.; Das Chagas, R.P. Chemical fixation of carbon dioxide to cyclic carbonates catalyzed by zinc(II) complex bearing 1,2-disubstituted benzimidazole ligand. Chin. J. Catal. 2018, 39, 245–249. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, R.; Yang, Z.; Zhou, X.; Ji, H. Imidazolium-based ionic liquid decorated zinc porphyrin catalyst for converting CO2 into five-membered heterocyclic molecules. Sustain. Energy Fuels 2018, 2, 125–132. [Google Scholar] [CrossRef]
- Sogukomerogullari, H.G.; Aytar, E.; Ulusoy, M.; Demir, S.; Dege, N.; Richeson, D.S.; Sönmez, M. Synthesis of complexes Fe, Co and Cu supported by “SNS” pincer ligands and their ability to catalytically form cyclic carbonates. Inorg. Chim. Acta 2018, 471, 290–296. [Google Scholar] [CrossRef]
- Bresciani, G.; Marco Bortoluzzi, M.; Marchetti, F.; Pampaloni, G. Iron(III) N,N-Dialkylcarbamate-Catalyzed Formation of Cyclic Carbonates from CO2 and Epoxides under Ambient Conditions by Dynamic CO2 Trapping as Carbamato Ligands. ChemSusChem 2018, 11, 2737–2743. [Google Scholar] [CrossRef]
- Arayachukiat, S.; Kongtes, C.; Barthel, A.; Vummaleti, S.V.C.; Poater, A.; Wannakao, S.; Cavallo, L.; D’Elia, V. Ascorbic Acid as a Bifunctional Hydrogen Bond Donor for the Synthesis of Cyclic Carbonates from CO2 under Ambient Conditions. ACS Sustain. Chem. Eng. 2017, 5, 6392–6397. [Google Scholar] [CrossRef]
- Sopeña, S.; Martin, E.; Escudero-Adán, E.C.; Kleij, A.W. Pushing the Limits with Squaramide-Based Organocatalysts in Cyclic Carbonate Synthesis. ACS Catal. 2017, 7, 3532–3539. [Google Scholar] [CrossRef]
- Kumatabara, Y.; Okada, M.; Shirakawa, S. Triethylamine Hydroiodide as a Simple Yet Effective Bifunctional Catalyst for CO2 Fixation Reactions with Epoxides under Mild Conditions. ACS Sustain. Chem. Eng. 2017, 5, 7295–7301. [Google Scholar] [CrossRef]
- Bettner, H.; Steinbauer, J.; Wulf, C.; Dindaroglu, M.; Schmalz, H.-G.; Werner, T. Organocatalyzed Synthesis of Oleochemical Carbonates from CO2 and Renewables. ChemSusChem 2017, 10, 1076–1079. [Google Scholar] [CrossRef]
- Saptal, V.B.; Bhanage, B.M. Bifunctional Ionic Liquids Derived from Biorenewable Sources as Sustainable Catalysts for Fixation of Carbon Dioxide. ChemSusChem 2017, 10, 1145–1151. [Google Scholar] [CrossRef]
- Ma, R.; He, L.-N.; Liu, X.-F.; Liu, X.; Wang, M.-I. DBU as activator for the N-iodosuccinimide promoted chemical fixation of carbon dioxide with epoxides. J. CO2 Util. 2017, 19, 28–32. [Google Scholar] [CrossRef]
- Zhang, Z.; Fan, F.; Xing, H.; Yang, Q.; Bao, Z.; Ren, Q. Efficient Synthesis of Cyclic Carbonates from Atmospheric CO2 Using a Positive Charge Delocalized Ionic Liquid Catalyst. ACS Sustain. Chem. Eng. 2017, 5, 2841–2846. [Google Scholar] [CrossRef]
- Hirose, T.; Qu, S.; Kodama, K.; Wang, X. Organocatalyst system for disubstituted carbonates from cycloaddition between CO2 and internal epoxides. J. CO2 Util. 2018, 24, 261–265. [Google Scholar] [CrossRef]
- Wang, T.; Zheng, D.; Ma, Y.; Guo, J.; He, Z.; Ma, B.; Liu, L.; Ren, T.; Wang, L.; Zhang, J. Benzyl substituted imidazolium ionic liquids as efficient solvent-free catalysts for the cycloaddition of CO2 with epoxides: Experimental and Theoretic study. J. CO2 Util. 2017, 22, 44–52. [Google Scholar] [CrossRef]
- Yue, S.; Hao, X.J.; Wang, P.-P.; Li, J. Amino acid-based ionic liquids for CO2 conversion to form cyclic carbonate under solvent-free conditions. Mol. Catal. 2017, 433, 420–429. [Google Scholar] [CrossRef]
- Jose, T.; Cañellas, S.; Pericàs, M.A.; Kleij, A.W. Polystyrene-supported bifunctional resorcinarenes as cheap, metal-free and recyclable catalysts for epoxide/CO2 coupling reactions. Green Chem. 2017, 19, 5488–5493. [Google Scholar] [CrossRef]
- Li, K.; Wu, X.; Gu, Q.; Zhao, X.; Yuan, M.; Ma, W.; Ni, W.; Hou, Z. Inclusion complexes of organic salts with β-cyclodextrin as organocatalysts for CO2 cycloaddition with epoxides. RSC Adv. 2017, 7, 14721–14732. [Google Scholar] [CrossRef]
- Hu, J.; Ma, J.; Liu, H.; Qian, Q.; Xiea, C.; Han, B. Dual-ionic liquid system: An efficient catalyst for chemical fixation of CO2 to cyclic carbonates under mild conditions. Green Chem. 2018, 20, 2990–2994. [Google Scholar] [CrossRef]
- Peng, J.; Wang, S.; Yang, H.-J.; Ban, B.; Wei, Z.; Wang, L.; Lei, B. Highly efficient fixation of carbon dioxide to cyclic carbonates with new multi-hydroxyl bis-(quaternary ammonium) ionic liquids as metal-free catalysts under mild conditions. Fuel 2018, 224, 481–488. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Zhao, Y.; Kodama, K.; Hirose, T. Efficient and practical organocatalytic system for the synthesis of cyclic carbonates from carbon dioxide and epoxides: 3-hydroxypyridine /tetra-n-butylammonium iodide. Tetrahedron 2017, 73, 1190–1195. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; Martínez, J.; de la Cruz-Martínez, F.; Caballero, M.P.; Fernández-Baeza, J.; Rodríguez-López, J.; Otero, A.; Lara-Sánchez, A.; Tejeda, J. Development of hydroxy-containing imidazole organocatalysts for CO2 fixation into cyclic carbonates. Catal. Sci. Technol. 2018, 8, 1981–1987. [Google Scholar] [CrossRef]
- Liu, A.-H.; Dang, Y.-L.; Zhou, H.; Zhang, J.-J.; Lu, X.-B. CO2 Adducts of Carbodicarbenes: Robust and Versatile Organocatalysts for Chemical Transformation of Carbon Dioxide into Heterocyclic Compounds. ChemCatChem 2018, 10, 2686–2692. [Google Scholar] [CrossRef]
- Maeda, C.; Sasaki, S.; Takaishi, K.; Ema, T. Calix[4]pyrroles as macrocyclic organocatalysts for the synthesis of cyclic carbonates from epoxides and carbon dioxide. Catal. Sci. Technol. 2018, 8, 4193–4198. [Google Scholar] [CrossRef]
- Hong, M.; Kim, Y.; Kim, H.; Cho, H.J.; Baik, M.-H.; Kim, Y. Scorpionate Catalysts for Coupling CO2 and Epoxides to Cyclic Carbonates: A Rational Design Approach for Organocatalysts. J. Org. Chem. 2018, 83, 9370–9380. [Google Scholar] [CrossRef]
- Liu, N.; Xie, Y.-F.; Wang, C.; Li, S.-J.; Wei, D.; Li, M.; Dai, B. Cooperative Multifunctional Organocatalysts for Ambient Conversion of Carbon Dioxide into Cyclic Carbonates. ACS Catal. 2018, 8, 9945–9957. [Google Scholar] [CrossRef]
- Mosteirin, N.F.; Jehanno, C.; Ruiperez, F.; Sardon, H.; Dove, A.P. Rational study of DBU salts for the CO2 insertion into epoxides for the synthesis of cyclic carbonates. ACS Sustain. Chem. Eng. 2019. [Google Scholar] [CrossRef]
- Wang, S.; Song, K.; Zhang, C.; Shu, Y.; Li, T.; Tan, B. A novel metalporphyrin-based microporous organic polymer with high CO2 uptake and efficient chemical conversion of CO2 under ambient conditions. J. Mater. Chem. A 2017, 5, 1509–1515. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, R.; Xu, Q.; Zhang, W.; Zhou, X.; Ji, H. State-of-the-Art Aluminum Porphyrin-based Heterogeneous Catalysts for the Chemical Fixation of CO2 into Cyclic Carbonates at Ambient Conditions. ChemCatChem 2017, 9, 767–773. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, R.; Xu, Q.; Jiang, J.; Zhou, X.; Ji, H. Charged Metalloporphyrin Polymers for Cooperative Synthesis of Cyclic Carbonates from CO2 under Ambient Conditions. ChemSusChem 2017, 10, 2534–2541. [Google Scholar] [CrossRef]
- Zhou, K.; Mousavi, B.; Luo, Z.; Phatanasri, S.; Chaemchuen, S.; Verpoort, F. Characterization and properties of Zn/Co zeolitic imidazolate frameworks vs. ZIF-8 and ZIF-67. J. Mater. Chem. A 2017, 5, 952–957. [Google Scholar] [CrossRef]
- Zanon, A.; Chaemchuen, S.; Mousavi, B.; Verpoort, F. 1 Zn-doped ZIF-67 as catalyst for the CO2 fixation into cyclic carbonates. J. CO2 Util. 2017, 20, 282–291. [Google Scholar] [CrossRef]
- Buyukcakir, O.; Je, S.H.; Talapaneni, S.N.; Kim, D.; Coskun, A. Charged Covalent Triazine Frameworks for CO2 Capture and Conversion. ACS Appl. Mater. Interfaces 2017, 9, 7209–7216. [Google Scholar] [CrossRef]
- Guo, Z.; Jiang, Q.; Shi, Y.; Li, J.; Yang, X.; Hou, W.; Zhou, Y.; Wang, J. Tethering Dual Hydroxyls into Mesoporous Poly(ionic liquid)s for Chemical Fixation of CO2 at Ambient Conditions: A Combined Experimental and Theoretical Study. ACS Catal. 2017, 7, 6770–6780. [Google Scholar] [CrossRef]
- Li, P.-Z.; Wang, X.-J.; Liu, J.; Phang, H.S.; Li, Y.; Zhao, Y. Highly Effective Carbon Fixation via Catalytic Conversion of CO2 by an Acylamide-Containing Metal−Organic Framework. Chem. Mater. 2017, 29, 9256–9261. [Google Scholar] [CrossRef]
- Lu, B.-B.; Yang, J.; Liu, Y.-Y.; Ma, J.F. A Polyoxovanadate−Resorcin[4]arene-Based Porous Metal−Organic Framework as an Efficient Multifunctional Catalyst for the Cycloaddition of CO2 with Epoxides and the Selective Oxidation of Sulfides. Inorg. Chem. 2017, 56, 11710–11720. [Google Scholar] [CrossRef]
- Mousavi, B.; Chaemchuen, S.; Moosavi, B.; Zhou, K.; Yusubov, M.; Verpoort, F. CO2 Cycloaddition to Epoxides by using M-DABCO Metal-Organic Frameworks and the Influence of the Synthetic Method on Catalytic Reactivity. ChemistryOpen 2017, 6, 674–680. [Google Scholar] [CrossRef]
- Ding, M.; Chen, S.; Liu, X.-Q.; Sun, L.-B.; Lu, J.; Jiang, H.-L. Metal–Organic Framework-Templated Catalyst: Synergy in Multiple Sites for Catalytic CO2 Fixation. ChemSusChem 2017, 10, 1898–1903. [Google Scholar] [CrossRef]
- Song, L.; Zhang, X.; Chen, C.; Liu, X.; Zhang, N. UTSA-16 as an efficient microporous catalyst for CO2 conversion to cyclic carbonates. Microporous Mesoporous Mater. 2017, 241, 36–42. [Google Scholar] [CrossRef]
- Liu, M.; Lan, J.; Liang, L.; Sun, J.; Arai, M. Heterogeneous catalytic conversion of CO2 and epoxides to cyclic carbonates over multifunctional tri-s-triazine terminal-linked ionic liquids. J. Catal. 2017, 347, 138–147. [Google Scholar] [CrossRef]
