Base Metal Catalysts for Deoxygenative Reduction of Amides to Amines


Abstract
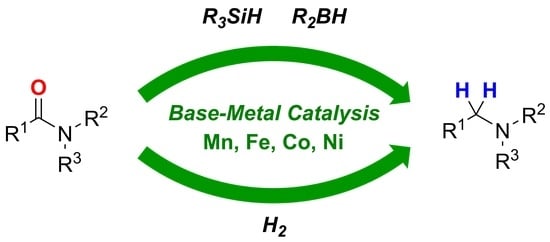
1. Introduction
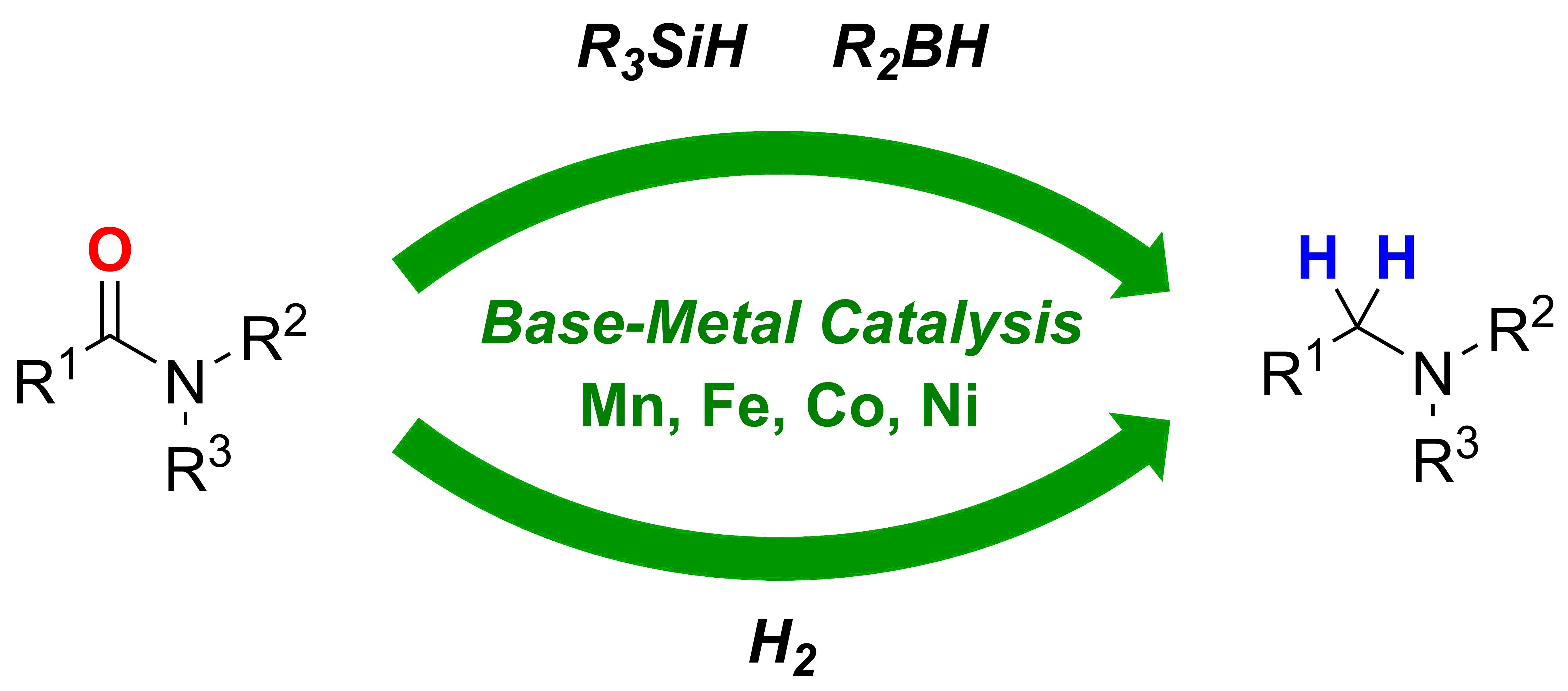
2. Deoxygenative