- Zhong, W.; Bobbink, F.D.; Fei, Z.; Dyson, P.J. Polyimidazolium Salts: Robust Catalysts for the Cycloaddition of Carbon Dioxide into Carbonates in Solvent-Free Conditions. ChemSusChem 2017, 10, 2728–2735. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Van Muyden, A.P.; Gopakumar, A.; Fei, Z.; Dyson, P.J. Synthesis of Cross-linked Ionic Poly(styrenes) and their Application as Catalysts for the Synthesis of Carbonates from CO2 and Epoxides. ChemPlusChem 2017, 82, 144–151. [Google Scholar] [CrossRef]
- Zhang, X.; Su, D.; Xiao, L.; Wu, W. Immobilized protic ionic liquids: Efficient catalysts for CO2 fixation with epoxides. J. CO2 Util. 2017, 17, 37–42. [Google Scholar] [CrossRef]
- Ravi, S.; Puthiaraj, P.; Ahn, W.-S. Cyclic carbonate synthesis from CO2 and epoxides over diamine functionalized porous organic frameworks. J. CO2 Util. 2017, 21, 450–458. [Google Scholar] [CrossRef]
- Steinbauer, J.; Longwitz, L.; Frank, M.; Epping, J.; Kragld, U.; Werner, T. Immobilized bifunctional phosphonium salts as recyclable organocatalysts in the cycloaddition of CO2 and epoxides. Green. Chem. 2017, 19, 4435–4445. [Google Scholar] [CrossRef]
- Zhi, Y.; Shao, P.; Feng, X.; Xia, H.; Zhang, Y.; Shi, Z.; Mua, Y.; Liu, X. Covalent organic frameworks: Efficient, metal-free, heterogeneous organocatalysts for chemical fixation of CO2 under mild conditions. J. Mater. Chem. A 2018, 6, 374–382. [Google Scholar] [CrossRef]
- Maya, M.M.; Rangel-Rangel, E.; Díaz, U.; Iglesias, M. Efficient cycloaddition of CO2 to epoxides using novel heterogeneous organocatalysts based on tetramethylguanidine-functionalized porous polyphenylenes. J. CO2 Util. 2018, 25, 170–179. [Google Scholar] [CrossRef]
- Song, X.; Wu, Y.; Pan, D.; Wei, R.; Gao, L.; Zhang, J.; Xiao, G. Melem based multifunctional catalyst for chemical fixation of carbon dioxide into cyclic carbonate. J. CO2 Util. 2018, 24, 287–297. [Google Scholar] [CrossRef]
- Subramanian, S.; Park, J.; Byun, J.; Jung, Y.; Yavuz, C.T. Highly Efficient Catalytic Cyclic Carbonate Formation by Pyridyl Salicylimines. ACS Appl. Mater. Interfaces 2018, 10, 9478–9484. [Google Scholar] [CrossRef]
- Wang, X.; Liu, M.S.; Yang, L.; Lan, J.W.; Chen, Y.L.; Sun, J.M. Synthesis of Zn Modified Carbon Nitrides Heterogeneous Catalyst for the Cycloaddition of CO2 to Epoxides. ChemistrySelect 2018, 3, 4101–4109. [Google Scholar] [CrossRef]
- Liu, D.; Li, G.; Liu, H. Functionalized MIL-101 with imidazolium-based ionic liquids for the cycloaddition of CO2 and epoxides under mild condition. Appl. Surf. Sci. 2018, 428, 218–225. [Google Scholar] [CrossRef]
- Shi, X.-L.; Chen, Y.; Duan, P.; Zhang, W.; Hu, Q. Conversion of CO2 into Organic Carbonates over a Fiber-Supported Ionic Liquid Catalyst in Impellers of the Agitation System. ACS Sustain. Chem. Eng. 2018, 6, 7119–7127. [Google Scholar] [CrossRef]
- Jayakumar, S.; Li, H.; Tao, L.; Li, C.; Liu, L.; Chen, J.; Yang, Q. Cationic Zn-Porphyrin Immobilized in Mesoporous Silicas as Bifunctional Catalyst for CO2 Cycloaddition Reaction under Cocatalyst Free Conditions. ACS Sustain. Chem. Eng. 2018, 6, 9237–9245. [Google Scholar] [CrossRef]
- Jayakumar, S.; Li, H.; Chen, J.; Yang, Q. Cationic Zn−Porphyrin Polymer Coated onto CNTs as a Cooperative Catalyst for the Synthesis of Cyclic Carbonates. ACS Appl. Mater. Interfaces 2018, 10, 2546–2555. [Google Scholar] [CrossRef]
- Tiffner, M.; Häring, M.; Díaz Díaz, D.; Waser, M. Cationic Polymers Bearing Quaternary Ammonium Groups-Catalyzed CO2 Fixation with Epoxides. Top. Catal. 2018, 61, 1545–1550. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, S.; Wu, Q.; Wang, X.; Wu, H.; Jiang, Z. Heterobimetallic Metal-Organic Framework Nanocages as Highly Efficient Catalysts for CO2 Conversion under Mild Condition. J. Mater. Chem. A 2018, 6, 2964–2973. [Google Scholar] [CrossRef]
- Li, Y.-Z.; Wang, H.-H.; Yang, H.-Y.; Hou, L.; Wang, Y.-Y.; Zhu, Z. An Uncommon Carboxyl-Decorated Metal–Organic Framework with Selective Gas Adsorption and Catalytic Conversion of CO2. Chem. Eur. J. 2018, 24, 865–871. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, H.; Wu, H.; Qian, Y.; Chen, L.; Chen, J. Chemical Fixation of CO2 by Using Carbon Material-Grafted N-Heterocyclic Carbene Silver and Copper Complexes. ACS Appl. Nano Mater. 2018, 1, 6463–6476. [Google Scholar] [CrossRef]
- Calabrese, C.; Liotta, L.F.; Giacalone, F.; Gruttadauria, M.; Aprile, C. Supported Polyhedral Oligomeric Silsesquioxane-Based (POSS) Materials as Highly Active Organocatalysts for the Conversion of CO2. ChemCatChem 2019, 11, 560–567. [Google Scholar] [CrossRef]
- Rintjema, J.; Kleij, A.W. Aluminum-mediated formation of cyclic carbonates: Benchmarking catalytic performance metrics. ChemSusChem 2017, 10, 1274–1282. [Google Scholar] [CrossRef]
- Yingcharoen, P.; Kongtes, C.; Arayachukiat, S.; Suvarnapunya, K.; Vummaleti, S.V.C.; Wannakao, S.; Cavallo, L.; Poater, A.; D’Elia, V. Assessing the pKa-Dependent Activity of Hydroxyl Hydrogen Bond Donors in the Organocatalyzed Cycloaddition of Carbon Dioxide to Epoxides: Experimental and Theoretical Study. Adv. Synth. Catal. 2019, 361, 366–373. [Google Scholar] [CrossRef]
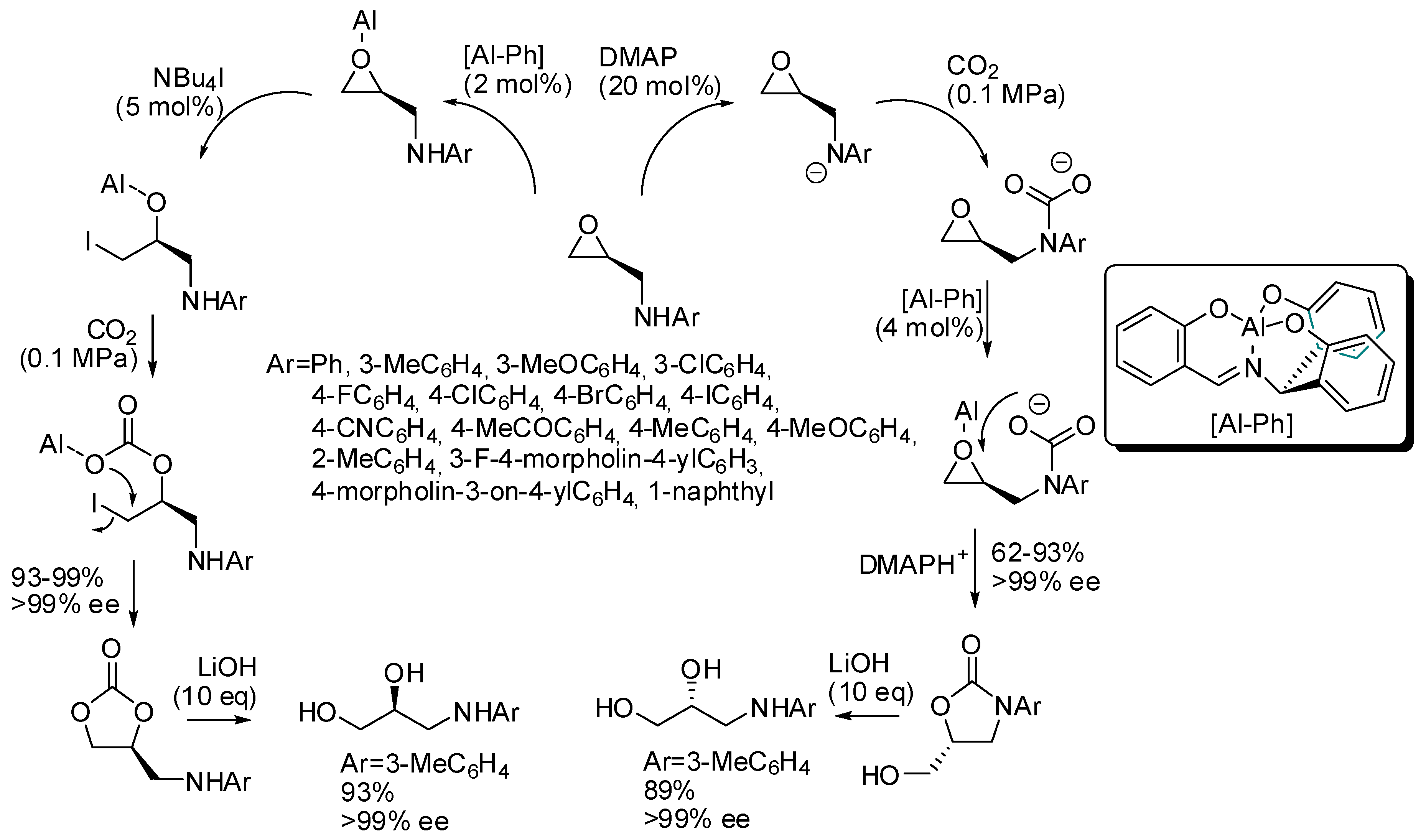
- Laserna, V.; Martin, E.; Escudero-Adán, E.C.; Kleij, A.W. Substrate-Triggered Stereoselective Preparation of Highly Substituted Organic Carbonates. ACS Catal. 2017, 7, 5478–5482. [Google Scholar] [CrossRef]
- Lee, Y.; Choi, J.; Kim, H. Stereocontrolled, Divergent, Al(lll)-Catalyzed Coupling of Chiral N-Aryl Epoxy Amines and CO2. Org. Lett. 2018, 20, 5036–5039. [Google Scholar] [CrossRef]
- Jawad, A.; Rezaei, F.; Rownaghi, A.A. Porous polymeric hollow fibers as bifunctional catalysts for CO2 conversion to cyclic carbonates. J. CO2 Util. 2017, 21, 589–596. [Google Scholar] [CrossRef]
- Liu, F.; Huang, K.; Wu, Q.; Dai, S. Solvent-Free Self-Assembly to the Synthesis of Nitrogen-Doped Ordered Mesoporous Polymers for Highly Selective Capture and Conversion of CO2. Adv. Mater. 2017, 29, 1700445. [Google Scholar] [CrossRef]
- Kim, K.; Kim, S.; Talapaneni, S.N.; Buyukcakir, O.; Almutawa, A.M.I.; Polychronopoulou, K.; Coskun, A. Transition metal complex directed synthesis of porous cationic polymers for efficient CO2 capture and conversion. Polymer 2017, 126, 296–302. [Google Scholar] [CrossRef]
- Mirabaud, A.; Mulatier, J.-C.; Martinez, A.; Dutasta, J.-P.; Dufaud, V. Merging host-guest chemistry and organocatalysis for the chemical valorization of CO2. Catal. Today 2017, 281, 387–391. [Google Scholar] [CrossRef]
- Bresciani, G.; Marchetti, F.; Rizzi, G.; Gabbani, A.; Pineider, F.; Pampaloni, G. Metal N,N-dialkylcarbamates as easily available catalytic precursors for the carbon dioxide/propylene oxide coupling under ambient conditions. J. CO2 Util. 2018, 28, 168–173. [Google Scholar] [CrossRef]
- Samanta, S.; Srivastava, R. A novel method to introduce acidic and basic bifunctional sites in graphitic carbon nitride for sustainable catalysis: Cycloaddition, esterification, and transesterification reactions. Sustain. Energy Fuels 2017, 1, 1390–1404. [Google Scholar] [CrossRef]
- Kim, H.G.; Limb, C.S.; Kim, D.-W.; Cho, D.H.; Lee, D.K.; Chung, J.S. Multifunctional alkanolamine as a catalyst for CO2 and propylene oxide cycloaddition. Mol. Catal. 2017, 438, 121–129. [Google Scholar] [CrossRef]
- Onyenkeadi, V.; Kellici, S.; Saha, B. Greener synthesis of 1,2-butylene carbonate from CO2 using graphene-inorganic nanocomposite catalyst. Energy 2018, 165A, 867–876. [Google Scholar] [CrossRef]
- Liu, W.; Lu, G.; Xiao, B.; Xie, C. Potassium iodide–polyethylene glycol catalyzed cycloaddition reaction of epoxidized soybean oil fatty acid methyl esters with CO2. RSC Adv. 2018, 8, 30860–30867. [Google Scholar] [CrossRef]
- Liu, S.; Suematsu, N.; Maruoka, K.; Shirakawa, S. Design of bifunctional quaternary phosphonium salt catalysts for CO2 fixation reaction with epoxides under mild conditions. Green Chem. 2016, 18, 4611–4615. [Google Scholar] [CrossRef]
- Ema, T.; Yokoyama, M.; Watanabe, S.; Sasaki, S.; Ota, H.; Takaishi, K. Chiral Macrocyclic Organocatalysts for Kinetic Resolution of Disubstituted Epoxides with Carbon Dioxide. Org. Lett. 2017, 19, 4070–4073. [Google Scholar] [CrossRef]
- Maeda, C.; Mitsuzane, M.; Ema, T. Chiral Bifunctional Metalloporphyrin Catalysts for Kinetic Resolution of Epoxides with Carbon Dioxide. Org. Lett. 2019, 21, 1397–1401. [Google Scholar] [CrossRef]
- Qin, J.; Larionov, V.A.; Harms, K.; Meggers, E. Kinetic Resolution of Epoxides with CO2 Catalyzed by a Chiral-at-Iridium Complex. ChemSusChem 2019, 12, 320–325. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Boyaval, A.; Méreau, R.; De Winter, J.; Gerbaux, P.; Detrembleur, C.; Tassaing, T.; Jérôme, C. Organocatalytic Coupling of CO2 with Oxetane. ChemSusChem 2017, 10, 1128–1138. [Google Scholar] [CrossRef]
- Huang, J.; de Winter, J.; Dove, A.P.; Coulembier, O. Metal-free synthesis of poly(trimethylene carbonate) by efficient valorization of carbon dioxide. Green Chem. 2019, 21, 472–477. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, G.-X.; Lu, X.-B. CO2 Adducts of α-Carbon Alkylated N-Heterocyclic Olefins: Highly Active Organocatalysts for CO2 Chemical Transformation. Asian. J. Org. Chem. 2017, 6, 1264–1269. [Google Scholar] [CrossRef]
- Qiu, J.; Zhao, Y.; Li, Z.; Wang, H.; Fan, M.; Wang, J. Efficient Ionic-Liquid-Promoted Chemical Fixation of CO2 into α-Alkylidene Cyclic Carbonates. ChemSusChem 2017, 10, 1120–1127. [Google Scholar] [CrossRef]
- Panwar, V.; Jain, S.L. Zinc grafted to magnetic nanostarch for cyclic carbonate synthesis from propargylic alcohols and CO2 at room temperature. J. CO2 Util. 2018, 24, 306–314. [Google Scholar] [CrossRef]
- Grignard, B.; Ngassamtounzoua, C.; Gennen, S.; Gilbert, B.; Méreau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Boosting the Catalytic Performance of Organic Salts for the Fast and Selective Synthesis of α-Alkylidene Cyclic Carbonates from Carbon Dioxide and Propargylic Alcohols. ChemCatChem 2018, 10, 2584–2592. [Google Scholar] [CrossRef]