Hydrosilylation of Amides
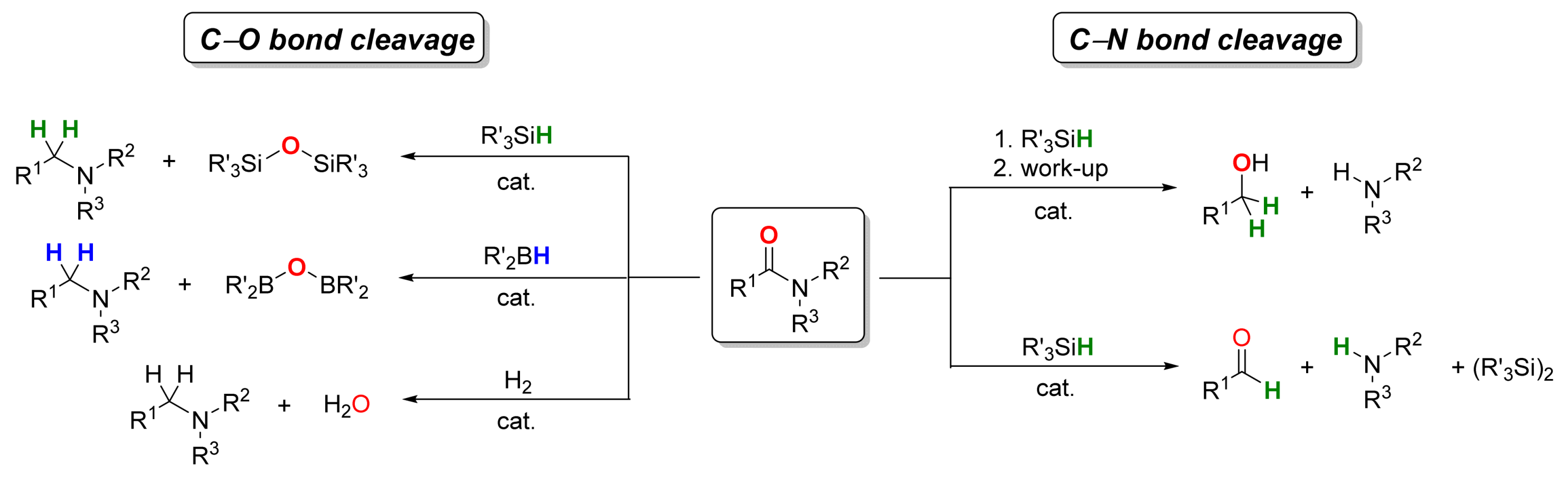
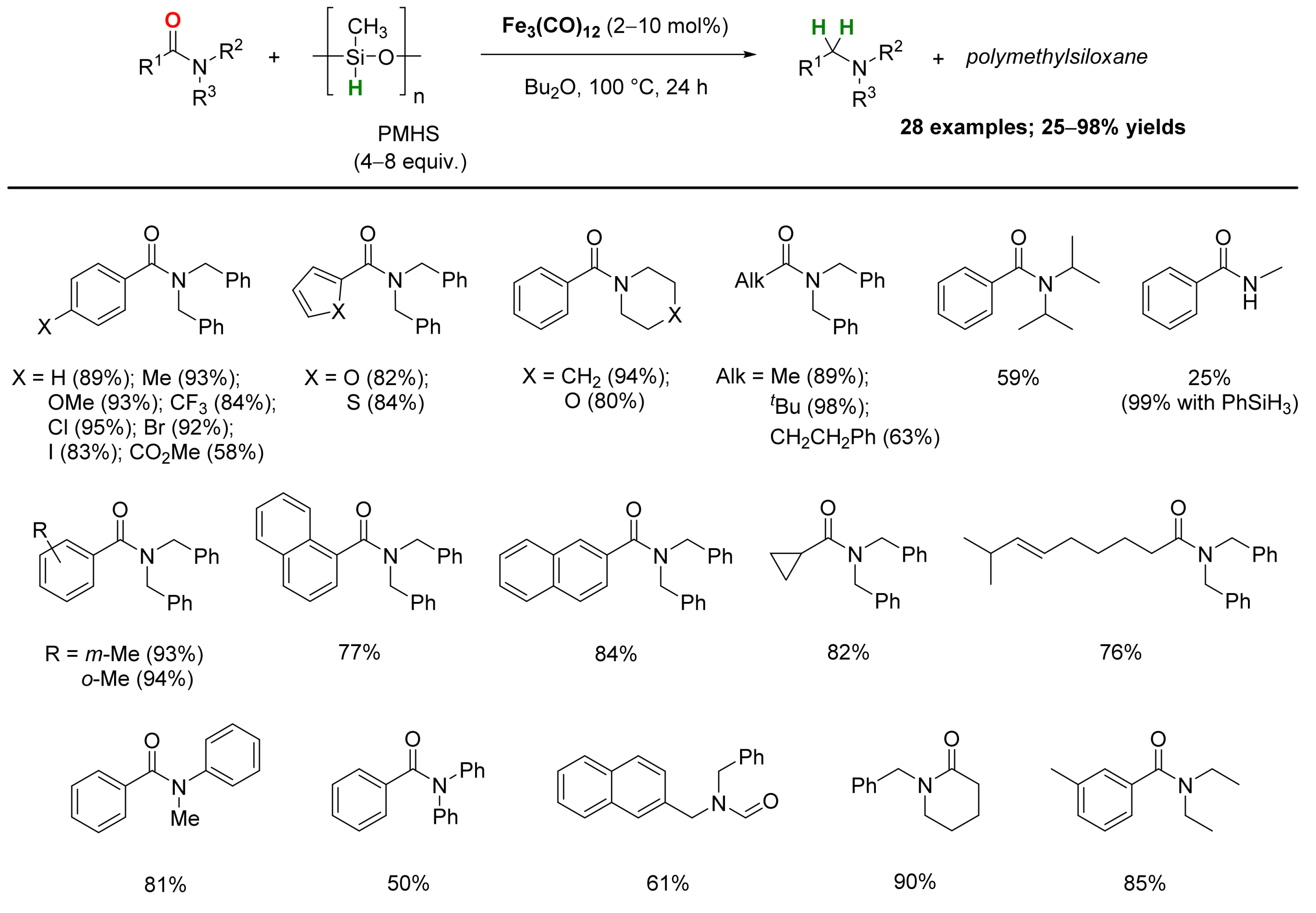
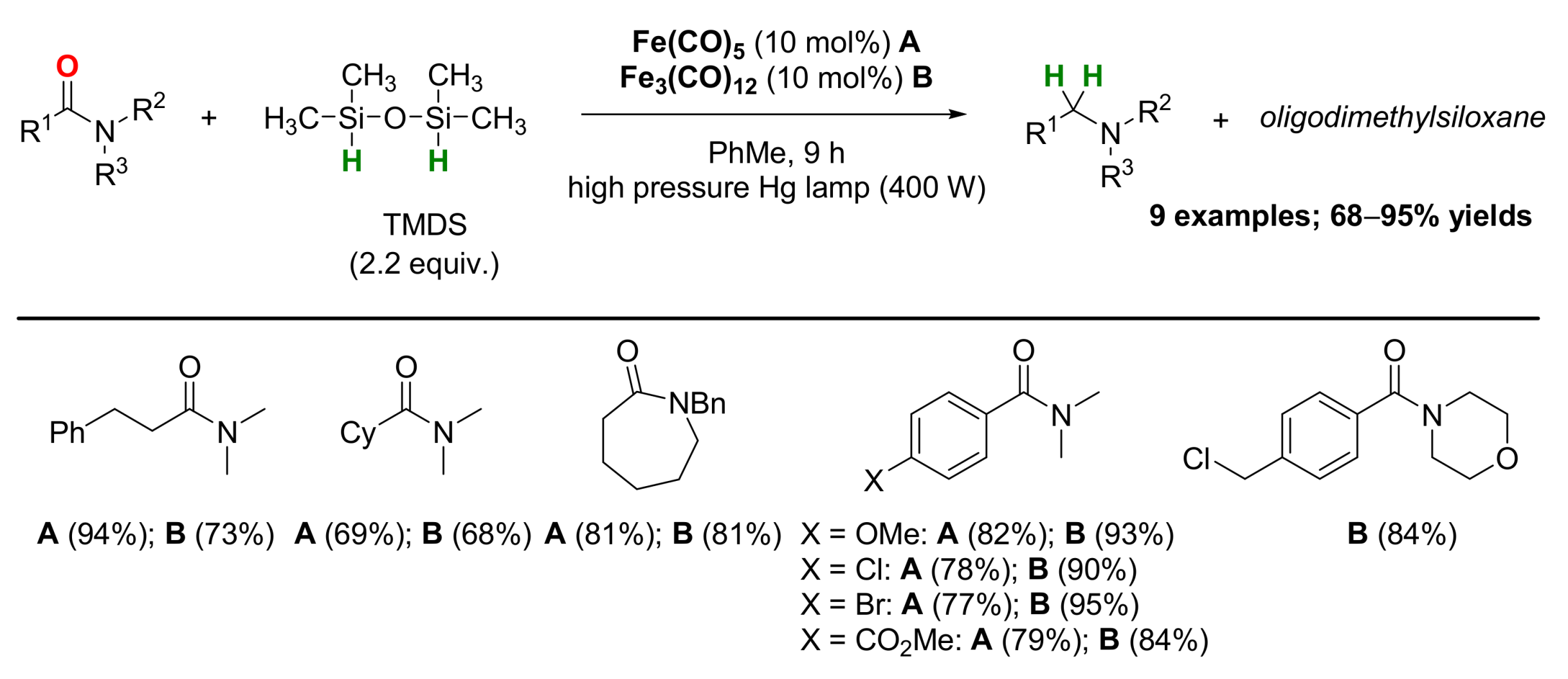
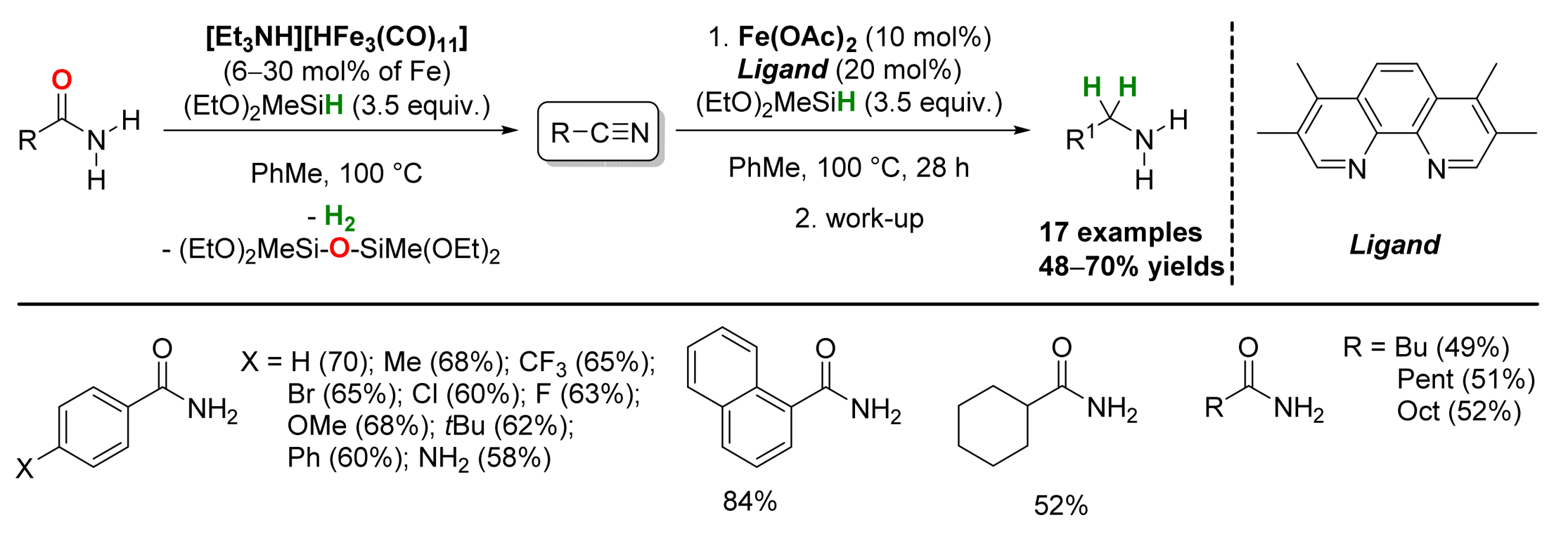