- Jung, M.E.; Piizi, G. gem-Disubstituent Effect: Theoretical Basis and Synthetic Applications. Chem. Rev. 2005, 105, 1735–1766. [Google Scholar] [CrossRef]
- Boyaval, A.; Méreau, R.; Grignard, B.; Detrembleur, C.; Jerome, C.; Tassaing, T. Organocatalytic Coupling of CO2 with a Propargylic Alcohol: A Comprehensive Mechanistic Study. ChemSusChem 2017, 10, 1241–1248. [Google Scholar] [CrossRef]
- Della Ca’, N.; Gabriele, B.; Ruffolo, G.; Veltri, L.; Zanetta, T.; Costa, M. Effective Guanidine-Catalyzed Synthesis of Carbonate and Carbamate Derivatives from Propargyl Alcohols in Supercritical Carbon Dioxide. Adv. Synth. Catal. 2011, 353, 133–146. [Google Scholar] [CrossRef]
- Méreau, R.; Grignard, B.; Boyaval, A.; Detrembleur, C.; Jerome, C.; Tassaing, T. Tetrabutylammonium Salts: Cheap Catalysts for the Facile and Selective Synthesis of α-Alkylidene Cyclic Carbonates from Carbon Dioxide and Alkynols. ChemCatChem 2018, 10, 956–960. [Google Scholar] [CrossRef]
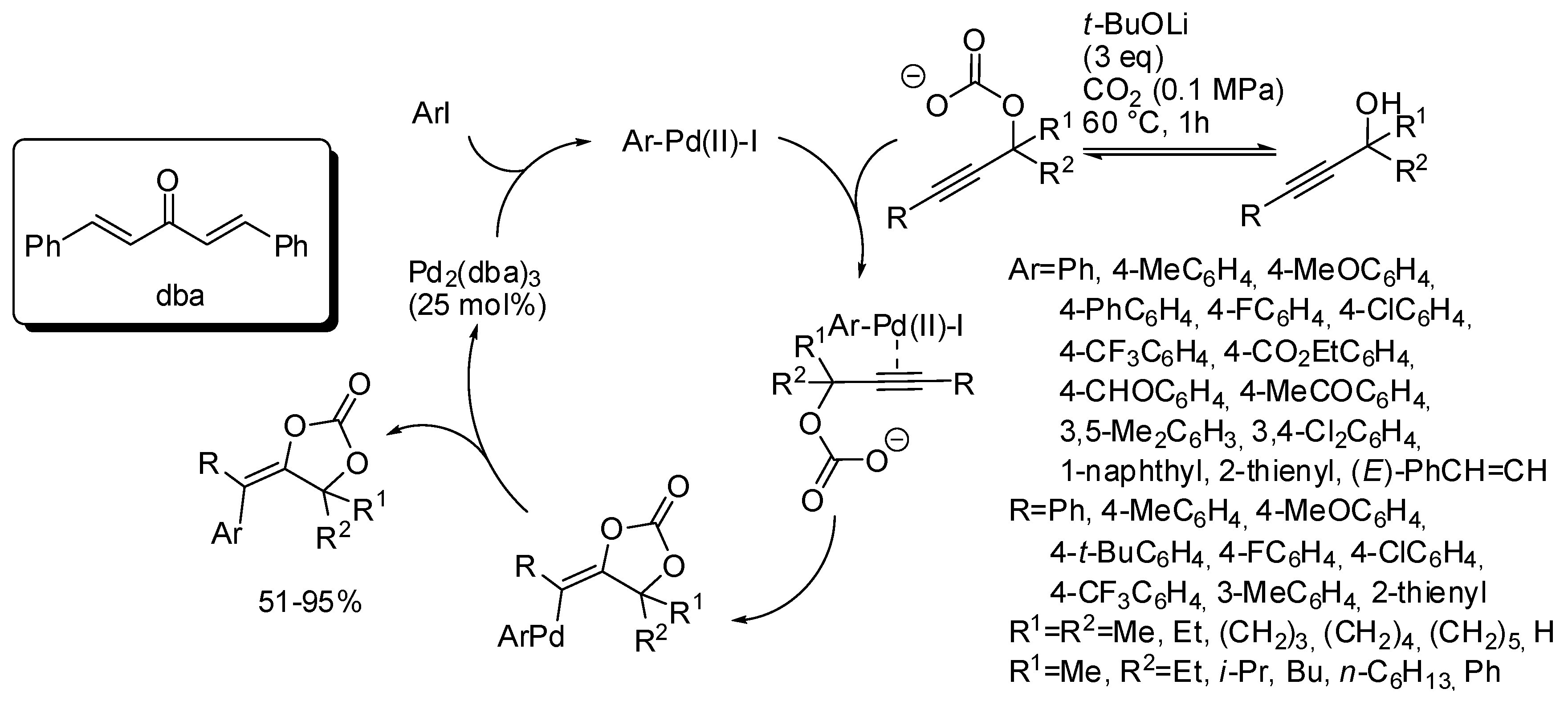
- Sun, S.; Wang, B.; Gu, N.; Yu, J.-T.; Cheng, J. Palladium-Catalyzed Arylcarboxylation of Propargylic Alcohols with CO2 and Aryl Halides: Access to Functionalized α-Alkylidene Cyclic Carbonates. Org. Lett. 2017, 19, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
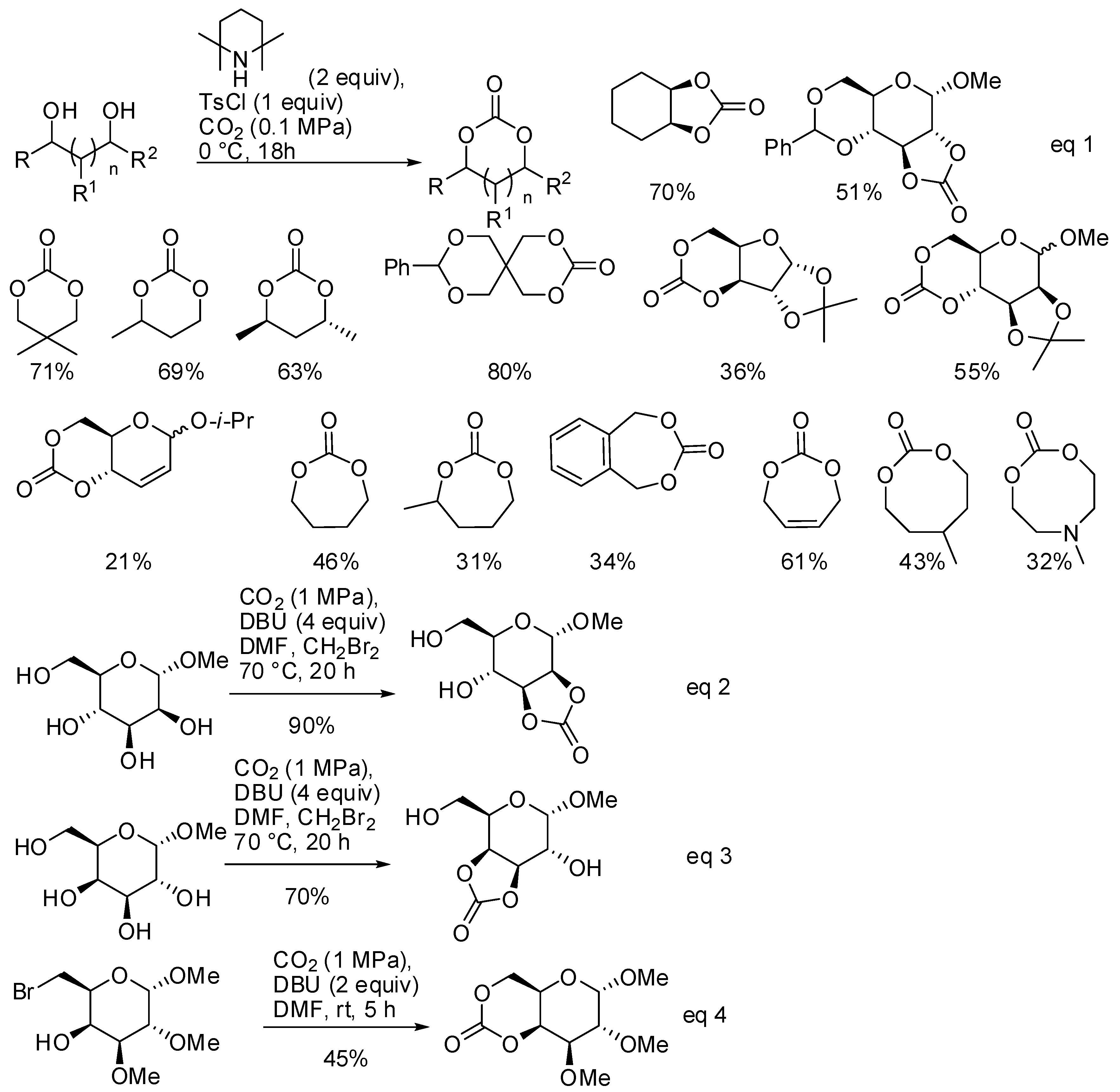
- Gregory, G.L.; Ulmann, M.; Buchard, A. Synthesis of 6-membered cyclic carbonates from 1,3-diols and low CO2 pressure: A novel mild strategy to replace phosgene reagents. RSC Adv. 2015, 5, 39404–39408. [Google Scholar] [CrossRef]
- McGuire, T.M.; López-Vidal, E.M.; Gregory, G.L.; Buchard, A. Synthesis of 5- to 8-membered cyclic carbonates from diols and CO2: A onestep, atmospheric pressure and ambient temperature procedure. J. CO2 Util. 2018, 27, 283–288. [Google Scholar] [CrossRef]
- Pati, D.; Chen, Z.; Feng, X.; Hadjichristidis, N.; Gnanou, Y. Synthesis of polyglycocarbonates through polycondensation of glucopyranosides with CO2. Polym. Chem. 2017, 8, 2640–2646. [Google Scholar] [CrossRef]
- Pati, D.; Feng, X.; Hadjichristidis, N.; Gnanou, Y. CO2 as versatile carbonation agent of glycosides: Synthesis of 5- and 6-membered cyclic glycocarbonates and investigation of their ring-opening. J. CO2 Util. 2018, 24, 564–571. [Google Scholar] [CrossRef]
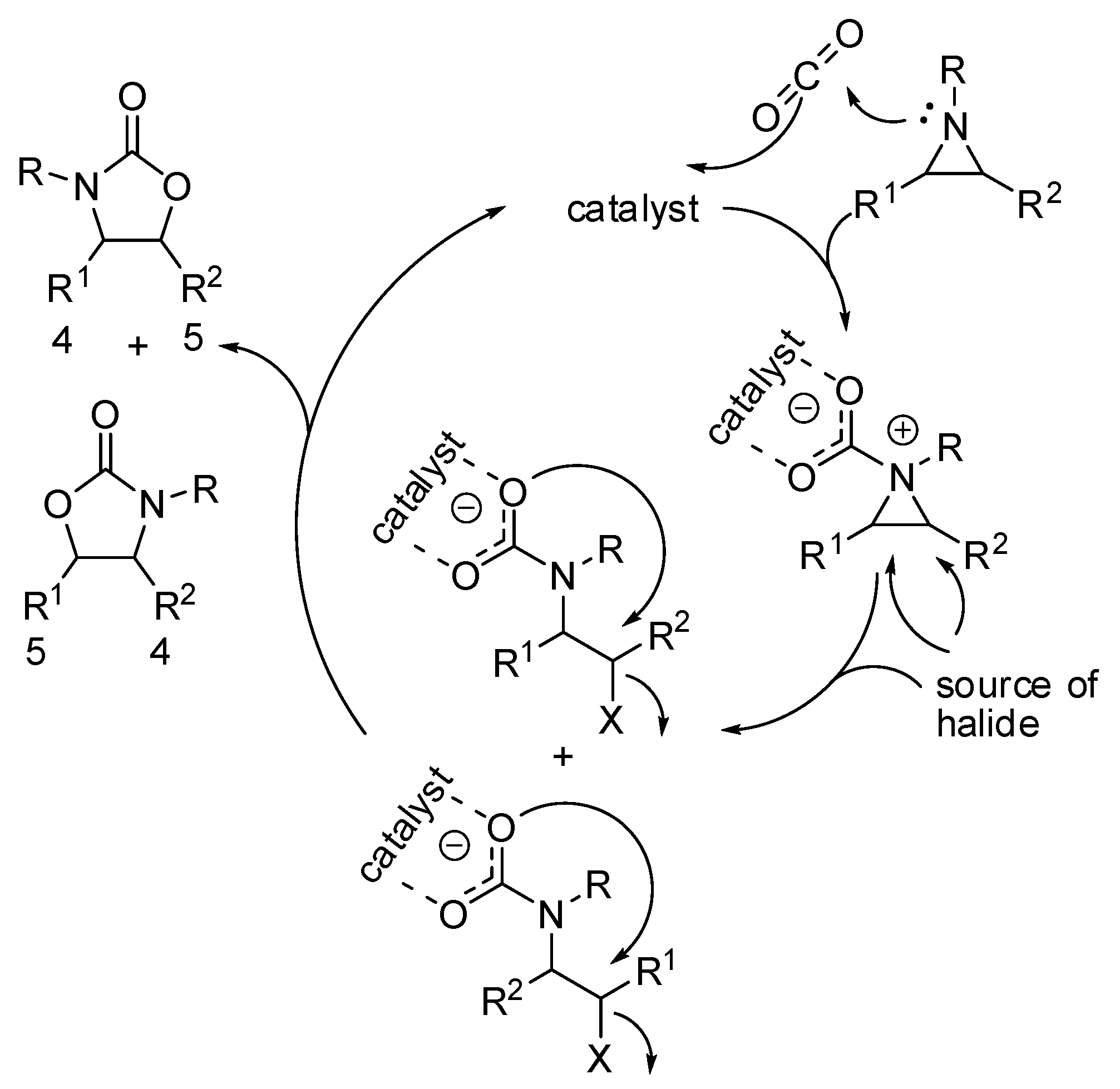
- Xie, Y.; Lu, C.; Zhao, B.; Wang, Q.; Yao, Y. Cycloaddition of Aziridine with CO2/CS2 Catalyzed by Amidato Divalent Lanthanide Complexes. J. Org. Chem. 2019, 84, 1951–1958. [Google Scholar] [CrossRef]
- Phung, C.; Tantillo, D.J.; Hein, J.E.; Pinhas, A.R. The mechanism of the reaction between an aziridine and carbon dioxide with no added catalyst. J. Phys. Org. Chem. 2018, 31, e3735. [Google Scholar] [CrossRef]
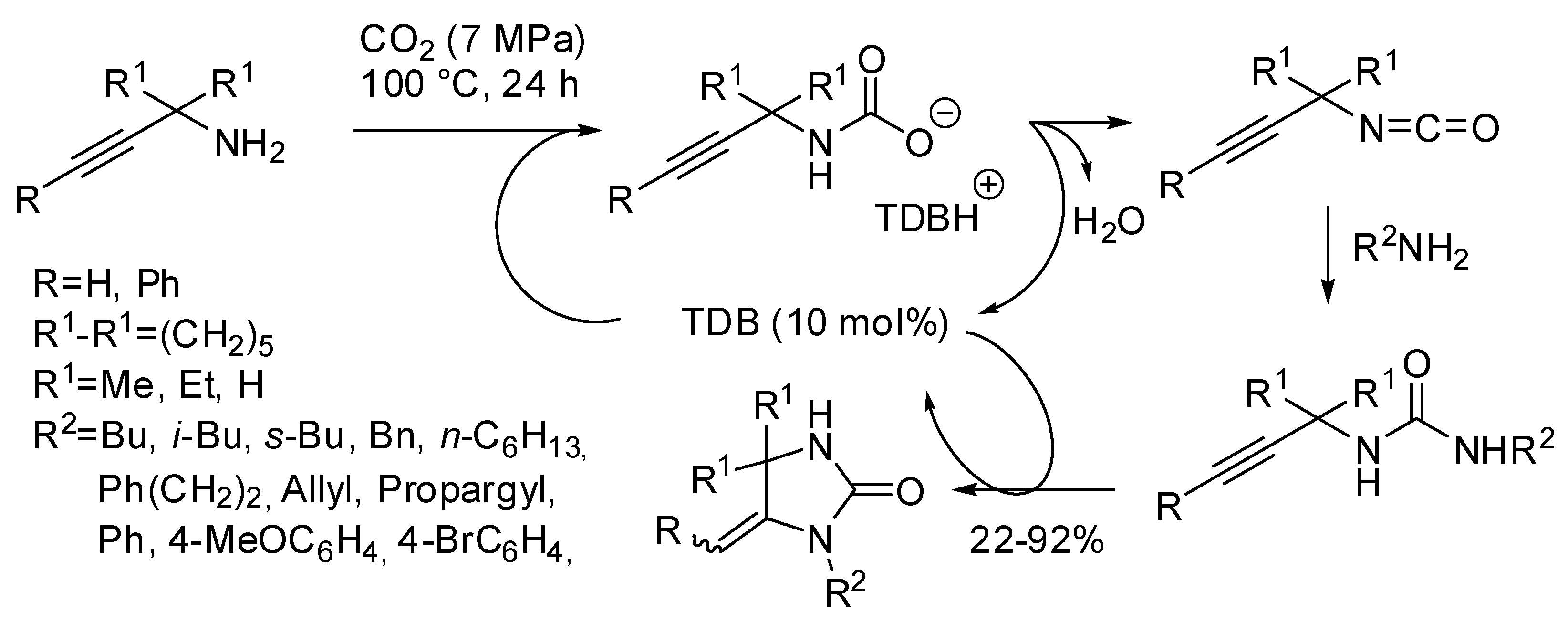
- Zhang, Z.; Ye, J.-H.; Wu, D.-S.; Zhou, Y.-Q.; Yu, D.-G. Synthesis of Oxazolidin-2-ones from Unsaturated Amines with CO2 by Using Homogeneous Catalysis. Chem. Asian. J. 2018, 13, 2292–2306. [Google Scholar] [CrossRef]
- Yuan, R.; Wei, B.; Fu, G. How the Coordinated Structures of Ag(I) Catalysts Affect the Outcomes of Carbon Dioxide Incorporation into Propargylic Amine: A DFT Study. J. Org. Chem. 2017, 82, 3639–3647. [Google Scholar] [CrossRef]
- Brunel, P.; Monot, J.; Kefalidis, C.E.; Maron, L.; Martin-Vaca, B.; Bourissou, D. Valorization of CO2: Preparation of 2-Oxazolidinones by Metal-Ligand Cooperative Catalysis with SCS Indenediide Pd Complexes. ACS Catal. 2017, 7, 2652–2660. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, X.-H.; Zhu, C.; Kang, Y.-S.; Wang, P.; Shi, Z.; Lu, Y.; Sun, W.-Y. Efficient and Reusable Metal–Organic Framework Catalysts for Carboxylative Cyclization of Propargylamines with Carbon Dioxide. ChemCatChem 2017, 9, 4598–4606. [Google Scholar] [CrossRef]
- Yu, B.; Kim, D.; Kim, S.; Hong, S.H. Cyanuric Acid-Based Organocatalyst for Utilization of Carbon Dioxide at Atmospheric Pressure. ChemSusChem 2017, 10, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, M.-Y.; Wang, S.-Y.; Wang, Q.; He, L.-N. In Situ Generated Zinc(II) Catalyst for Incorporation of CO2 into 2-Oxazolidinones with Propargylic Amines at Atmospheric Pressure. ChemSusChem 2017, 10, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qiu, J.; Li, Z.; Wang, H.; Fan, M.; Wang, J. An Experimental and Theoretical Study on the Unexpected Catalytic Activity of Triethanolamine for the Carboxylative Cyclization of Propargylic Amines with CO2. ChemSusChem 2017, 10, 2001–2007. [Google Scholar] [CrossRef] [PubMed]
- Fujii, A.; Choi, J.-C.; Fujita, K.-i. Quaternary ammonium salt-catalyzed carboxylative cyclization of propargylic amines with CO2. Tetrahedron Lett. 2017, 58, 4483–4486. [Google Scholar] [CrossRef]
- Fujii, A.; Matsuo, H.; Choi, J.-C.; Fujitani, T.; Fujita, K.-i. Efficient synthesis of 2-oxazolidinones and quinazoline-2,4(1H,3H)-diones from CO2 catalyzed by tetrabutylammonium fluoride. Tetrahedron 2018, 74, 2914–2920. [Google Scholar] [CrossRef]
- Inagaki, F.; Maeda, K.; Nakazawa, K.; Mukai, C. Construction of the Oxazolidinone Framework from Propargylamine and CO2 in Air at Ambient Temperature: Catalytic Effect of a Gold Complex Featuring an L2/Z-Type Ligand. Eur. J. Org. Chem. 2018, 2972–2976. [Google Scholar] [CrossRef]
- Sadeghzadeh, S.M.; Zhiani, R.; Emrani, S. Ni@Pd nanoparticles supported on ionic liquid-functionalized KCC-1 as robust and recyclable nanocatalysts for cycloaddition of propargylic amines and CO2. Appl. Organometal. Chem. 2018, 32, e3941. [Google Scholar] [CrossRef]
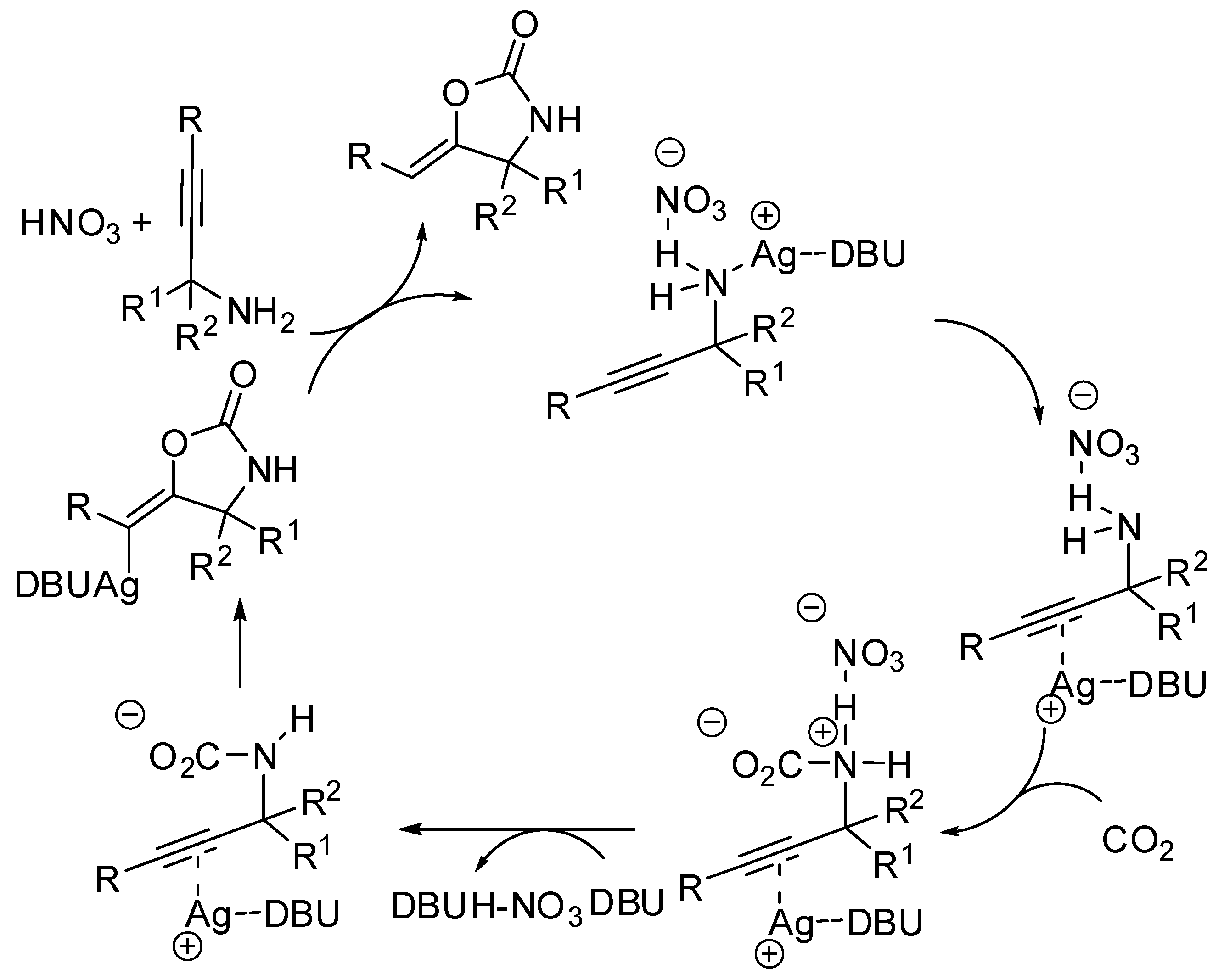
- Yoshida, M.; Mizuguchi, T.; Shishido, K. Synthesis of Oxazolidinones by Efficient Fixation of Atmospheric CO2 with Propargylic Amines by using a Silver/1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) Dual-Catalyst System. Chem. Eur. J. 2012, 18, 15578–15581. [Google Scholar] [CrossRef]
- Inagaki, F.; Okada, Y.; Matsumoto, C.; Yamada, M.; Nakazawa, K.; Mukai, C. Energyless CO2 Absorption, Generation, and Fixation Using Atmospheric CO2. Chem. Pharm. Bull. 2016, 64, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-T.; Gan, C.-C.; Liu, S.-I.; Zhou, F.; Wu, H.-H.; Zhou, J. Utilization of CO2 as a C1 Building Block in a Tandem Asymmetric A3 Coupling-Carboxylative Cyclization Sequence to 2-Oxazolidinones. ACS Catal. 2017, 7, 8588–8593. [Google Scholar] [CrossRef]
- Kim, N.-K.; Sogawa, H.; Yamamoto, K.; Hayashi, Y.; Kawauchi, S.; Takata, T. DBU-mediated Highly Efficient CO2-fixation to Propargylamines and Polypropargylamine. Chem. Lett. 2018, 47, 1063–1066. [Google Scholar] [CrossRef]
- Kim, N.-K.; Sogawa, H.; Felicia, M.D.; Takata, T. DBU-catalyzed CO2 fixation in polypropargylamines under solvent-free conditions. Polym. J. 2019, 51, 351–357. [Google Scholar] [CrossRef]
- Sugiyama, N.; Ohseki, M.; Kobayashi, R.; Sekine, K.; Saito, K.; Yamada, T. Silver-catalyzed Three-component Reaction of Propargylic Amines, Carbon Dioxide, and N-Bromosuccinimide for Stereoselective Preparation of (E)-Bromovinyloxazolidinones. Chem. Lett. 2017, 46, 1323–1326. [Google Scholar] [CrossRef]
- Yin, Z.-B.; Ye, J.-H.; Zhou, W.-J.; Zhang, Y.-H.; Ding, L.; Gui, Y.-Y.; Yan, S.-S.; Li, J.; Yu, D.-G. Oxy-Difluoroalkylation of Allylamines with CO2 via Visible-Light Photoredox Catalysis. Org. Lett. 2018, 20, 190–193. [Google Scholar] [CrossRef]
- Sun, L.; Ye, J.-H.; Zhou, W.-J.; Zeng, X.; Yu, D.-G. Oxy-Alkylation of Allylamines with Unactivated Alkyl Bromides and CO2 via Visible-Light-Driven Palladium Catalysis. Org. Lett. 2018, 20, 3049–3052. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Cao, Y.; Liu, X.; Wang, N.; He, L.-N.; Li, S.-H. Photoinduced radical-initiated carboxylative cyclization of allyl amines with carbon dioxide. Green Chem. 2017, 19, 1240–1244. [Google Scholar] [CrossRef]
- Seo, U.R.; Chung, Y.K. Potassium phosphate-catalyzed one-pot synthesis of 3-aryl-2-oxazolidinones from epoxides, amines, and atmospheric carbon dioxide. Green Chem. 2017, 19, 803–808. [Google Scholar] [CrossRef]
- Lv, M.; Wang, P.; Yuan, D.; Yao, Y. Conversion of Carbon Dioxide into Oxazolidinones Mediated by Quaternary Ammonium Salts and DBU. ChemCatChem 2017, 9, 4451–4455. [Google Scholar] [CrossRef]
- Chen, F.; Li, M.; Wang, J.; Dai, B.; Liu, N. Fe(II) complexes: Reservoirs for Lewis acids and carbenes and their utility in the conversion of CO2 to oxazolidinones. J. CO2 Util. 2018, 28, 181–188. [Google Scholar] [CrossRef]
- Sadeghzadeh, S.M.; Zhiani, R.; Moradi, M. KCC-1 Supported Cu(II)-β-Cyclodextrin Complex as a Reusable Catalyst for the Synthesis of 3-Aryl-2-oxazolidinones from Carbon Dioxide, Epoxide, Anilines. ChemistrySelect 2018, 3, 3516–3522. [Google Scholar] [CrossRef]
- Mancuso, R.; Raut, D.S.; Della Ca’, N.; Fini, F.; Carfagna, C.; Gabriele, B. Catalytic oxidative carbonylation of amino moieties to ureas, oxamides, 2-oxazolidinones, and benzoxazolones. ChemSusChem 2015, 8, 2204–2211. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Song, Q.-W.; Lang, X.-D.; Chang, Y.; He, L.-N. AgI/TMG-Promoted Cascade Reaction of Propargyl Alcohols, Carbon Dioxide, and 2-Aminoethanols to 2-Oxazolidinones. ChemPhysChem 2017, 18, 3182–3188. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Cao, Y.; Ma, R.; He, L.-N. Thermodynamically favorable protocol for the synthesis of 2-oxazolidinones via Cu(I)-catalyzed three-component reaction of propargylic alcohols, CO2 and 2-aminoethanols. J. CO2 Util. 2018, 25, 338–345. [Google Scholar] [CrossRef]
- Xia, S.; Song, Y.; Li, X.; Li, H.; He, L.-N. Ionic Liquid-Promoted Three-Component Domino Reaction of Propargyl Alcohols, Carbon Dioxide and 2-Aminoethanols: A Thermodynamically Favorable Synthesis of 2-Oxazolidinones. Molecules 2018, 23, 3033. [Google Scholar] [CrossRef] [PubMed]