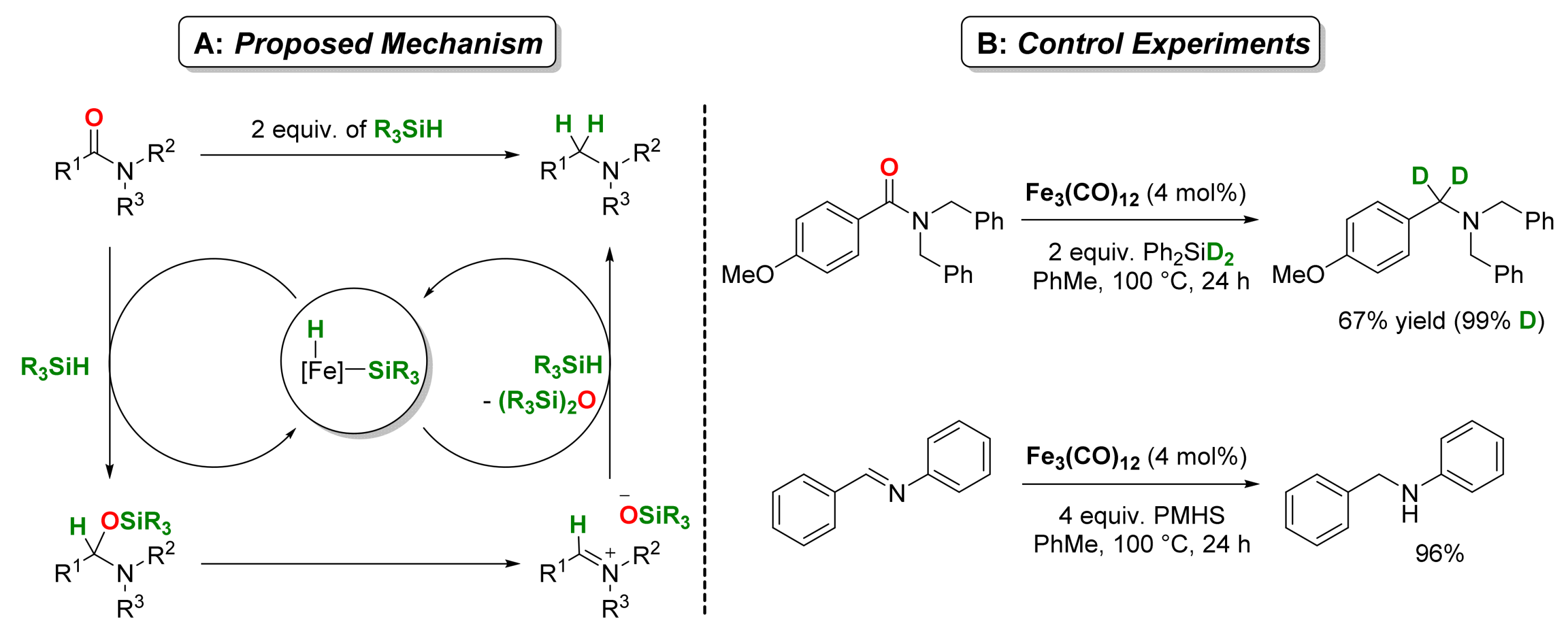
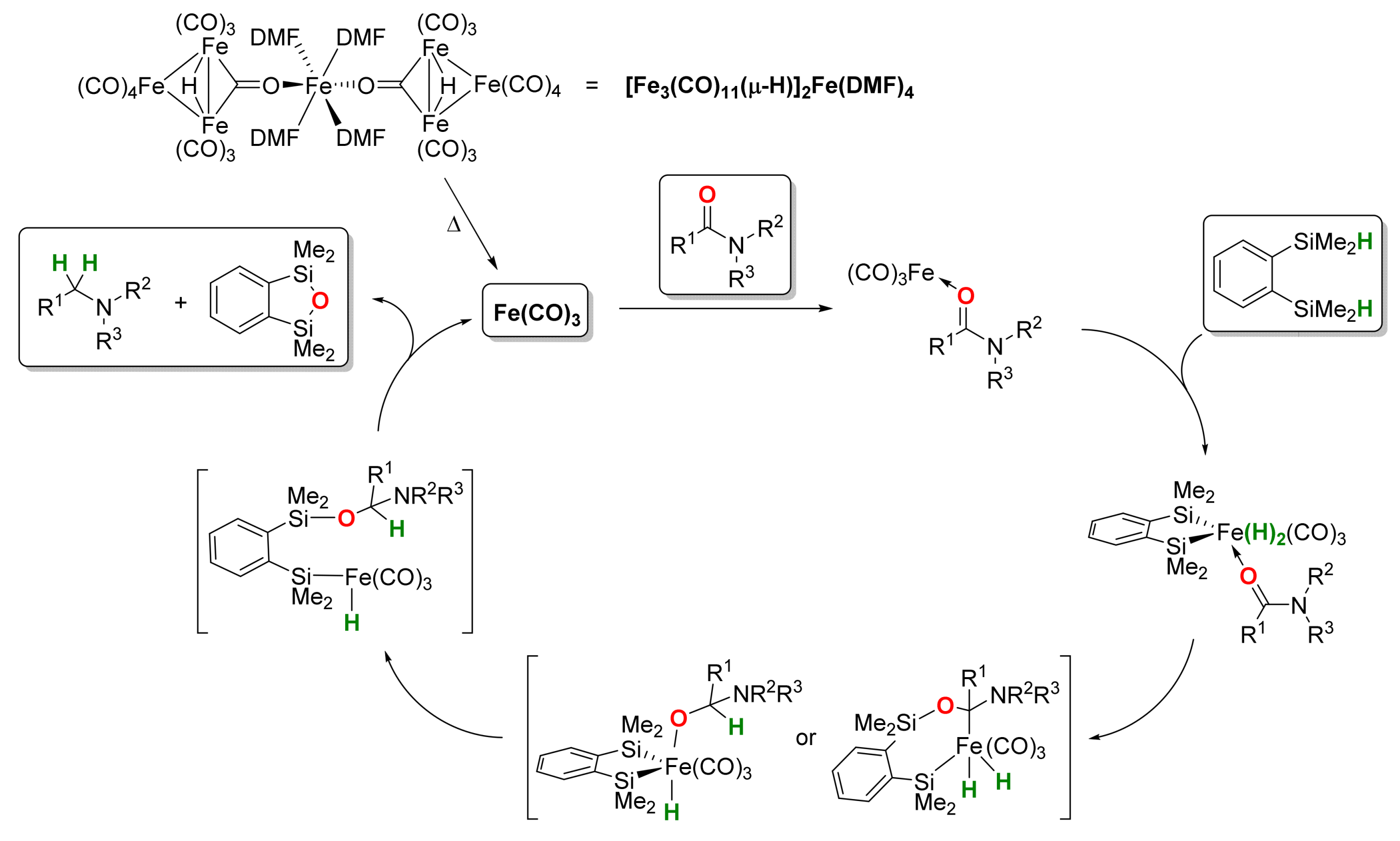
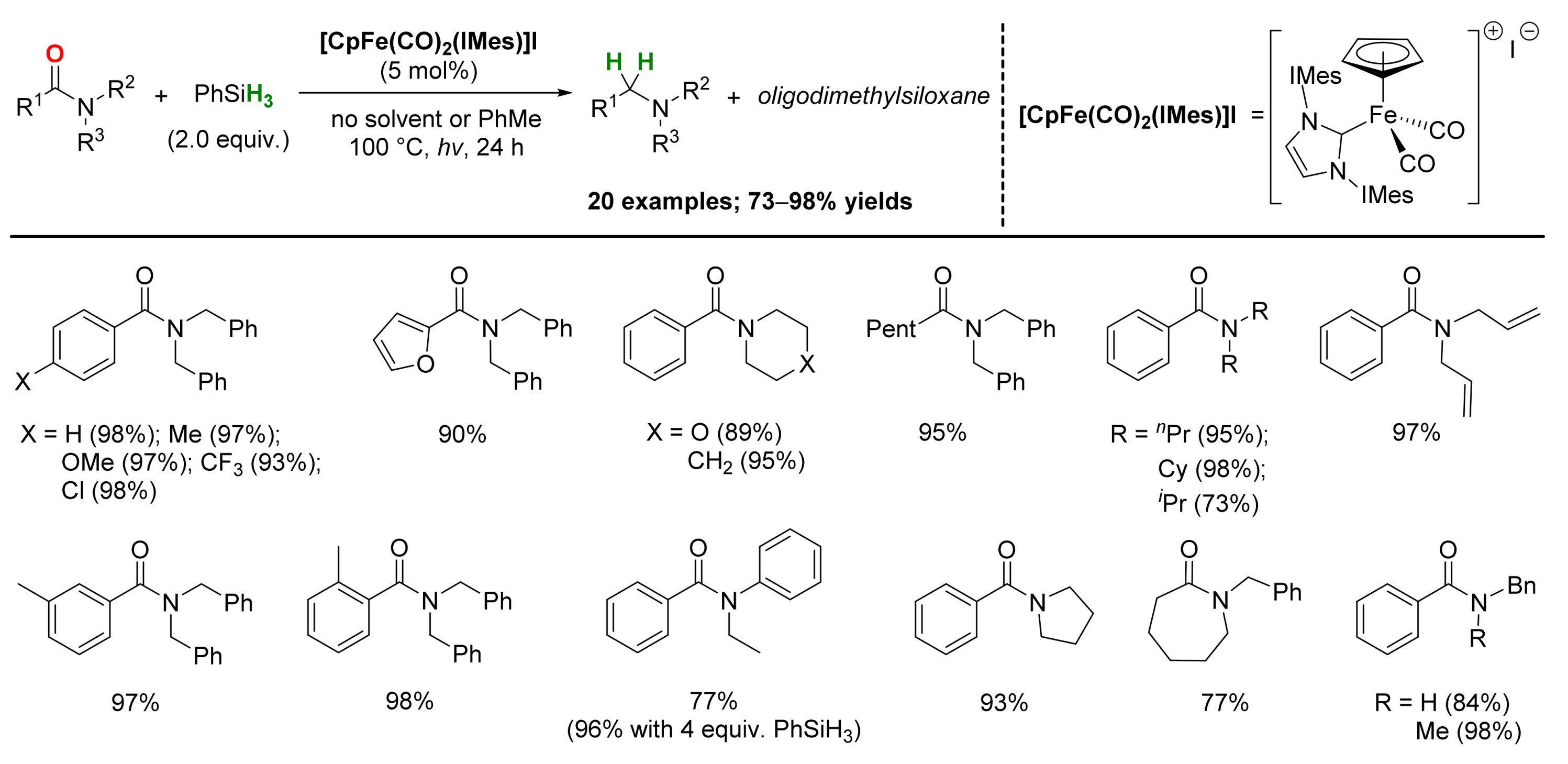