- Niemi, T.; Fernández, I.; Steadman, B.; Mannisto, J.K.; Repo, T. Carbon dioxide-based facile synthesis of cyclic carbamates from amino alcohols. Chem. Commun. 2018, 54, 3166–3169. [Google Scholar] [CrossRef] [PubMed]
- Mei, C.; Zhao, Y.; Chen, Q.; Cao, C.; Pang, G.; Shi, Y. Synthesis of Oxazolidinones and Derivatives through Three-Component Fixation of Carbon Dioxide. ChemCatChem 2018, 10, 3057–3068. [Google Scholar] [CrossRef]
- Yousefi, R.; Struble, T.J.; Payne, J.L.; Vishe, M.; Schley, N.D.; Johnston, J.N. Catalytic, Enantioselective Synthesis of Cyclic Carbamates from Dialkyl Amines by CO2-Capture: Discovery, Development, and Mechanism. J. Am. Chem. Soc. 2019, 141, 618–625. [Google Scholar] [CrossRef]
- Nikpassand, M.; Fekri, L.Z.; Pourahmad, A. One-pot Synthesis of new azo-linked 4H-benzo[d][1,3]oxazine-2,4-diones from carbon dioxide using CuO@RHA/MCM-41 nanocomposite in green media. J. CO2 Util. 2018, 27, 320–325. [Google Scholar] [CrossRef]
- Guo, C.-X.; Zhang, W.-Z.; Zhang, N.; Lu, X.-B. 1,3-Dipolar Cycloaddition of Nitrile Imine with Carbon Dioxide: Access to 1,3,4-Oxadiazole-2(3H)-ones. J. Org. Chem. 2017, 82, 7637–7642. [Google Scholar] [CrossRef]
- Murillo, F.; Barroso, J.; de los Santos, M.G.; Ávila, G.; Pan, S.; Fernández-Herrera, M.A.; Merino, G. Revisiting the Formation Mechanism of 1,3,4-Oxadiazole-2(3H)-ones from Hydrazonyl Chloride and Carbon Dioxide. J. Org. Chem. 2018, 83, 13045–13050. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Zhang, H.; Yuan, G. KI-catalyzed reactions of aryl hydrazines with α-oxocarboxylic acids in the presence of CO2: Access to 1,3,4-oxadiazol-2(3H)-ones. Org. Chem. Front. 2019, 6, 532–536. [Google Scholar] [CrossRef]
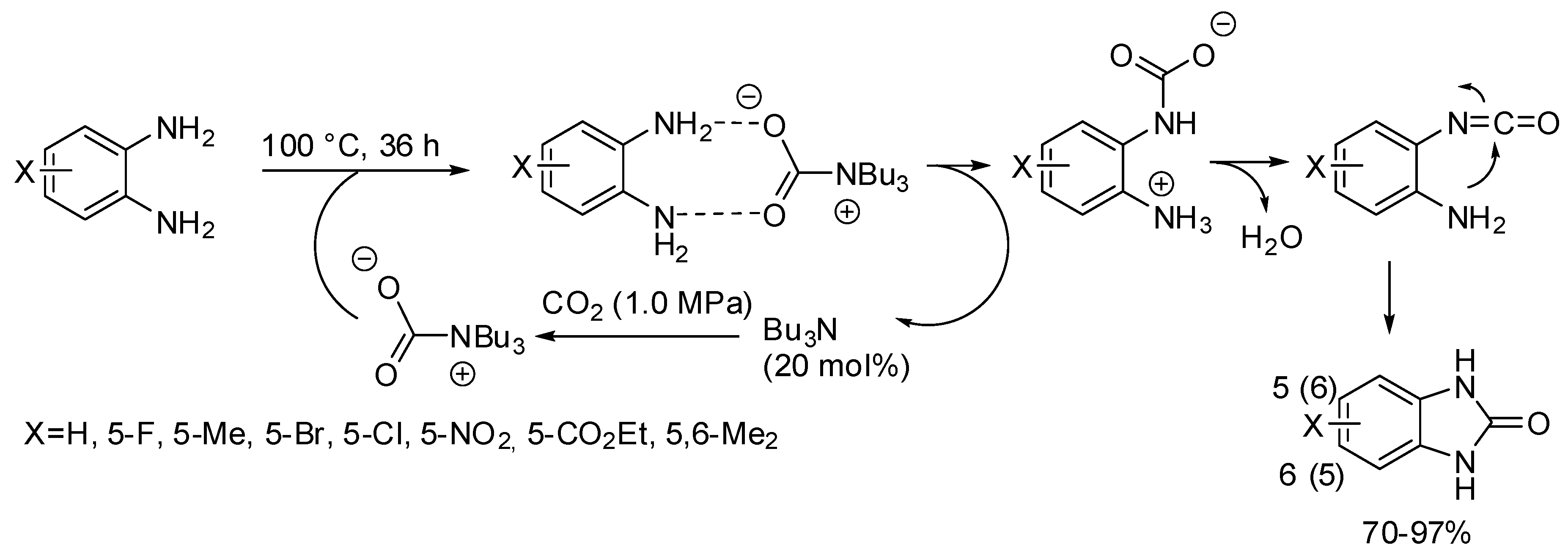
- Brahmayya, M.; Dai, S.A.; Suen, S.-Y. Facile synthesis of 2-benzimidazolones via carbonylation of o-phenylenediamines with CO2. J. CO2 Util. 2017, 22, 135–142. [Google Scholar] [CrossRef]
- Wang, H.; Xin, Z.; Li, Y. Synthesis of Ureas from CO2. Top. Curr. Chem. 2017, 375, 49. [Google Scholar] [CrossRef]
- He, X.; Li, X.-Y.; Song, Y.; Xia, S.-M.; Lang, X.-D.; He, L.-N. Synthesis of Urea Derivatives using Carbon Dioxide as Carbonylation Reagent in Ionic Liquids. Curr. Organocat. 2017, 4, 112–121. [Google Scholar] [CrossRef]
- Hulla, M.; Chamam, S.M.A.; Laurenczy, G.; Das, S.; Dyson, P.J. Delineating the Mechanism of Ionic Liquids in the Synthesis of Quinazoline-2,4(1H,3H)-dione from 2-Aminobenzonitrile and CO2. Angew. Chem. Int. Ed. 2017, 56, 10559–10563. [Google Scholar] [CrossRef] [PubMed]
- Sabet-Sarvestani, H.; Eshghi, H.; Izadyar, M. A theoretical study on the efficiency and role of guanidines-based organic superbases on carbon dioxide utilization in quinazoline-2,4(1H, 3H)-diones synthesis. Struct. Chem. 2017, 28, 675–686. [Google Scholar] [CrossRef]
- Yan, C.; Ren, Y.; Jia, J.-F.; Wu, H.-S. Mechanism of the chemical fixation of carbon dioxide with 2-aminobenzonitrile catalyzed by cesium carbonate: A computational study. Mol. Catal. 2017, 432, 172–186. [Google Scholar] [CrossRef]
- Li, C.; Lu, X.; Yang, Y.; Yang, S.; Zhang, L. Ionic liquid promoted synthesis of heterocycle-fused pyrimidine-2,4-(1H,3H)-diones utilising CO2. Tetrahedron Lett. 2018, 59, 2463–2466. [Google Scholar] [CrossRef]
- Shi, G.; Chen, K.; Wang, Y.; Li, H.; Wang, C. Highly Efficient Synthesis of Quinazoline-2,4(1H,3H)-diones from CO2 by Hydroxyl Functionalized Aprotic Ionic Liquids. ACS Sustain. Chem. Eng. 2018, 6, 5760–5765. [Google Scholar] [CrossRef]
- Hu, J.; Chen, S.; Guo, Y.; Li, L.; Deng, T. Basic Salt-Lake Brine: An Efficient Catalyst for the Transformation of CO2 into Quinazoline-2,4(1H,3H)-diones. ChemSusChem 2018, 11, 4219–4225. [Google Scholar] [CrossRef] [PubMed]
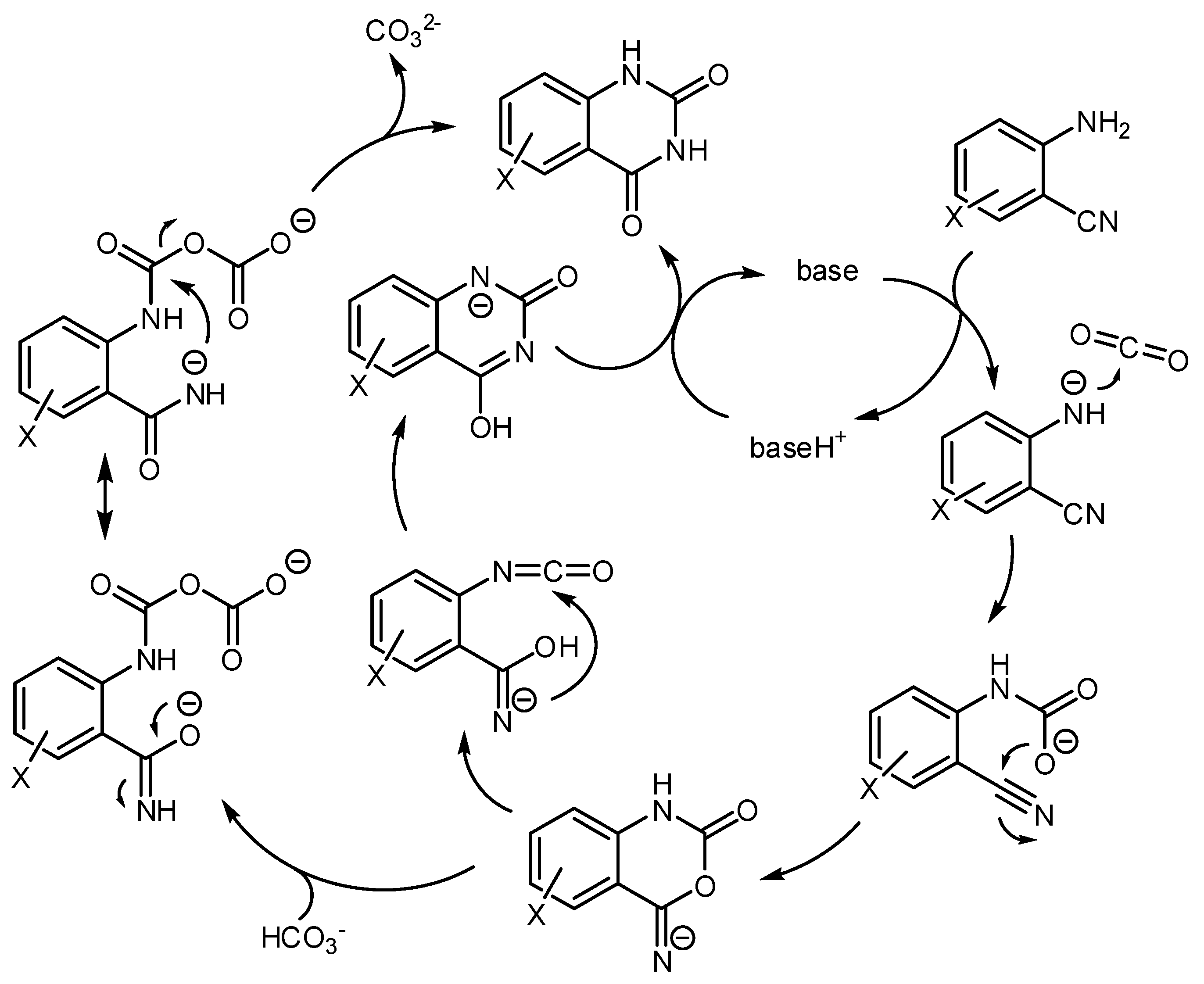
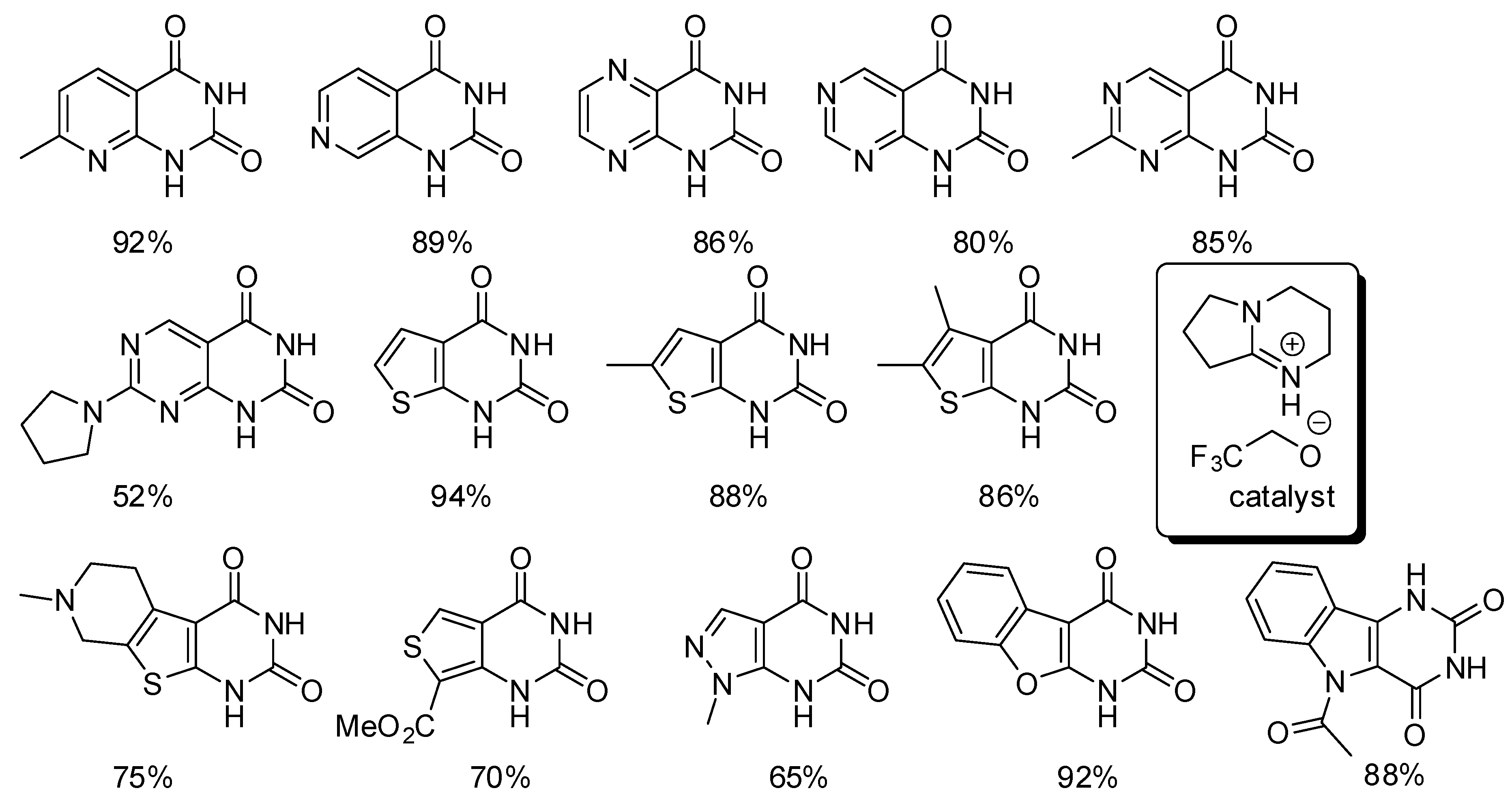
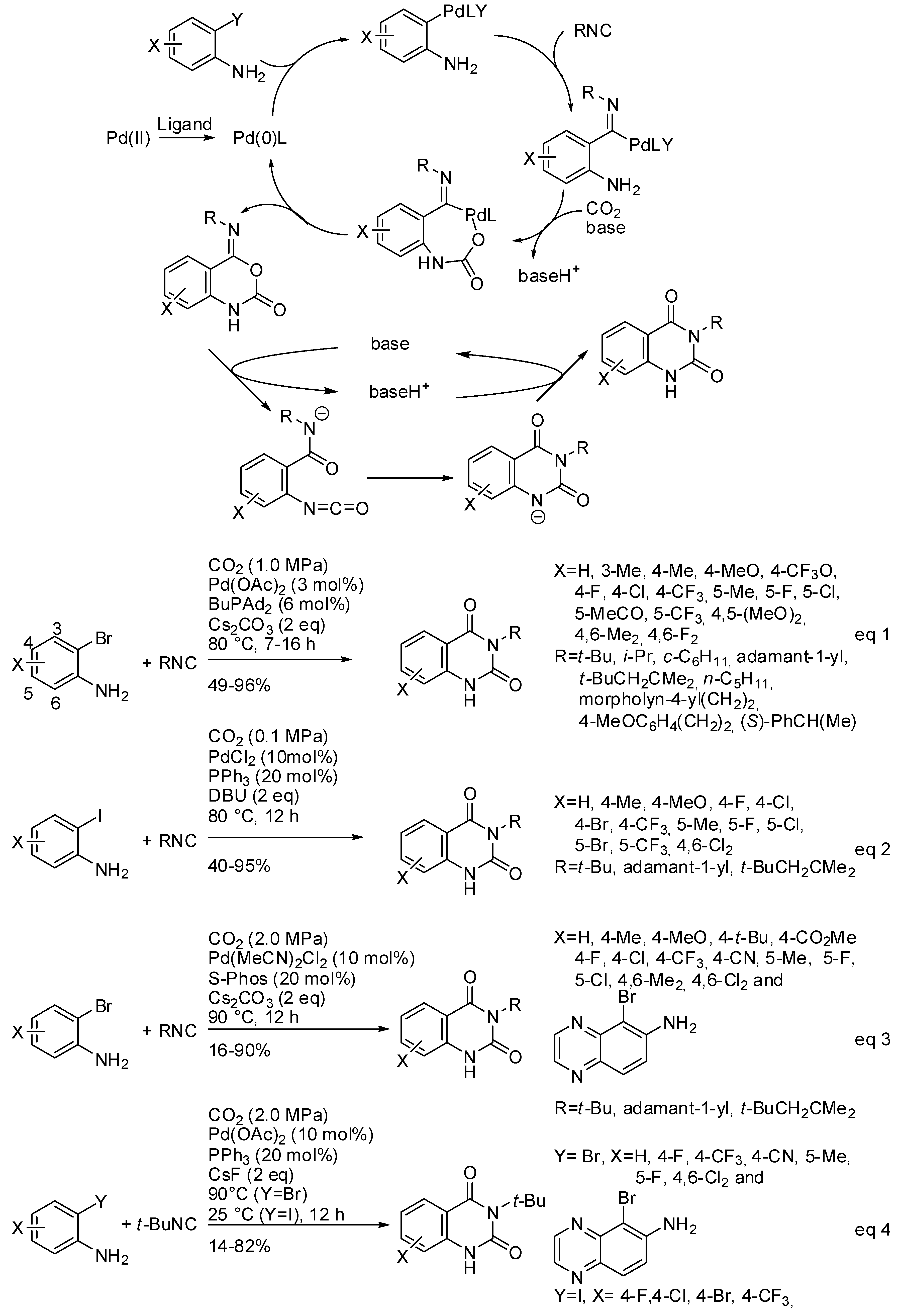
- Mampuys, P.; Neumann, H.; Sergeyev, S.; Orru, R.V.A.; Jiao, H.; Spannenberg, A.; Maes, B.U.W.; Beller, M. Combining Isocyanides with Carbon Dioxide in Palladium-Catalyzed Heterocycle Synthesis: N3-Substituted Quinazoline-2,4-(1H,3H)-diones via a Three-Component Reaction. ACS Catal. 2017, 7, 5549–5556. [Google Scholar] [CrossRef]
- Xu, P.; Wang, F.; Wei, T.-Q.; Yin, L.; Wang, S.-Y.; Ji, S.J. Palladium-Catalyzed Incorporation of Two C1 Building Blocks: The Reaction of Atmospheric CO2 and Isocyanides with 2-Iodoanilines Leading to the Synthesis of Quinazoline-2,4(1H,3H)-diones. Org. Lett. 2017, 19, 4484–4487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-Z.; Li, H.; Zeng, Y.; Tao, X.; Lu, X. Palladium-Catalyzed Cyclization Reaction of o-Haloanilines, CO2 and Isocyanides: Access to Quinazoline-2,4(1H,3H)-diones. Chin. J. Chem. 2018, 36, 112–118. [Google Scholar] [CrossRef]
- Jin, S.-J.; Khan, Y.; Maeng, J.-H.; Kim, Y.J.; Hwang, J.; Cheong, M.; Lee, J.S.; Kim, H.S. Efficient catalytic systems for the carboxylation of diamines to cyclic ureas using ethylene urea as a promoter. Appl. Catal. B Environ. 2017, 209, 139–145. [Google Scholar] [CrossRef]
- Hwang, J.; Han, D.; Oh, J.J.; Cheong, M.; Koo, H.-J.; Lee, J.S.; Kim, H.S. Efficient Non-Catalytic Carboxylation of Diamines to Cyclic Ureas Using 2-Pyrrolidone as a Solvent and a Promoter. Adv. Synth. Catal. 2019, 361, 297–306. [Google Scholar] [CrossRef]
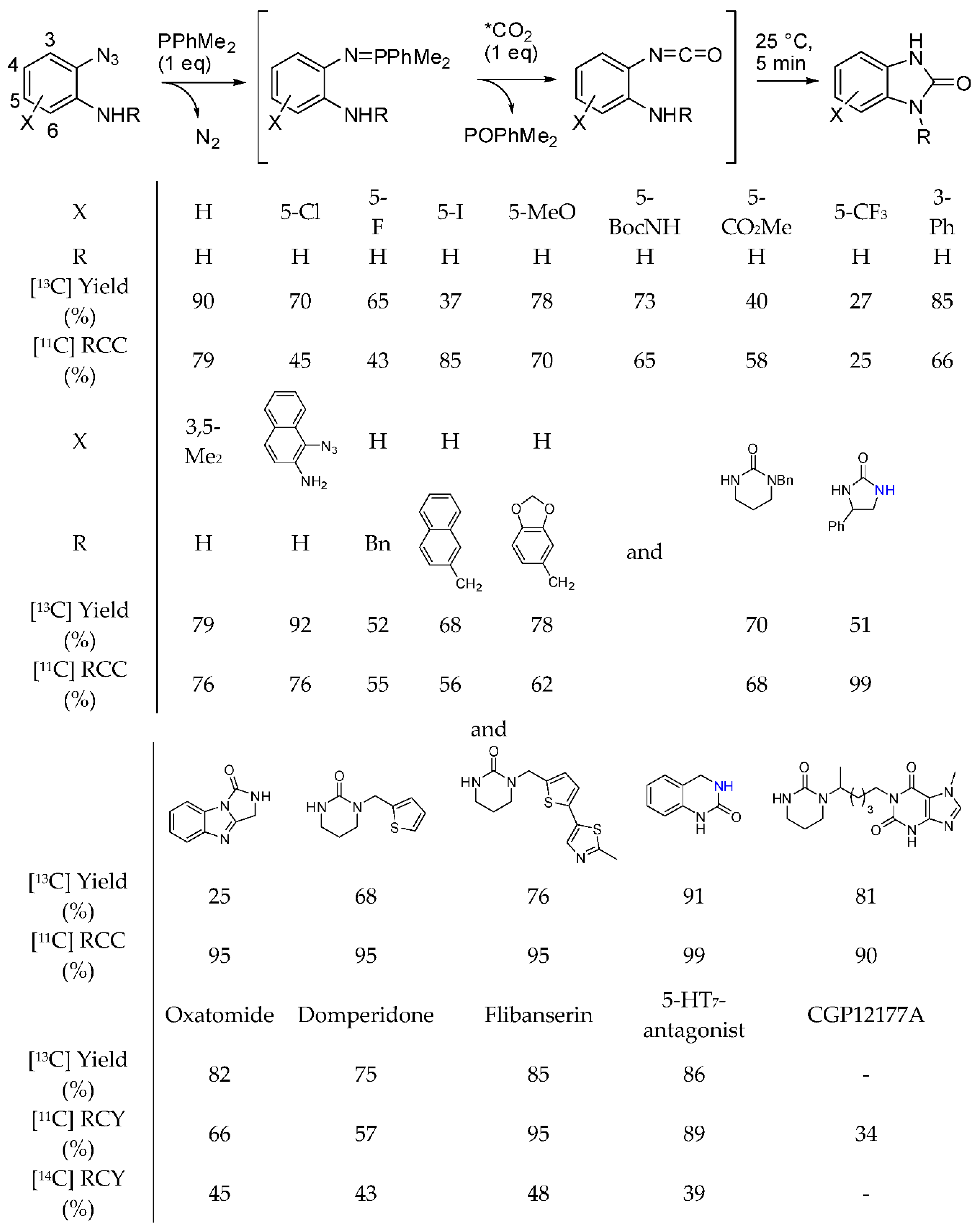
- Del Vecchio, A.; Caillé, F.; Chevalier, A.; Loreau, O.; Horkka, K.; Halldin, C.; Schou, M.; Camus, N.; Kessler, P.; Kuhnast, B.; et al. Late-Stage Isotopic Carbon Labeling of Pharmaceutically Relevant Cyclic Ureas Directly from CO2. Angew. Chem. Int. Ed. 2018, 57, 9744–9748. [Google Scholar] [CrossRef] [PubMed]
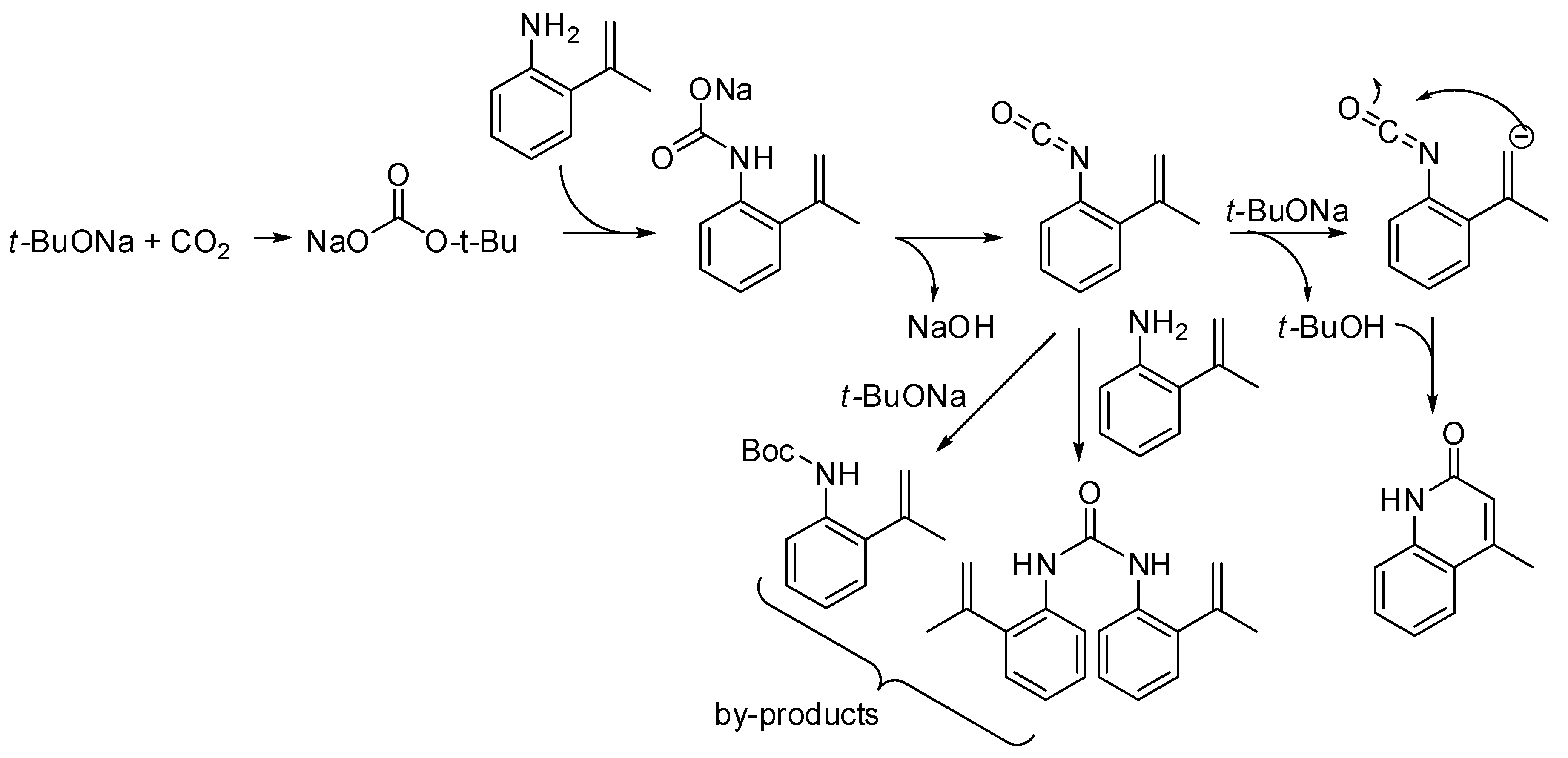
- Streng, E.S.; Lee, D.S.; George, M.W.; Poliakoff, M. Continuous N-alkylation reactions of amino alcohols using γ-Al2O3 and supercritical CO2: Unexpected formation of cyclic ureas and urethanes by reaction with CO2. Beilstein J. Org. Chem. 2017, 13, 329–337. [Google Scholar] [CrossRef]
- Marchegiani, M.; Nodari, M.; Tansini, F.; Massera, C.; Mancuso, R.; Gabriele, B.; Costa, M.; Della Cà, N. Urea derivatives from carbon dioxide and amines by guanidine catalysis: Easy access to imidazolidin-2-ones under solvent-free conditions. J. CO2 Util. 2017, 21, 553–561. [Google Scholar] [CrossRef]
- Chun, S.; Yang, S.; Chung, Y.K. Synthesis of benzothiazoles from 2-aminobenzenethiols in the presence of a reusable polythiazolium precatalyst under atmospheric pressure of carbon dioxide. Tetrahedron 2017, 73, 3438–3442. [Google Scholar] [CrossRef]
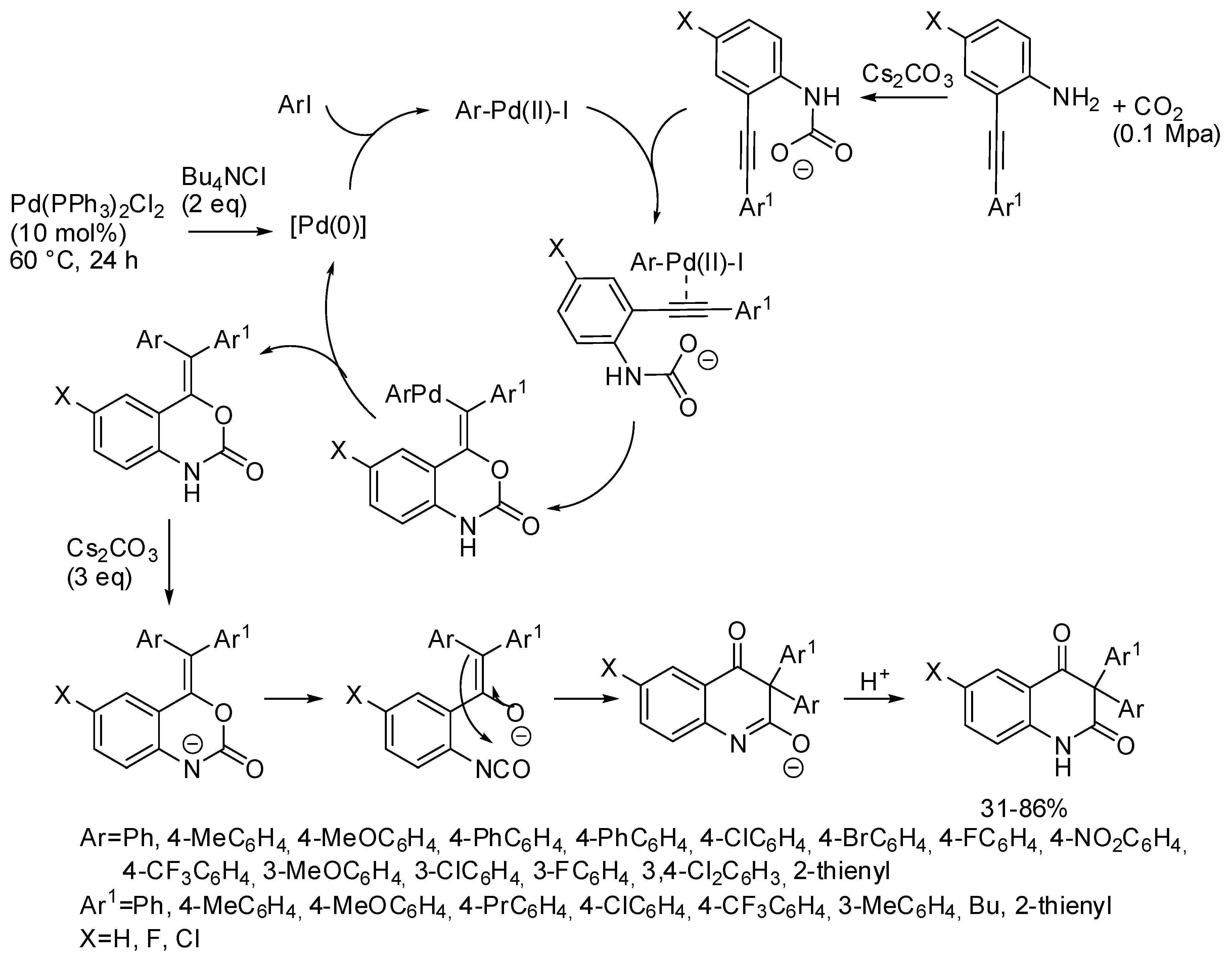
- Wang, B.; Sun, S.; Yu, J.-T.; Jiang, Y.; Cheng, J. Palladium-Catalyzed Multicomponent Reactions of o-Alkynylanilines, Aryl Iodides, and CO2 toward 3,3-Diaryl 2,4-Quinolinediones. Org. Lett. 2017, 19, 4319–4322. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, C.; Lyu, Y. Lactamization of sp2 C–H bonds with CO2 under transition-metal-free and redox-neutral conditions: A computational mechanistic study. Org. Chem. Front. 2018, 5, 2189–2201. [Google Scholar] [CrossRef]
- Wang, S.; Xi, C. Recent advances in nucleophile-triggered CO2-incorporated cyclization leading to heterocycles. Chem. Soc. Rev. 2019, 48, 382–404. [Google Scholar] [CrossRef] [PubMed]
- Niemi, T.; Repo, T. Antibiotics from Carbon Dioxide: Sustainable Pathways to Pharmaceutically Relevant Cyclic Carbamates. Eur. J. Org. Chem. 2019, 1180–1188. [Google Scholar] [CrossRef]
- Vaitla, J.; Guttormsen, Y.; Mannisto, J.K.; Nova, A.; Repo, T.; Bayer, A.; Hopmann, K.H. Enantioselective Incorporation of CO2: Status and Potential. ACS Catal. 2017, 7, 7231–7244. [Google Scholar] [CrossRef]





































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R, R1 | PCO2 (MPa) | Conditions | Yield (%) | Ref |
|---|---|---|---|---|---|
| Homogenous metal catalysts | |||||
| 1 | R=H, R1=Me, Ph, CH2Cl, CH2OAllyl R=R1=(CH2)4 | 1.2 |  or or  Bu4NBr (0.3 mol% each), 80 °C, 6 h | 16–92 | [46] |
| 2 | R=H, R1=Me, Ph R=R1=(CH2)4 | 2.0 |  (0.2 mol%) Bu4NBr (0.4 mol%), 120 °C, 5–18 h | 87–99 | [47] |
| 3 | R=H, R1=Me, Et, Bu, Ph, n-C6H13, CH2Cl,  , Me2, CH2OPh, CH2OBn, CH2O(9H-carbazol-4-yl), , Me2, CH2OPh, CH2OBn, CH2O(9H-carbazol-4-yl),R=R1=(CH2)4, CH2CH(vinyl)(CH2)2 | 0.5 |  (0.3 mol%), Bu4NBr (3 mol%), 25 °C (80 °C for internal epoxides), 24 h | 49–99 | [48] |
| 4 | R=H, R1=Ph, Me, Bu, n-C8H17, n-C10H23, CH2OH, CH2OPh, CH2Cl, 4-ClC6H4, 4-BrC6H4 | 1.0 |  Bu4NBr, 50 °C, 24 h aliphatic epoxides (0.1:0.2 mol%) aromatic epoxides (0.25:0.5 mol%) | 50–94 | [49] |
| 5 | R=H, R1=Me2, R=Ph, R1=Me, Ph (only trans) R=R1=Me, (CH2)4, (CH2)3 |  Bu4NBr, (1:2 mol%), 50 °C, 24 h | 17–60 | ||
| 6 | R=H, R1=Me, Et, Ph, n-C6H13, n-C8H17, CH2Cl, CH2Br, 4-ClC6H4, CH2Ometacrylate, CH2OAllyl, | 1.0 |  ISA6 (1 mol%) 50 °C, 2–12 h | 90–99 | [50] |
| 7 | R=H, R1=Ph, Me, Et, Bu, Bn, 4-MeC6H4, 4-ClC6H4, CH2Cl, CH2O-i-Pr, CH2OBn, CH2OAllyl, CH2OPropargyl | 0.1 |  SalenCoI (1 mol%), Ph3P=CHCHO (1 mol%), 25 °C, 24–36 h | 79–95 | [51] |
| 8 | R=H, R1=Ph, 4-ClC6H4, 4-FC6H4, n-C6H13, Pr, (CH2)2CH=CH2, CH2OAllyl, CH2OBn, CH2Cl, CH2NMe2 | 0.1 |  (10 mol%), 40 °C, 24 h | 86–99 | [52] |
| 9 | R=H, R1=Et, Me2, (R)-Me, (R)-Ph, 4-FC6H4, n-C6H13, (CH2)2CH=CH2, vinyl, CH2Cl, CH2OH, CH2Ot-Bu, CH2OPropargyl, CH2OCH2furyl, CH2Ometacrylate  | 1.0 | CaI2/PEG-500 (5 mol%), 25 °C, 24 h | 84–99 | [53] |
| 10 | R=R1=(CH2)3, (CH2)4, (CH2)5, (CH2)6, Me, CH2OCH2, CH2CH=CHCH2, CH2CH(Me)(CH2)2, CH2CH(vinyl)(CH2)2,  R=C8H17, R1=(CH2)7CO2Me | A: 2.0 B: 5.0 | CaI2/PEG-500 (5 mol%) A: 70 °C, 24 h B: 90 °C, 48 h | 29–98 cis:trans 53:47 to >99:1 cis:trans 11:89 (R=R1= t-Me) | |
| 11 | R=H, R1=CH2O-t-Bu, CH2OH, CH2OMe, CH2OPropargyl, CH2Cl, CH2OAllyl, CH2OCH2furyl, CH2Ometacrylate, CH2OSiMe2t-Bu, CH2OCH2CF2CHF2, CH2OCH2C2F5, CH2OCH2(CF2)3CHF2, Me, Et, Bu, n-C6H13, Ph,  , ,  (R2= Me, CH2Cl, Ph, CO2Me) (R2= Me, CH2Cl, Ph, CO2Me) | 0.1 |  (5 mol%), 23 °C, 24 h | 23–99 | [54] |
| 12 | R=R1=Me, (CH2)3, (CH2)4, (CH2)5, (CH2)6, CH2OCH2, CH=CH(CH2)2, CH2CH(Me)(CH2)2, CH2CH(vinyl)(CH2)2, Me, Ph; R=Ph, R1=Me, CO2Et; R=4-MeOC6H4, R1= CO2Me | 1.0 |  (5 mol%), 45 °C, 48 h | 4–98% | |
| 13 | R=C8H17, R1=(CH2)7CO2Me, (CH2)7CO2Et, (CH2)7CO2(CH2)5-i-Pr, (CH2)11CO2Me,  , R=H, R1=(CH2)8CO2Me, epoxidized methyl O-acetyl ricinoleate, methyl soyate, sunflower, soybean, linseed oils , R=H, R1=(CH2)8CO2Me, epoxidized methyl O-acetyl ricinoleate, methyl soyate, sunflower, soybean, linseed oils | 0.5 |  (5 mol%), Ph3P (5 mol%), 45 °C, 24 h | 55–98 | [55] |
| 14 | (+)-limonene oxide, (+)-limonene dioxide, epoxidized citronellyl propionate,  | 5.0 |  (5 mol%), Ph3P (5 mol%), 45 °C, 48 h | 19–81 | |
| 15 | R=H, R1=Me, Et, Bu, Ph, n-C6H13, CH2Cl, CH2OH, CH2OPh, CH2OBn, CH2OAllyl,  R=R1=(CH2)4 R=R1=(CH2)4 | 0.1 |  (10 mol%), 50 °C, 6 h | 25–99 | [56] |
| 16 | R=H, R1=Ph, CH2O-i-Pr, CH2OAllyl, CH2OPh, CH2Cl | 0.1 | Nanocrytalline MgO (12.5 mg per mmole epoxide), Bu4NBr (50 mol%), r. t., 4–8 h | 42–99 | [57] |
| 17 | R=H, R1=Me, Et, Ph, CH2Cl | 1.6 |  (0.1 mol%), Dimethylaminopyridine (0.2 mol%), 100 °C, 2 h | 54–97 | [58] |
| 18 | R=H, R1=Me, Bu, Ph, n-C10H23, CH2OAllyl, CH2OMe, CH2Cl, CH2OCH2furyl R=R1=(CH2)3, c-Me | 0.1 |  (0.2 mol%), 35 °C, 6 h | 26–65 | [59] |
| 19 | R=H, R1=Ph, 4-ClC6H4, Bu, CH2Cl | 5.0 |  (2 mol%), 50 °C, 24 h | 98 | [60] |
| 20 | R=H, R1=Me, Ph, Et, CH2OAllyl, CH2OH, CH2OPh, CH2Cl,  | 3.0 |  (0.04 mol%), Bu4NBr (0.4 mol%), 100 °C, 6 h | 27–100 | [61] |
| 21 | R=H, R1=Me, Et, Bu, Ph, n-C8H17, CH2Cl, CH2OAllyl | 2.0 |  IL-Zn-TPP (0.1 mol%), 60 °C, 8–30 h | 72–98 | [62] |
| 22 | R=H, R1=Me, Et, Ph, CH2Cl R=R1=(CH2)4, | 1.6 |  (0.1 mol%), Dimethylaminopyridine (0.2 mol%), 100 °C, 2 h | 6–89 | [63] |
| 23 | R=H, R1=Me, Et, Ph, | 0.1 | Fe(O2CNEt3)3 (1 mol%), Bu4NBr (2 mol%), 25 °C, 24 h | 87–90 | [64] |
| Homogenous organocatalysts | |||||
| 24 | R=H, R1= Me, Et, Ph, CH2Cl, CH2=CH | 0.1–0.5 | Ascorbic acid (2–4 mol%), Bu4NI (4–8mol%), r.t.−60 °C, 23 h | 82–97 | [65] |
| 25 | R=R1=(CH2)4, (CH2)3, CH2OCH2, CH2N(Bn)CH2, Me (c:t=94:6),  R=Ph, R1=Me, Ph, CH2OMe, CO2Et (c:t=12:88 to 1:99) | 3.0 |  (3 mol%), Et4NBr (6 mol%), 80 °C, 18 h | 53–90 | [66] |
| 26 | R=H, R1=Ph, Pr, 4-ClC6H4, 4-FC6H4, (CH2)2CH=CH2, CH2OAllyl, CH2OBn, CH2Cl | 0.1 | NEt3•HI (10 mol%), 40 °C, 24 h | 82–99 | [67] |
| 27 | R=H, R1=(CH2)8CO2Me, R=C8H17, R1=(CH2)7CO2Me, (CH2)7CO2Et, (CH2)7CO2(CH2)5-i-Pr, (CH2)9CO2Me, R=CH2CHOH(CH2)5Me, CH2CHOAc(CH2)5Me R1=(CH2)7CO2Me,  epoxidized sunflower, soybean, linseed oils, methyl soyate epoxidized sunflower, soybean, linseed oils, methyl soyate | 2.5 |  (5 mol%), 80 °C, 24 h (5 mol%), 80 °C, 24 h | 62–99 | [68] |
| 28 | R=H, R1=Me, Et, Ph, CH2Cl, 4-FC6H4, 4-ClC6H4, 4-BrC6H4 R=R1=(CH2)4, (CH2)3 | 0.1 |  (10 mol%), Bu4NI (10 mol%), 80 °C, 30 h | 78–92 | [69] |
| 29 | R=H, R1=Me, Me2, Bu, CH2OPh, CH2Cl R=R1=(CH2)4 | 0.1 | 1,5-diazabiciclo(5.4.0)undec-7-ene (DBU) (5 mol%), N-iodosuccinimide (NIS) (5 mol%) 60 °C, 12 h | 71–99 | [70] |
| 30 | R=H, R1=Ph, CH2Cl, CH2Br, CH2OAllyl, CH2O-t-Bu, CH2OPh | 0.1 | 4-(Dimethylammino)pyridinium bromide (DMAPHBr) (1 mol%) 120 °C, 4 h | 94–98 | [71] |
| 31 | R=R1=(CH2)3, (CH2)4, CH2CH(vinyl)(CH2)2, CH2OCH2, c-Ph, t-Ph, c-Me, t-Me | 0.1–0.4 | DBU (5 mol%), nBu4NCl (10 mol%), 120 °C, 24 h. | 41–96 | [72] |
| 32 | R=H, R1=Me, Ph, CH2Cl R=R1=H, (CH2)4 | 2.0 |  (0.25 mol%), 130 °C, 4 h | 52–96 | [73] |
| 33 | R=H, R1=Me, Ph, Bu, n-C6H13, CH2Cl | 0.5 |  (0.3 mol%), 130 °C, 12 h | 96–99 | [74] |
| 34 | R=H, R1=Me, Bu, Ph, n-C6H13, CH2Cl, CH2OH, CH2OPh, CH2OMe, CH2OAllyl R=R1=(CH2)4, (CH2)3, CH2OCH2 | 0.5 |  (1 mol%), 80 °C, 18 h | 41–97 | [75] |
| 35 | R=H, R1=Me, Ph, Bu, CH2Cl, CH2OPh R=R1=(CH2)4, | 3.0 |  / /β-cyclodextrin (1.5 mol%), 130 °C, 10 h | 63–98 | [76] |
| 36 | R=H, R1=Ph, CH2Cl, CH2Br, CH2OPh | 0.1 |  (25 mol%), 30–60 °C, 12*30 h | 94–95 | [77] |
| 37 | R=H, R1=Me, Ph, CH2Cl, CH2O-t-Bu, CH2OBu, CH2OPh, CH2OAllyl,  R=R1=(CH2)4 | 2.0 |  (0.25 mol%), 120 °C, 3 h | 67–99 | [78] |
| 38 | R=H, R1=CH2Cl, (CH2)2CH=CH2, CH2OAllyl, CH2OPh, Me, Bu, n-C6H13, Ph R=R1=(CH2)4 | 0.1 |  (5 mol%), Bu4NI (5 mol%), 25–60 °C, 24 h | 14–95 | [79] |
| 39 | R=H, R1=Bu, Me, Et, n-C8H17,Ph, 4-ClC6H4, 4-BrC6H4, CH2Cl, CH2OH, CH2OPh | 1.0 |  (1 mol%) Bu4NI (1 mol%), 90 °C, 2 h | R2=Ph 52–96 R2=Bu 97–99 | [80] |
| 40 | R=H, R1=Bu, Me, Et, n-C8H17,Ph, 4-ClC6H4, 4-BrC6H4, CH2Cl, CH2OH, CH2OPh, CH2OCH2(2-furyl), CH2OCO(2-furyl),  R=R1=(CH2)3, (CH2)4 |  (0.75 mol%), 80 °C, 1 h | 49–99 | ||
| 41 | R=H, R1=Bu, Me, Ph, CH2OAllyl, CH2OPh, CH2OBn, CH2Cl, CH2Br, CH2Morpholin-4-yl | 2.0 |  CDC–CO2 (5 mol%) 100 °C, 12 h | 77–96 | [81] |
| 42 | R=H, R1=Bu, Ph, C8H17, C12H25, CH2Cl, CH2OEt, CH2OPh, CH2OCOC(Me)=CH2 R=R1=t-Me, (CH2)3, (CH2)4 | 1.7 |  (1 mol%) Bu4NI (1 mol%), 100 °C, 15 h | 48–98 | [82] |
| 43 | R=H, R1=Me, CH2Cl, CH2OH, CH2OMe, CH2O-t-Bu, CH2OPh | 1.0 |  (2 mol%) r. t., 6–12 h | 90–99 | [83] |
| 44 | R=H, R1=Me, Et, Bu, (CH2)2CH=CH2, n-C6H13, Ph, CH2Cl, CH2OBu, CH2OPh, CH2OAllyl, Me2, Me(CH2Cl), CH2O(9H-carbazol-4-yl) | 0.1 |  (4 mol%), r,t., 24 h | 95–73 | [84] |
| 45 | R=R1=Me, (CH2)3, (CH2)4, CH2OCH2, R=C8H17, R1=(CH2)7CO2Me, (CH2)7CO2Et, (CH2)11CO2Me, | 0.1 |  (4 mol%), 80 °C, 24 h | 43–95 | |
| 46 | R=H, R1=Ph, n-C6H13, Bu, CH2OAllyl, CH2OBn, CH2Cl, CH2OH, | 0.1 |  (10 mol%), 70 °C, 4 h | 65–87 | [85] |
| Heterogeneous catalysts | |||||
| 47 | R=H, R1=Me, Et, Ph, CH2Cl, CH2Br, | 0.1 | HUST-1-Co (0.8 mg/mmol), Bu4NBr (7 mol%), rt, 30–48 h | 93–97 | [86] |
| 48 | R=H, R1=Me, Et, Ph, CH2Cl, n-C6H13, n-C10H21, CH2OAllyl | 1.0 | Al-HPC (0.25 mmol%), Bu4NBr (2 mol%), 40 °C, 1–24 h | 85–99 | [87] |
| 49 | R=H, R1=Me, Et, Ph, CH2Cl, n-C6H13, n-C10H21, CH2OAllyl R=R1=(CH2)4 | 1.0 | Al-iPOP-1 or Al-iPOP-2 (0.1 mol%) 40 °C, 3–36 h | 8–99 (1) 14–99 (2) | [88] |
| 50 | R=H, R1=CH2Cl | 0.1 | Zn-Co/ZIF, 80 °C, 24 h | 57 | [89] |
| 51 | R=H, R1=Me, Ph, CH2OPh, CH2Cl, CH2OAllyl, R=R1=(CH2)4, (CH2)6 | 0.7 | Zn-Co/ZIF-67 (Zn:Co 1:9) (50 mg), 100 °C, 2–18 h | 8–99 | [90] |
| 52 | R=H, R1=Me, Ph, Bn, CH2Cl | 1.0 | cCTF-500 (4% wt), 90 °C, 12 h | 36–99 | [91] |
| 53 | R=H, R1=Ph, Bu, CH2OPh, CH2Cl, CH2OAllyl R=R1=(CH2)4 | 0.4 | PGDBr-5–2OH (1.9 mol %), 70 °C, 4–48 h | 90–98 | [92] |
| 0.1 | PGDBr-5–2OH (1.9 mol %), 70 °C, 24–96 h | 90–97 | |||
| 0.1 | PGDBr-5–2OH (1.9 mol %), n-Bu4NI (8 mol %), r. t., 18–120 h | 80–93 | |||
| 54 | R=H, R1=Me, Et, CH2Cl, CH2Br | 0.1 | Cu2[(C20H12N2O2)(COO)4] (0.2 mol%), Bu4NBr (8 mol%), r.t., 48 h | 88–96 | [93] |
| 55 | R=H, R1=Ph, Bu, CH2OPh, CH2OBu, CH2OBn, CH2Cl,  | 0.1 | [Co2(resorcin-4-arene0.5)V4O12]·3DMF 5H2O (0.2 mol%, based on V), Bu4NBr (5 mol%), 80 °C, 12 h | 87–99 | [94] |
| 56 | R=H, R1=Me, Ph, CH2Cl, CH2OPh | 0.8 | Zn2[1,4-(CO2)2C6H4](DABCO) (17 mg per mmol substrate), 100 °C, 12–30 h | 90–99 | [95] |
| 57 | R=H, R1=Me, Pr, Ph, CH2Cl, CH2Br, CH2OPh | 0.1 | ZnO@NPC-Ox-700 (50 mg per mmol substrate), Bu4NBr (20 mol%), 25–60 °C, 1–3 h | 85–99 | [96] |
| 58 | R=H, R1=CH2Cl | 1.0 | KCo3(C6H4O7) (C6H5O7) (H2O)2 (UTSA-16) (0.15 mmol), 120 °C, 6 h | 98 | [97] |
| 59 | R=H, R1=Me, Et, Ph, CH2Cl | 1.5 | UDIL-I-60%U (5% wt) 120 °C, 3 h | 83–99 | [98] |
| 60 | R=H, R1=Bu | 0.5 |  (0.89 mol%), 80 °C, 18 h | >99 | [65] |
| 61 | R=H, R1=CH2Cl, CH2OAllyl, CH2OPh, n-C6H13 | 0.1 |  (5 mol %) 100 °C, 24 h | 78–99 | [99] |
| 62 | R=H, R1=Ph, CH2Cl, CH2OAllyl, n-C6H13R=R1=(CH2)4 | 2.5 |  (0.5 mol%) 130 °C, 15 h (140 °C, 86 h for cyclohexene oxide) | 94–99 | [100] |
| 63 | R=H, R1=Me, Ph, CH2Cl, CH2OPh R=R1=(CH2)4 | 2.5 |  (1.4 mol%), 140 °C, 3 h (24 h for cyclohexene oxide) | 66–98 | [101] |
| 64 | R=H, R1=Me, Ph, CH2Cl, CH2OAllyl, (CH2)2CH=CH2 | 1.0 | CBAP-1(EDA) (2 mol% of N sites), 130 °C, 4 h | 77–98 | [102] |
| 65 | CBAP-1(EDA) (2 mol% of N sites), Bu4NBr (1.8 mol%), 25 °C, 36 h | 81–95 | |||
| 66 | R=H, R1=Me, Et, Bu, Ph, n-C6H13, t-BuO, (CH2)8CO2Me, CH2Cl, (CH2)2CH=CH2, i-PrO, CH2OAllyl, Me(CH2Cl) R=R1=(CH2)4, R=C8H17, R1=(CH2)7CO2Me | 1.0 |  (2 mol%), 90 °C, 6 h | 23–98 | [103] |
| 67 | R=H, R1=Et, Bu, Ph, CH2Cl, CH2Br, CH2OPh | 0.1 | COF-JLU7 (0.5 mol%), Bu4NBr (0.5 mol%l), 40 °C, 48 h | 61–99 | [104] |
| 68 | R=H, R1=Ph, CH2Cl | 0.7 |  (0.1 mol%), 100 °C, 4–10 h | 90–100 | [105] |
| 69 | R=H, R1=Me, Et, Ph, CH2Cl, CH2OAllyl | 2.0 |  (2% wt), 120 °C, 2–15 h | 80–95 | [106] |
| 70 | R=H, R1=Me, Ph, 4-FC6H4, CH2Cl, CH2Br | 0.1 |  (6 mg per mmol of substrate), 100 °C, 24 h | 95–98 | [107] |
| 71 | R=H, R1=Me, Et, Ph, CH2Cl | 2.0 | Zn-C3N4(25) (3.5 mg per mmol of substrate), KI (1.5 mol%), 130–150 *C, 5 h | 92–99 | [108] |
| 72 | R=H, R1=Me, Ph, CH2Cl, Me2 | 0.8 |  (5 mg per mmol of substrate), 80 °C, 4 h | 33–93 | [109] |
| 73 | R=H, R1=Me, Ph, Bu, CH2Cl, CH2OPh R=R1=(CH2)4 | 1.0 |  (1 mol%), 100 °C, 2.5 H | 81–99 | [110] |
| 74 | R=H, R1=Me, Ph, Bu, CH2Cl, CH2OBu R=R1=(CH2)4 | 1.5 | SBA-Zn-TPy+PBr− (0.1 mol%), 120 °C, 4.5–5 h | 23–99 | [111] |
| 75 | R=H, R1=Me, Ph, Bu, CH2Cl, CH2OBu R=R1=(CH2)4 | 1.5 | ZnTPy-BIM4/CNTs-3, (POSS-Imi, 0.07 mol%) 120 °C, 2.5–24 h | 51–98 | [112] |
| 76 | R=H, R1=Ph, Bn, 4-ClC6H4, 4-FC6H4, CH2Cl, CH2OPh, CH2OBn | 0.1 |  (0.5 mol%), 120 °C, 10 h | 94–96 | [113] |
| 77 | R=H, R1=Me, Et, Ph, CH2Cl R=R1=(CH2)4, (CH2)3 | 3.0 | RH or RD Au/Zn-MOF nanocages (3.2.mg/mmol), 70 °C, 6 h | 95–99 | [114] |
| 78 | R==H, R1=Me, Ph, CH2Cl, CH2Br, CH2OBu | 0.1 | [Ni(4,6-bis(triazol-1-yl)isophthalate)(4,6-bis(triazol-1-yl)isophthalic acid)] 2DMF·2H2O (1 mol%), Bu4NBr (10 mol%), 25 °C, 48 h | 40–99 | [115] |
| 79 | R=H, R1=Me, Et, Ph, CH2Cl, CH2Br, CH2OPh | 3.0 | CNT-NHC-Ag (8 mg/mmol) 4-dimethylaminopyridine (0.5 mol%) 120 °C, 8 h | 30–92 | [116] |
| 80 | R=H, R1=Me, Ph, CH2Cl, CH2OH,  R=R1=(CH2)4 | 4.0 | POSS-Imi (0.013–0.133 mol%), 150 °C, 3–16 h | 30–99 | [117] |