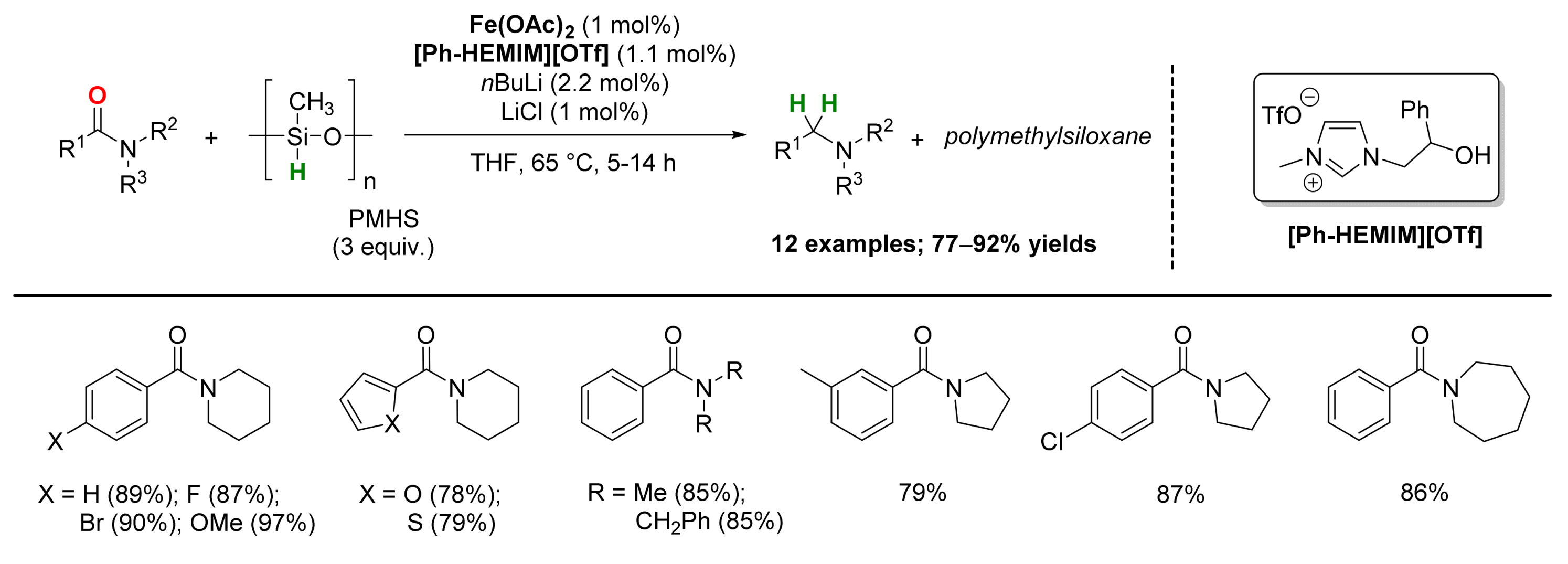
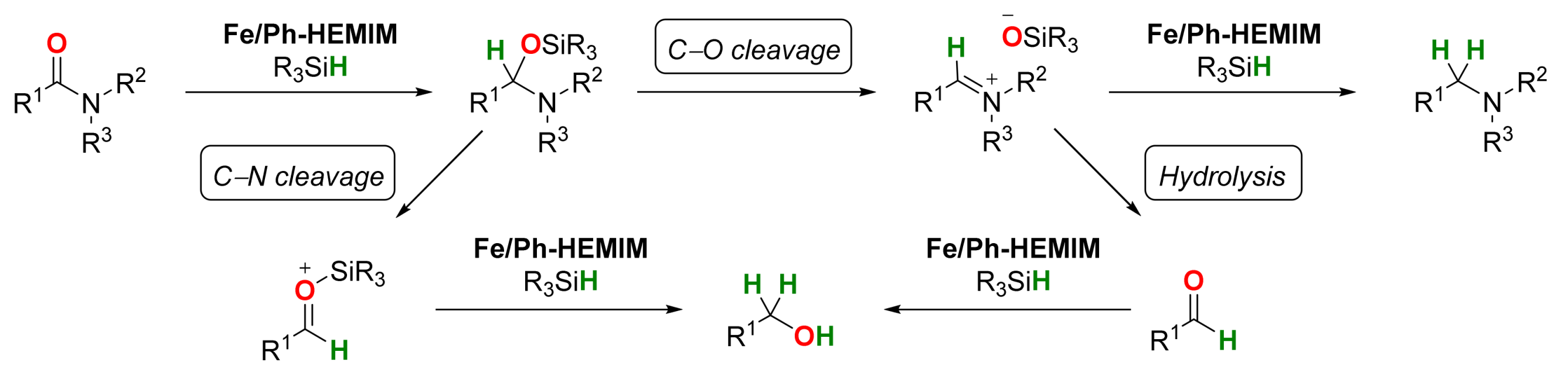