 Gave also a rearranged product (81% overall yield, 12:88 ratio). Entry 13: mixtures of cis:trans cyclic carbonates or of all possible diastereomers were always obtained. Entry 14: cyclic carbonate from limonene oxides was recovered as a c:t = 14:86 mixture. Entry 15: epichlorohydrin and cyclohexene oxide gave the worse yields, the former because of substitution of the chlorine with N-methyldiethanolamine, the latter for steric hindrance. The reaction was scaled up to 100 mmol. (R)-styrene oxide was converted into cyclic (R)-carbonate in 97% yield with 99% ee. Catalyst could be separated by precipitation in Et2O and recycled six times with very low decrease in activity. Entry 17: cyclohexene oxide gave only 4% yield. Other cobaloximes were tested with lower results. Entry 18: conversions, not yields (selectivity >99%), cis-butene oxide gave cyclic carbonate in c:t = 98:2 ratio. Epichlorohydrin and disubstituted substrates required a catalyst loading of 0.4 mol%. Entry 19: it should be noted that the catalyst recovered by CCl4 precipitation and reused produced only 33–40% yield of styrene carbonate, but increased the yield of hexane oxide from 40% to 98%. Very likely some water molecules remained coordinated to the catalyst. The catalyst then remained active in, at least, five consecutive runs without loss of its activity. Entry 20: conversion not yields, with >99% selectivity. Cyclohexene oxide did not react. Entry 21: cyclohexene oxide gave only 21% yield after 48 h. Catalyst could be recovered by solvent precipitation and reused in ten consecutive runs without loss of its activity. Entry 22: complexes with Fe and Cu or the presence of a second ortho-methyl group on the S-phenyl group were less efficient. Interestingly when this second ligand was allowed to react with copper, it is reduced from Cu(II) to Cu(I). Entry 23: epichlorohydrin and cyclohexene oxide were also tested but the isolated yields were not determined. Styrene oxide required 48 h. Entry 24: the best reaction conditions depended on the substrate. Cyclohexene epoxide was also tested and required 2.0 MPa at 100 °C. A DFT calculation of the reaction pathway was performed. Entry 25: all reactions occurred with retention of configuration, according to a double-inversion mechanism as depicted in Scheme 2. Even styrene carbonate from (S)-styrene oxide was obtained with 99% ee, (45 °C, 1.0 MPa). Entry 26: substituted styrene oxides required 60 °C. The reactions with 1,2-disubstituted epoxides led to very low yields (5–8%), with retention of configuration. Enantiopure epoxides gave cyclic carbonates with no loss of enantiomeric purity. The reaction is scalable up to 50 mmol. The catalyst was reused five times, by distilling off the product, with slow decrease of activity, because of partial sublimation property under the distillation conditions. Entry 27: c:t ratios ranged from 65:35 to 50:50. For epoxidized oils: 5 mol% with respect to oxirane number. Entry 29: DBU/NIS adduct was insoluble in ether, thus, after the extraction of products with ether the catalyst could be dried and reused for five times with no significant loss in its catalytic activity. To achieve good yields 2,2-dimethyloxirane and cyclohexene oxide required also Bu4NI (5 mol%). Entry 30: cyclohexene oxide required 12 h and was recovered in 33% yield. A mixture of 15% CO2 and 85% N2 afforded styrene carbonate in 91% yield after 14 h. Catalyst could be separated from the aqueous phase after pouring the reaction mixture in water. After drying, it was reused five times without decrease of activity. Entry 31: low-boiling epoxides gave higher yields in a 20 mmol scale with respect to a 5 mmol scale with a refluxing condenser. Higher CO2 pressure was requested by more sterically hindered epoxides. Stereochemistry was generally maintained but at harsher conditions it decreased, very likely by partial Sn1 reaction. Cyclooctene oxide gave only a 6% yield. Entry 32: the catalyst could be separated from the reaction mixture by distillation under reduced pressure and reused five times with no significant loss in its catalytic activity. Entry 33: the catalyst could be separated by vacuum distillation, dried and reused five times with no significant loss in its catalytic activity. Entry 34: disubstituted epoxides required 64 h, 100 °C, and 3 mol% of catalyst, to avoid diol formation. Entry 35: the catalyst was precipitated with Et2O, dried and reused four times with low decrease in its catalytic activity, due to some leaching of the salt from dextrin cavity. Entry 36: only epoxides with strong electron donating groups reacted and the less the electron donating power were, the harsher the reaction conditions must be. Propene oxide was already unreactive. Entry 37: isobutylene oxide gave only 10% yield. The authors affirmed in the text that also cyclohexene oxide gave low yield but 91% was reported in the table. Most epoxides exhibited good conversion also under atmospheric pressure. Entry 38: higher temperatures were requested by less reactive epoxides such as styrene and cyclohexene oxides. Enantiopure epoxides gave carbonate in 64–92% ee and enantiomeric excess decreased with increasing temperature. Entry 39: higher yields with R2=Bu were due to the high solubility of the catalyst. Entry 40: other imidazolium salt gave lower yields. Products could be purified by distillation and the residue containing the catalyst could be used five times with no significant loss in its catalytic activity. Entry 41: the actual catalytic species was
Gave also a rearranged product (81% overall yield, 12:88 ratio). Entry 13: mixtures of cis:trans cyclic carbonates or of all possible diastereomers were always obtained. Entry 14: cyclic carbonate from limonene oxides was recovered as a c:t = 14:86 mixture. Entry 15: epichlorohydrin and cyclohexene oxide gave the worse yields, the former because of substitution of the chlorine with N-methyldiethanolamine, the latter for steric hindrance. The reaction was scaled up to 100 mmol. (R)-styrene oxide was converted into cyclic (R)-carbonate in 97% yield with 99% ee. Catalyst could be separated by precipitation in Et2O and recycled six times with very low decrease in activity. Entry 17: cyclohexene oxide gave only 4% yield. Other cobaloximes were tested with lower results. Entry 18: conversions, not yields (selectivity >99%), cis-butene oxide gave cyclic carbonate in c:t = 98:2 ratio. Epichlorohydrin and disubstituted substrates required a catalyst loading of 0.4 mol%. Entry 19: it should be noted that the catalyst recovered by CCl4 precipitation and reused produced only 33–40% yield of styrene carbonate, but increased the yield of hexane oxide from 40% to 98%. Very likely some water molecules remained coordinated to the catalyst. The catalyst then remained active in, at least, five consecutive runs without loss of its activity. Entry 20: conversion not yields, with >99% selectivity. Cyclohexene oxide did not react. Entry 21: cyclohexene oxide gave only 21% yield after 48 h. Catalyst could be recovered by solvent precipitation and reused in ten consecutive runs without loss of its activity. Entry 22: complexes with Fe and Cu or the presence of a second ortho-methyl group on the S-phenyl group were less efficient. Interestingly when this second ligand was allowed to react with copper, it is reduced from Cu(II) to Cu(I). Entry 23: epichlorohydrin and cyclohexene oxide were also tested but the isolated yields were not determined. Styrene oxide required 48 h. Entry 24: the best reaction conditions depended on the substrate. Cyclohexene epoxide was also tested and required 2.0 MPa at 100 °C. A DFT calculation of the reaction pathway was performed. Entry 25: all reactions occurred with retention of configuration, according to a double-inversion mechanism as depicted in Scheme 2. Even styrene carbonate from (S)-styrene oxide was obtained with 99% ee, (45 °C, 1.0 MPa). Entry 26: substituted styrene oxides required 60 °C. The reactions with 1,2-disubstituted epoxides led to very low yields (5–8%), with retention of configuration. Enantiopure epoxides gave cyclic carbonates with no loss of enantiomeric purity. The reaction is scalable up to 50 mmol. The catalyst was reused five times, by distilling off the product, with slow decrease of activity, because of partial sublimation property under the distillation conditions. Entry 27: c:t ratios ranged from 65:35 to 50:50. For epoxidized oils: 5 mol% with respect to oxirane number. Entry 29: DBU/NIS adduct was insoluble in ether, thus, after the extraction of products with ether the catalyst could be dried and reused for five times with no significant loss in its catalytic activity. To achieve good yields 2,2-dimethyloxirane and cyclohexene oxide required also Bu4NI (5 mol%). Entry 30: cyclohexene oxide required 12 h and was recovered in 33% yield. A mixture of 15% CO2 and 85% N2 afforded styrene carbonate in 91% yield after 14 h. Catalyst could be separated from the aqueous phase after pouring the reaction mixture in water. After drying, it was reused five times without decrease of activity. Entry 31: low-boiling epoxides gave higher yields in a 20 mmol scale with respect to a 5 mmol scale with a refluxing condenser. Higher CO2 pressure was requested by more sterically hindered epoxides. Stereochemistry was generally maintained but at harsher conditions it decreased, very likely by partial Sn1 reaction. Cyclooctene oxide gave only a 6% yield. Entry 32: the catalyst could be separated from the reaction mixture by distillation under reduced pressure and reused five times with no significant loss in its catalytic activity. Entry 33: the catalyst could be separated by vacuum distillation, dried and reused five times with no significant loss in its catalytic activity. Entry 34: disubstituted epoxides required 64 h, 100 °C, and 3 mol% of catalyst, to avoid diol formation. Entry 35: the catalyst was precipitated with Et2O, dried and reused four times with low decrease in its catalytic activity, due to some leaching of the salt from dextrin cavity. Entry 36: only epoxides with strong electron donating groups reacted and the less the electron donating power were, the harsher the reaction conditions must be. Propene oxide was already unreactive. Entry 37: isobutylene oxide gave only 10% yield. The authors affirmed in the text that also cyclohexene oxide gave low yield but 91% was reported in the table. Most epoxides exhibited good conversion also under atmospheric pressure. Entry 38: higher temperatures were requested by less reactive epoxides such as styrene and cyclohexene oxides. Enantiopure epoxides gave carbonate in 64–92% ee and enantiomeric excess decreased with increasing temperature. Entry 39: higher yields with R2=Bu were due to the high solubility of the catalyst. Entry 40: other imidazolium salt gave lower yields. Products could be purified by distillation and the residue containing the catalyst could be used five times with no significant loss in its catalytic activity. Entry 41: the actual catalytic species was  , which acted as a nucleophile on the epoxide. Entry 42: disubstituted epoxides needed a higher catalyst loading (3%) and longer reaction times (48 h). Entry 43: the catalyst could be precipitated and used five times with no significant loss in its catalytic activity. Entry 44: styrene, isobutene oxides, and carbazolylglicidol required 40 °C. (R)-styrene oxide gave (R)-carbonate with 70% ee. Entry 45: fatty acid oxides required 0.5 MPa of CO2 and 100 °C and trans-carbonate was the most abundant isomer (83:17 to 99:1). Entry 46: when [HDBU]I was insoluble in epoxide dimethylformamide was added as the solvent. Cyclohexene oxide reacted at 140 °C, with 3 MPa after 48 h in dimethylformamide leading to the product in 76% yield. The catalyst could be recovered after reduced pressure distillation of the products and reused five times with no significant loss in its catalytic activity. DFT calculations were performed to elucidate the mechanism. Entry 47: HUST-1-Co was a crosslinked cobalt porphyrin obtained from 5,10,15,20-tetraphenylporphyrin, dichloromethane as cross-linker and cobalt acetate. The catalyst could be used fifteen times with no significant loss in its catalytic activity. Entry 48: Al-HPC was a crosslinked aluminum porphyrin obtained from 5,10,15,20-tetraphenylporphyrin, fomaldehyde dimethyl acetal as cross-linker and diethylaluminum chloride. The catalyst could be used ten times with no significant loss in its catalytic activity. Cyclohexene oxide gave only 28% yield. Entry 49: Al-iPOP was a crosslinked aluminum porphyrin obtained from 5,10,15,20-tetra(4-bromophenyl)porphyrin linked by Yamamoto–Ullmann coupling, coupled with a polymeric ionic liquid from (4-bromophenyl)- (1) and (4-bromobenzyl)imidazolium bromide (2). Al-iPOP-2 was generally more efficient. The catalysts could be used six times with no significant loss in its catalytic activity. Entry 50: the catalyst was obtained from 2-methylimidazole (8 mmol), Co(NO3).6H2O (0.5 mmol) and Zn(NO3)2.6H2O (0.5 mmol) in methanol. The catalyst could be used four times with no significant loss in its catalytic activity. Entry 51: the catalyst was obtained from 2-methylimidazole, Co(NO3).6H2O and Zn(NO3)2.4H2O in water. The amount refers to a 9 mmol scale. It was also recycled four times with little deactivation (from >99 to 91% yield). The substrates were lacking in the paper table and were furnished by private communication. Entry 52: porous charged covalent triazine framework obtained at 500 °C by reaction of 1,1′-bis(4-cyanophenyl)-[4,4′-bipyridine]-1,1′-diium dichloride in melted anhydrous ZnCl2. It was also recycled 4 times without deactivation. Entry 53: a mesoporous poly(ionic liquid) obtained from 1-glycidyl-3-vinylimidazolium bromide and divinylbenzene then opened in hot water. The catalyst was recycled 10 times without deactivation. Entry 54: larger R1 groups gave very poor yields (6–10%). The catalyst was recycled 5 times without deactivation. Entry 55: samples of Cu-DABCO, Ni-DABCO, and Co-DABCO were also tested but with worse results. The catalyst was used three times without deactivation. Entry 57: ZnO@NPC-Ox was obtained from ZIF-8 (see entries 42–43) which was pyrolyzed and subsequently oxidized by NaOCl to produce ZnO nanoparticles (NPs) encapsulated into N-doped porous carbon. The catalyst was used 10 times without deactivation. Entry 58: the catalyst was used five times without deactivation, but it required a tedious purification procedure from the reaction mixture. Other epoxides gave very low conversions (24–26%), albeit with high selectivity (99%) and styrene oxide did not react. Entry 59: UDIL= urea-derivative-based ionic liquids, I=iodide, 60%U is the relative mass of urea added to UDIL. Cyclohexene oxide gave 45% yield, at 130 °C, after 9 h and with 3.0 MPa of CO2. The catalyst was used five times without deactivation and was tolerant of the presence of water. Entry 60: The catalyst was used for 12 runs. Quantitative yield was recovered in the first four runs, then a slight decrease (up to 85% in the twelfth run) together a loss of catalyst (0.69 mol% recovered from the last run) was observed. Entry 61 other imidazolium-based ionic polymers gave worse results. The catalyst was used 10 times without deactivation. Entry 62: the catalyst was used four times without deactivation. Entry 63: the catalyst was used 10 times without deactivation and was also used in a continuous flow reactor for 120 h without deactivation. Entry 64: CBAP-1(EDA) was prepared from 1,3,5-triphenylbenzene and terephthaloyl chloride. The obtained polymer was then reductively aminated with ethylene diamine. Cyclohexene oxide gave only 14% yield after 6 h at 140 °C. The catalyst was used 5 times without deactivation. Entry 65: CBAP-1(EDA-Zn) was obtained by treatment of CBAP-1(EDA) with Zn(OAc)2. Cyclohexene oxide gave 45% yield after 48 h at 60 °C. The catalyst was used five times without deactivation. Entry 66: most of reactions were carried out with the same catalyst sample recycled from the previous reaction. Its efficiency was verified repeating the reaction with butane oxide every seven times and comparing the yield. Catalyst efficiency significantly decreased after 14 runs. Oleate oxide gave 43:57 c:t mixture. Entry 67: COF-JLU7 was prepared from 2,4,6-tris(4-aminophenoxy)triazine and 2,5-dihydroxy-1,4-benzenedicarboxaldehyde under acidic catalysis. The reaction with (R)- or (S)-styrene oxide exhibited excellent enantioselectivity (97% and 93% ee, respectively). The catalyst was used five times without deactivation. Entry 68: cyclohexene oxide gave only 30% of conversion. The catalyst could be reused but poisoning of the active sites was observed. Thus, to maintain high conversion and selectivity, the catalyst had to be regenerated, by treatment with diluted NaOH. Entry 69: cyclohexene oxide gave only 9% yield. The yield of carbonate decreased sharply from 97% to 64% in the third cycle and then slowly (until 59% after other two runs). After the five cycles, less than the original catalyst amount was recovered, thus authors explained the lesser efficiency with this loss of catalyst amount. Entry 70: the catalyst was used 10 times without deactivation. Entry 71: Zn-C3N4(25) was prepared by thermal polymerization of Zn(OAc)2·2H2O/dicyandiamine in mass ratio of 25%. The catalyst was used five times without deactivation. Entry 72: cyclohexene oxide gave only 6% yield together many byproducts. The catalyst was used five times without deactivation. Entry 73: the addition of N-(3-aminopropyl)-imidazole increased the fiber weight of 20%. The fiber was intertwined on the stirring paddle of the reactor and could be used 21 times in gram-scale reactions. Entry 74: obtained by refluxing 5,10,15,20-tetrakis(4-pyridyl)porphyrin zinc(II), mesoporous silica SBA-15, and 3-(trimethoxysilyl)propyl bromide in dimethylformamide. Other solvents gave a catalyst with worse catalytic activity. The catalyst was used five times without deactivation. Entry 75: obtained by the reaction of 5,10,15,20-tetrakis(4-pyridyl)porphyrin zinc(II), di(1H-imidazol-1-yl)methane, and 1,4-bis(bromomethyl)benzene in the presence of carbon nanotubes (CNTs). The catalyst was used seven times without deactivation. Entry 76: the catalyst was used five times without deactivation. 4-nitrostyrene oxide and (but-3-en-1-yl)oxirane did not react or decomposed. Entry 77: regular hexahedral (RH) ZIF-8 and rhombic dodecahedral (RD) ZIF-8 homometallic nanoparticles (see entries 50-51) underwent cation exchange with Au3+ ions from NaAuCl4. The catalyst was used 6 times without deactivation. Entry 78: there is a discrepancy between text (chloro- and bromo-methyloxirane reported) and table (bromo- and chloro-oxirane reported). We think that the text is right. Catalyst was prepared from nickel wires and 4,6-bis(triazol-1-yl)isophthalic acid in acidic solution. Entry 79: the catalyst was obtained from reaction of carbon nanotubes (CNT) with N-vinyl-N’-allylimidazole silver complex (NHC-Ag). Cyclohexene and isobutene oxides gave poor yields (10 and 11% respectively). The reusability was tested with propargyl alcohols (see footnote Table 2, entry 5). Entry 80: obtained by reaction between triethoxy-3-(2-imidazolin-1-yl)propylsilane and octakis(3-bromopropyl)-octasilsesquioxane then grafted onto SiO2 and finally by reaction with 1-methylimidazole. The catalyst was used five times without significant deactivation.