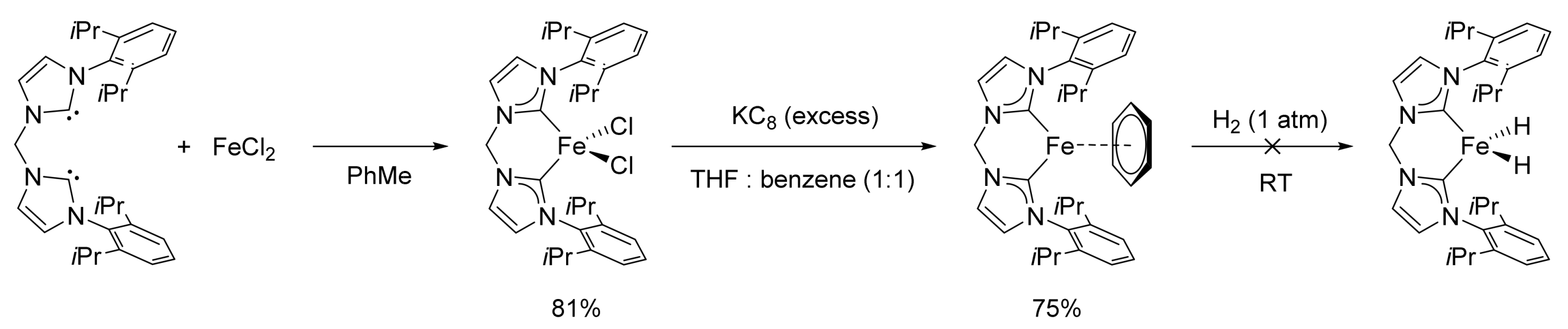
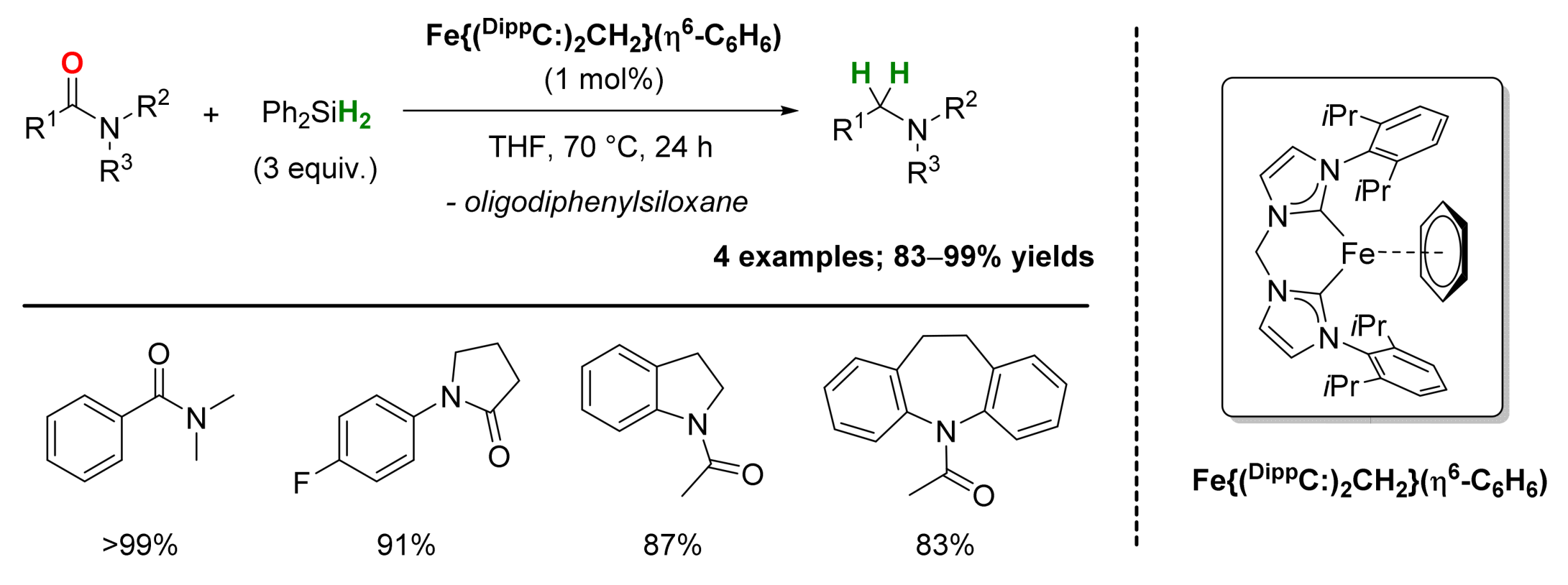