, which acted as a nucleophile on the epoxide. Entry 42: disubstituted epoxides needed a higher catalyst loading (3%) and longer reaction times (48 h). Entry 43: the catalyst could be precipitated and used five times with no significant loss in its catalytic activity. Entry 44: styrene, isobutene oxides, and carbazolylglicidol required 40 °C. (R)-styrene oxide gave (R)-carbonate with 70% ee. Entry 45: fatty acid oxides required 0.5 MPa of CO2 and 100 °C and trans-carbonate was the most abundant isomer (83:17 to 99:1). Entry 46: when [HDBU]I was insoluble in epoxide dimethylformamide was added as the solvent. Cyclohexene oxide reacted at 140 °C, with 3 MPa after 48 h in dimethylformamide leading to the product in 76% yield. The catalyst could be recovered after reduced pressure distillation of the products and reused five times with no significant loss in its catalytic activity. DFT calculations were performed to elucidate the mechanism. Entry 47: HUST-1-Co was a crosslinked cobalt porphyrin obtained from 5,10,15,20-tetraphenylporphyrin, dichloromethane as cross-linker and cobalt acetate. The catalyst could be used fifteen times with no significant loss in its catalytic activity. Entry 48: Al-HPC was a crosslinked aluminum porphyrin obtained from 5,10,15,20-tetraphenylporphyrin, fomaldehyde dimethyl acetal as cross-linker and diethylaluminum chloride. The catalyst could be used ten times with no significant loss in its catalytic activity. Cyclohexene oxide gave only 28% yield. Entry 49: Al-iPOP was a crosslinked aluminum porphyrin obtained from 5,10,15,20-tetra(4-bromophenyl)porphyrin linked by Yamamoto–Ullmann coupling, coupled with a polymeric ionic liquid from (4-bromophenyl)- (1) and (4-bromobenzyl)imidazolium bromide (2). Al-iPOP-2 was generally more efficient. The catalysts could be used six times with no significant loss in its catalytic activity. Entry 50: the catalyst was obtained from 2-methylimidazole (8 mmol), Co(NO3).6H2O (0.5 mmol) and Zn(NO3)2.6H2O (0.5 mmol) in methanol. The catalyst could be used four times with no significant loss in its catalytic activity. Entry 51: the catalyst was obtained from 2-methylimidazole, Co(NO3).6H2O and Zn(NO3)2.4H2O in water. The amount refers to a 9 mmol scale. It was also recycled four times with little deactivation (from >99 to 91% yield). The substrates were lacking in the paper table and were furnished by private communication. Entry 52: porous charged covalent triazine framework obtained at 500 °C by reaction of 1,1′-bis(4-cyanophenyl)-[4,4′-bipyridine]-1,1′-diium dichloride in melted anhydrous ZnCl2. It was also recycled 4 times without deactivation. Entry 53: a mesoporous poly(ionic liquid) obtained from 1-glycidyl-3-vinylimidazolium bromide and divinylbenzene then opened in hot water. The catalyst was recycled 10 times without deactivation. Entry 54: larger R1 groups gave very poor yields (6–10%). The catalyst was recycled 5 times without deactivation. Entry 55: samples of Cu-DABCO, Ni-DABCO, and Co-DABCO were also tested but with worse results. The catalyst was used three times without deactivation. Entry 57: ZnO@NPC-Ox was obtained from ZIF-8 (see entries 42–43) which was pyrolyzed and subsequently oxidized by NaOCl to produce ZnO nanoparticles (NPs) encapsulated into N-doped porous carbon. The catalyst was used 10 times without deactivation. Entry 58: the catalyst was used five times without deactivation, but it required a tedious purification procedure from the reaction mixture. Other epoxides gave very low conversions (24–26%), albeit with high selectivity (99%) and styrene oxide did not react. Entry 59: UDIL= urea-derivative-based ionic liquids, I=iodide, 60%U is the relative mass of urea added to UDIL. Cyclohexene oxide gave 45% yield, at 130 °C, after 9 h and with 3.0 MPa of CO2. The catalyst was used five times without deactivation and was tolerant of the presence of water. Entry 60: The catalyst was used for 12 runs. Quantitative yield was recovered in the first four runs, then a slight decrease (up to 85% in the twelfth run) together a loss of catalyst (0.69 mol% recovered from the last run) was observed. Entry 61 other imidazolium-based ionic polymers gave worse results. The catalyst was used 10 times without deactivation. Entry 62: the catalyst was used four times without deactivation. Entry 63: the catalyst was used 10 times without deactivation and was also used in a continuous flow reactor for 120 h without deactivation. Entry 64: CBAP-1(EDA) was prepared from 1,3,5-triphenylbenzene and terephthaloyl chloride. The obtained polymer was then reductively aminated with ethylene diamine. Cyclohexene oxide gave only 14% yield after 6 h at 140 °C. The catalyst was used 5 times without deactivation. Entry 65: CBAP-1(EDA-Zn) was obtained by treatment of CBAP-1(EDA) with Zn(OAc)2. Cyclohexene oxide gave 45% yield after 48 h at 60 °C. The catalyst was used five times without deactivation. Entry 66: most of reactions were carried out with the same catalyst sample recycled from the previous reaction. Its efficiency was verified repeating the reaction with butane oxide every seven times and comparing the yield. Catalyst efficiency significantly decreased after 14 runs. Oleate oxide gave 43:57 c:t mixture. Entry 67: COF-JLU7 was prepared from 2,4,6-tris(4-aminophenoxy)triazine and 2,5-dihydroxy-1,4-benzenedicarboxaldehyde under acidic catalysis. The reaction with (R)- or (S)-styrene oxide exhibited excellent enantioselectivity (97% and 93% ee, respectively). The catalyst was used five times without deactivation. Entry 68: cyclohexene oxide gave only 30% of conversion. The catalyst could be reused but poisoning of the active sites was observed. Thus, to maintain high conversion and selectivity, the catalyst had to be regenerated, by treatment with diluted NaOH. Entry 69: cyclohexene oxide gave only 9% yield. The yield of carbonate decreased sharply from 97% to 64% in the third cycle and then slowly (until 59% after other two runs). After the five cycles, less than the original catalyst amount was recovered, thus authors explained the lesser efficiency with this loss of catalyst amount. Entry 70: the catalyst was used 10 times without deactivation. Entry 71: Zn-C3N4(25) was prepared by thermal polymerization of Zn(OAc)2·2H2O/dicyandiamine in mass ratio of 25%. The catalyst was used five times without deactivation. Entry 72: cyclohexene oxide gave only 6% yield together many byproducts. The catalyst was used five times without deactivation. Entry 73: the addition of N-(3-aminopropyl)-imidazole increased the fiber weight of 20%. The fiber was intertwined on the stirring paddle of the reactor and could be used 21 times in gram-scale reactions. Entry 74: obtained by refluxing 5,10,15,20-tetrakis(4-pyridyl)porphyrin zinc(II), mesoporous silica SBA-15, and 3-(trimethoxysilyl)propyl bromide in dimethylformamide. Other solvents gave a catalyst with worse catalytic activity. The catalyst was used five times without deactivation. Entry 75: obtained by the reaction of 5,10,15,20-tetrakis(4-pyridyl)porphyrin zinc(II), di(1H-imidazol-1-yl)methane, and 1,4-bis(bromomethyl)benzene in the presence of carbon nanotubes (CNTs). The catalyst was used seven times without deactivation. Entry 76: the catalyst was used five times without deactivation. 4-nitrostyrene oxide and (but-3-en-1-yl)oxirane did not react or decomposed. Entry 77: regular hexahedral (RH) ZIF-8 and rhombic dodecahedral (RD) ZIF-8 homometallic nanoparticles (see entries 50-51) underwent cation exchange with Au3+ ions from NaAuCl4. The catalyst was used 6 times without deactivation. Entry 78: there is a discrepancy between text (chloro- and bromo-methyloxirane reported) and table (bromo- and chloro-oxirane reported). We think that the text is right. Catalyst was prepared from nickel wires and 4,6-bis(triazol-1-yl)isophthalic acid in acidic solution. Entry 79: the catalyst was obtained from reaction of carbon nanotubes (CNT) with N-vinyl-N’-allylimidazole silver complex (NHC-Ag). Cyclohexene and isobutene oxides gave poor yields (10 and 11% respectively). The reusability was tested with propargyl alcohols (see footnote Table 2, entry 5). Entry 80: obtained by reaction between triethoxy-3-(2-imidazolin-1-yl)propylsilane and octakis(3-bromopropyl)-octasilsesquioxane then grafted onto SiO2 and finally by reaction with 1-methylimidazole. The catalyst was used five times without significant deactivation.| Entry | R | R1, R2 | PCO2 (MPa) | Conditions | Yield (%) | Ref |
|---|---|---|---|---|---|---|
| 1 | Ph, 4-MeC6H4, 4-MeOC6H4, 4-ClC6H4, 4-MeCOC6H4, 4-CF3C6H4, 3-piridyl | R1=R2=Me, (CH2)4 R1=Me, R2=Et, i-Pr | 2.0 |  (5 mol%), 60 °C, 3 h | 55–96 | [137] |
| 2 | Ph, 4-MeC6H4, 4-FC6H4, 4-C6H13C6H4, 4-t-BuC6H4 | R1=R2=Me, Et i-Pr | 2.5 |  (200 mol%) 60 °C, 24 h | 68–99 | [138] |
| 3 | H, Ph, 4-MeC6H4, 4-ClC6H4, 4-CF3C6H4, 4-MeOC6H4, 4-pyridyl | R1=R2=Me, (CH2)3, (CH2)4 R1=Me, R2=Ph | 2.0 | CDC–CO2 (5 mol%), (entry 39, Table 1), 80 °C, 12 h | 51–94 | [80] |
| 4 | H | R1=R2=Me, Et, (CH2)4 R1=Me, R2=Et | 1.0 | Zn/Fe3O4/ECS (133 mg/mmol), NEt3 (Equation (1)) 30 °C, 12–20 h | 90–93 | [139] |
| 5 | H | R1=R2=(CH2)4 R1=Me, R2=Et, i-Bu | 3.0 | CNT-NHC-Ag (151 mg/mmol) (entry 71, Table 1), 80 °C, 24 h | 97–99 | [115] |
| 6 | H | R1=R2=Me, Ph R1=Me, R2=Et, Ph, i-Bu, Allyl | 5.0 | (n-Bu4N)2(CO2)2, (2.5–5 mol%) 80 °C, 6 h | 60–99 | [140] |
 . Entry 2: 1-(2-phenylethynyl)cyclohexanol was unreactive; 2-methylbut-3-yn-2-ol gave quantitatively 1,1-dimethyl-2-oxopropyl-1′,1′-dimethyl-2′-propynyl carbonate. The catalyst could be separated with water, then after drying reused four times without significant deactivation. Entry 3: the active catalyst
. Entry 2: 1-(2-phenylethynyl)cyclohexanol was unreactive; 2-methylbut-3-yn-2-ol gave quantitatively 1,1-dimethyl-2-oxopropyl-1′,1′-dimethyl-2′-propynyl carbonate. The catalyst could be separated with water, then after drying reused four times without significant deactivation. Entry 3: the active catalyst  acted as a base on the propargyl alcohol. Entry 4: Zn/Fe3O4/ECS was a magnetically separable catalyst prepared from corn starch, magnetite and ZnI2. 4-Phenyl-2-methylbut-3-yn-2-ol was unreactive. The catalyst was recovered with an external magnet, and used 4 times without significant deactivation. Entry 5: the NHC-Ag complex was also supported on graphene (GN) and the reaction gave similar results. CNT-NHC-Cu and GN-NHC-Cu complexes gave lesser yields. However, CNT-NHC-Cu was used in the recycle tests and reused eight times without significant deactivation. Entry 6: Propargyl alcohol and but-3-yn-2-ol were unreactive, owing to the well-known Thorpe–Ingold effect [141], 1-(2-Phenylethynyl)cyclohexanol and 3,6-dimethylocta-1,7-diyne-3,6-diol gave low conversion (12% and 35%, respectively), but selectivity near 100%. A higher catalyst loading was necessary for sterically hindered alcohols.
acted as a base on the propargyl alcohol. Entry 4: Zn/Fe3O4/ECS was a magnetically separable catalyst prepared from corn starch, magnetite and ZnI2. 4-Phenyl-2-methylbut-3-yn-2-ol was unreactive. The catalyst was recovered with an external magnet, and used 4 times without significant deactivation. Entry 5: the NHC-Ag complex was also supported on graphene (GN) and the reaction gave similar results. CNT-NHC-Cu and GN-NHC-Cu complexes gave lesser yields. However, CNT-NHC-Cu was used in the recycle tests and reused eight times without significant deactivation. Entry 6: Propargyl alcohol and but-3-yn-2-ol were unreactive, owing to the well-known Thorpe–Ingold effect [141], 1-(2-Phenylethynyl)cyclohexanol and 3,6-dimethylocta-1,7-diyne-3,6-diol gave low conversion (12% and 35%, respectively), but selectivity near 100%. A higher catalyst loading was necessary for sterically hindered alcohols.| Entry | R | R1,R2 | PCO2 (MPa) | Conditions | Yield (%) | Regio. (R25:R24) | Ref |
|---|---|---|---|---|---|---|---|
| 1 | R=Pr, Bu, n-C5H11, (CH2)2-i-Pr | R1=H, R2=Ph, 4-MeC6H4 | 1.0 | ISA 6 (1 mol%), (entry 6, Table 1), 50 °C, 2–8 h | 80–99 | 95:5 to 98:2 | [49] |
| 2 | R=H, Me, Et, Bn | R1=H, R2=Ph, 4-MeC6H4, 4-ClC6H4, 4-(MeOCH2)C6H4, 2-naphthyl | 0.1 | SalenCoI (2.5 mol%), Ph3P=CHCOPh (2.5 mol%), (entry 7, Table 1), 25 °C, 48 h | 38–90 | 100:0 | [50] |
| 3 | R=Pr, Bu, i-Bu, n-C5H11, | R1=H, R2=Ph, 4-MeC6H4 | 2.0 | IL-Zn-TPP (0.1 mol%), (entry 21, Table 1) 90 °C, 2–10 h | 82–96 | 97:3 to 98:2 | [61] |
| 4 | Ph, 4-MeC6H4, 4-ClC6H4, 4-BrC6H4, CH2OPh, 2-thienyl | R1=H, R2=Et, Pr, Bu, i-Pr, c-C6H11, Bn | 2.0 | CDC–CO2 (5 mol%), (entry 39, Table 1), 80 °C, 12 h | 52–99 | 90:10 to 97:3 | [115] |
| 5 | Ph, 4-MeC6H4, 4-ClC6H4, 4-BrC6H4, 4-MeOC6H4, 4-t-BuC6H4 | R1=H, R2=Et, Pr, Bu, i-Bu | 0.1 |  (1 mol%), DBU (2 mol%), 50 °C, 48 h | 43–92 | 93:7 to 99:1 | [150] |
| Entry | R | R1,R2 | NR3 | PCO2 (MPa) | Conditions | Yield (%) | Ref |
|---|---|---|---|---|---|---|---|
| 1 | H, Ph, Me | R1=R2=H, Me, (CH2)4 | H, Me, i-Pr, t-Bu, Bn, 4-MeOC6H4 | 0.1 |  (1 mol%), 40–80 °C, 0.16–90 h | 74–98 | [154] |
| 2 | H, Me, Ph, 4-MeC6H4, 4-MeOC6H4, 4-CNC6H4 | R1=R2=H, Me, Et, (CH2)4 | H, Me | 0.5 | MOF (0.4 mol%), 60 °C, 24 h | 31–99 | [155] |
| 3 | Ph, 4-MeC6H4, 4-BrC6H4 | R1=R2=Me, (CH2)4 R1=H, R2=c-C6H11 R1=Me, R2=Et | Bu, c-C5H9, Bn | 0.1 |  (11–22 mol%), 100 °C, 24 h | 89–93 | [156] |
| 4 | H, Ph | R1=R2=H R1=H, R2=Et | Bn, Bu, Ph, c-C6H11, PhCH(Me), Ph(CH2)2, 4-ClC6H4CH2, 4-MeOC6H4CH2 | 0.1 |  (5 mol%), 60 °C, 12 h | 8–96 | [157] |
| 5 | H, Ph, 4-MeC6H4 | R1=R2=H, Me R1=H, R2=Me, Et, Pr, i-Pr, | Bu, Bn | 0.1 |  (10 mol%), 90 °C, 10 h | 71–99 | [158] |
| 6 | H | R1=R2=H, Me, (CH2)4 R1=H, R2=Me | Me, Bn | 0.5 | Bu4NF (1 mol%), 110 °C, 12 h | 94–99 | [159] |
| 7 | Ph, 4-MeC6H4, 4-MeOC6H4, 4-CF3C6H4, 4-CNC6H4 | R1=R2=H | Me, Bn | 0.5 | Bu4NF (1–2 mol%), 110 °C, 24–48 h | 77–91 | [160] |
| 8 | H, Me, Et, Ph | R1=R2=H, Me | Bn, 4-MeOC6H4CH2 | Air |  (0.5 mol%) DBU (10 mol%) r. t., 48 h | 35–90 | [161] |
| 9 | H, Me, Ph, 4-MeC6H4 | R1=R2=H, R1=H, R2=Me, Ph, 4-MeC6H4 | Me, i-Pr, Bu, Bn, | 1.0 | KCC-1/IL/Ni@Pd NPs (0.1 mg/mmol), 15 W compact fluorescent lamp r. t., 3 h | 83–96 | [162] |
 , 1,4-benzenedicarboxylic acid and Cd(ClO4)2.6H2O. Increasing the substrate size the yields decreased and with R = 1-naphthyl no reaction occurred. Diprop-2-ynylamine gave 5-methylene-3-(prop-2-ynyl)oxazolidin-2-one in 62% isolated yield. The catalyst could be reused up to four times without deactivation. Entry 5: both amino-activated and CO2-activated mechanisms were investigated by DFT calculations and CO2-activated resulted in a low energy barrier in the first step. Entry 6: primary amines were unreactive. Entry 7: electron withdrawing groups in R favored tautomerization to 2-oxazolones. (oxazolidinone/oxazolone ratio 12:57 and 2:64 for R = 4-CF3C6H4, 4-CNC6H4, respectively. In these cases oxazolidinones were obtained at 70 °C in 24 and 3 h for R = 4-CF3C6H4, 4-CNC6H4, respectively. Entry 8: internal bulky alkynes gave the lowest yields. N-Phenyl amines were unreactive and primary amines gave only 4% yield. Entry 9: KCC-1/IL/Ni@Pd NPs was prepared in three steps: KCC-1 NPs from tetraethyl orthosilicate, cetylpyridinium bromide and urea; KCC-1/IL NPs from KCC-1 NPs, bis(trimethoxysilylpropyl)imidazolium iodide and NaH by sonication; finally KCC-1/IL/Ni@Pd NPs from KCC-1/IL NPs, Ni(OAc)2.4H2O and PdBr2. The catalyst could be reused up to ten times without deactivation.
, 1,4-benzenedicarboxylic acid and Cd(ClO4)2.6H2O. Increasing the substrate size the yields decreased and with R = 1-naphthyl no reaction occurred. Diprop-2-ynylamine gave 5-methylene-3-(prop-2-ynyl)oxazolidin-2-one in 62% isolated yield. The catalyst could be reused up to four times without deactivation. Entry 5: both amino-activated and CO2-activated mechanisms were investigated by DFT calculations and CO2-activated resulted in a low energy barrier in the first step. Entry 6: primary amines were unreactive. Entry 7: electron withdrawing groups in R favored tautomerization to 2-oxazolones. (oxazolidinone/oxazolone ratio 12:57 and 2:64 for R = 4-CF3C6H4, 4-CNC6H4, respectively. In these cases oxazolidinones were obtained at 70 °C in 24 and 3 h for R = 4-CF3C6H4, 4-CNC6H4, respectively. Entry 8: internal bulky alkynes gave the lowest yields. N-Phenyl amines were unreactive and primary amines gave only 4% yield. Entry 9: KCC-1/IL/Ni@Pd NPs was prepared in three steps: KCC-1 NPs from tetraethyl orthosilicate, cetylpyridinium bromide and urea; KCC-1/IL NPs from KCC-1 NPs, bis(trimethoxysilylpropyl)imidazolium iodide and NaH by sonication; finally KCC-1/IL/Ni@Pd NPs from KCC-1/IL NPs, Ni(OAc)2.4H2O and PdBr2. The catalyst could be reused up to ten times without deactivation.
| Entry | X | PCO2 (MPa) | Conditions | Yield (%) | Ref |
|---|---|---|---|---|---|
| 1 | H, 4-Cl, 4,5-(MeO)2 | 0.1 |  (22 mol%), 100 °C, 24 h | 78–86 | [156] |
| 2 | H, 4-Me, 4-NO2, 5-Cl | 2.0 |  (0.5 g/mmol), 100 °C, 20 h | 56–83 | [68] |
| 3 | H, 5-Me, 5-F, 5-Cl, 5-Br, 4,5-(MeO)2 | 2.0 | Bu4NF (1 mol%), 110 °C, 24 h | 96->99 | [160] |
| 4 | H | 1.0 | Bu3N (20 mol%), 100 °C, 36 h | 75 | [187] |
| 4 | H, 4-Cl, 4-Me, 5-F, 5-Me, 4,5-(MeO)2 | 0.1 |  (100% mol%), 80 °C, 24 h | 87–98 | [194] |
| 5 | H, 4-Cl, 5-Cl, 5-Br, 4,5-(MeO)2 | 1.0 | Zhabuye basic salt-lake brine (1.5 mL/mmol), 140 °C, 8–30 h | 96–98 | [195] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalpozzo, R.; Della Ca’, N.; Gabriele, B.; Mancuso, R. Recent Advances in the Chemical Fixation of Carbon Dioxide: A Green Route to Carbonylated Heterocycle Synthesis. Catalysts 2019, 9, 511. https://doi.org/10.3390/catal9060511
Dalpozzo R, Della Ca’ N, Gabriele B, Mancuso R. Recent Advances in the Chemical Fixation of Carbon Dioxide: A Green Route to Carbonylated Heterocycle Synthesis. Catalysts. 2019; 9(6):511. https://doi.org/10.3390/catal9060511
Chicago/Turabian StyleDalpozzo, Renato, Nicola Della Ca’, Bartolo Gabriele, and Raffaella Mancuso. 2019. "Recent Advances in the Chemical Fixation of Carbon Dioxide: A Green Route to Carbonylated Heterocycle Synthesis" Catalysts 9, no. 6: 511. https://doi.org/10.3390/catal9060511
APA StyleDalpozzo, R., Della Ca’, N., Gabriele, B., & Mancuso, R. (2019). Recent Advances in the Chemical Fixation of Carbon Dioxide: A Green Route to Carbonylated Heterocycle Synthesis. Catalysts, 9(6), 511. https://doi.org/10.3390/catal9060511