2.1. Iron-Catalyzed Reactions
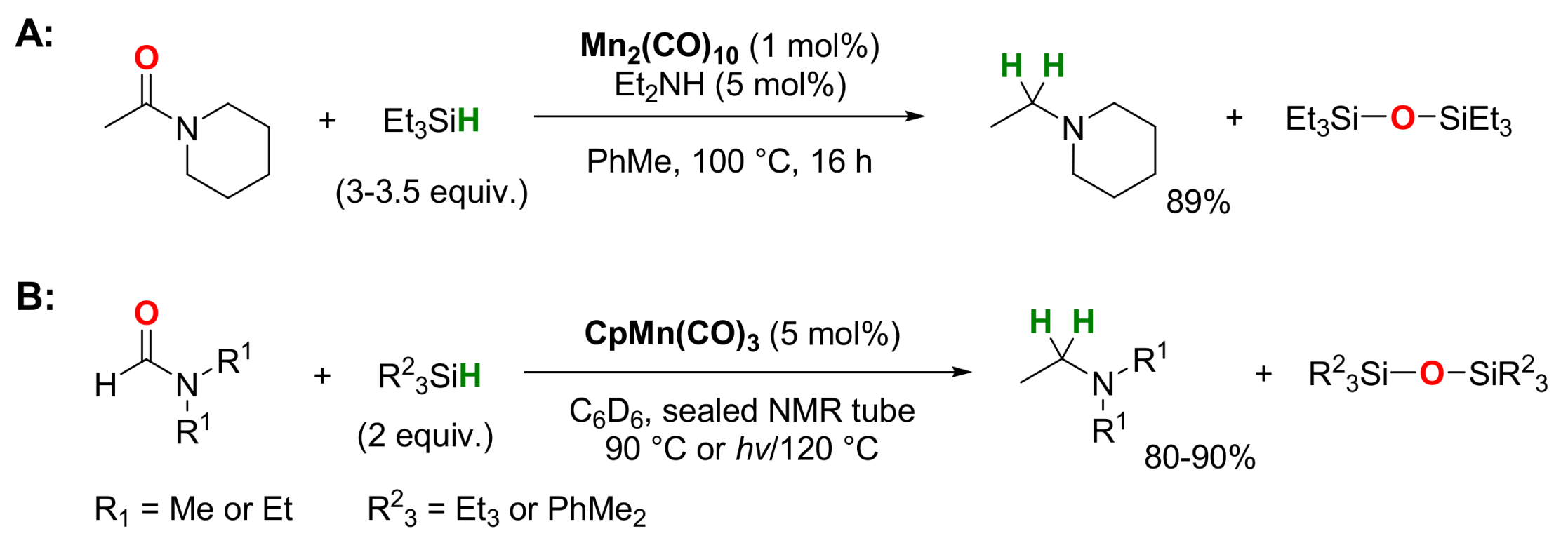
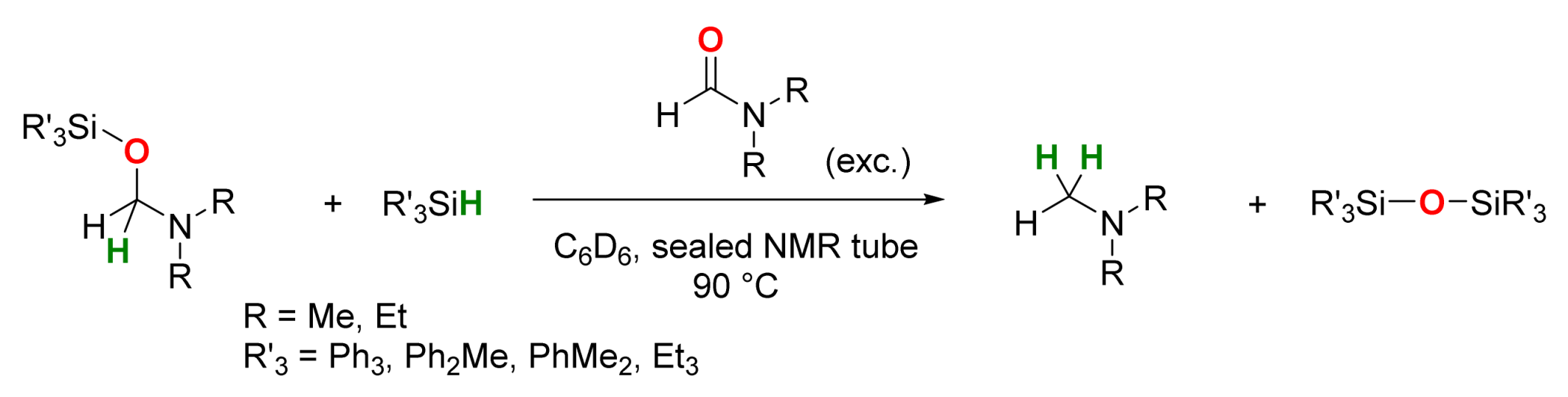
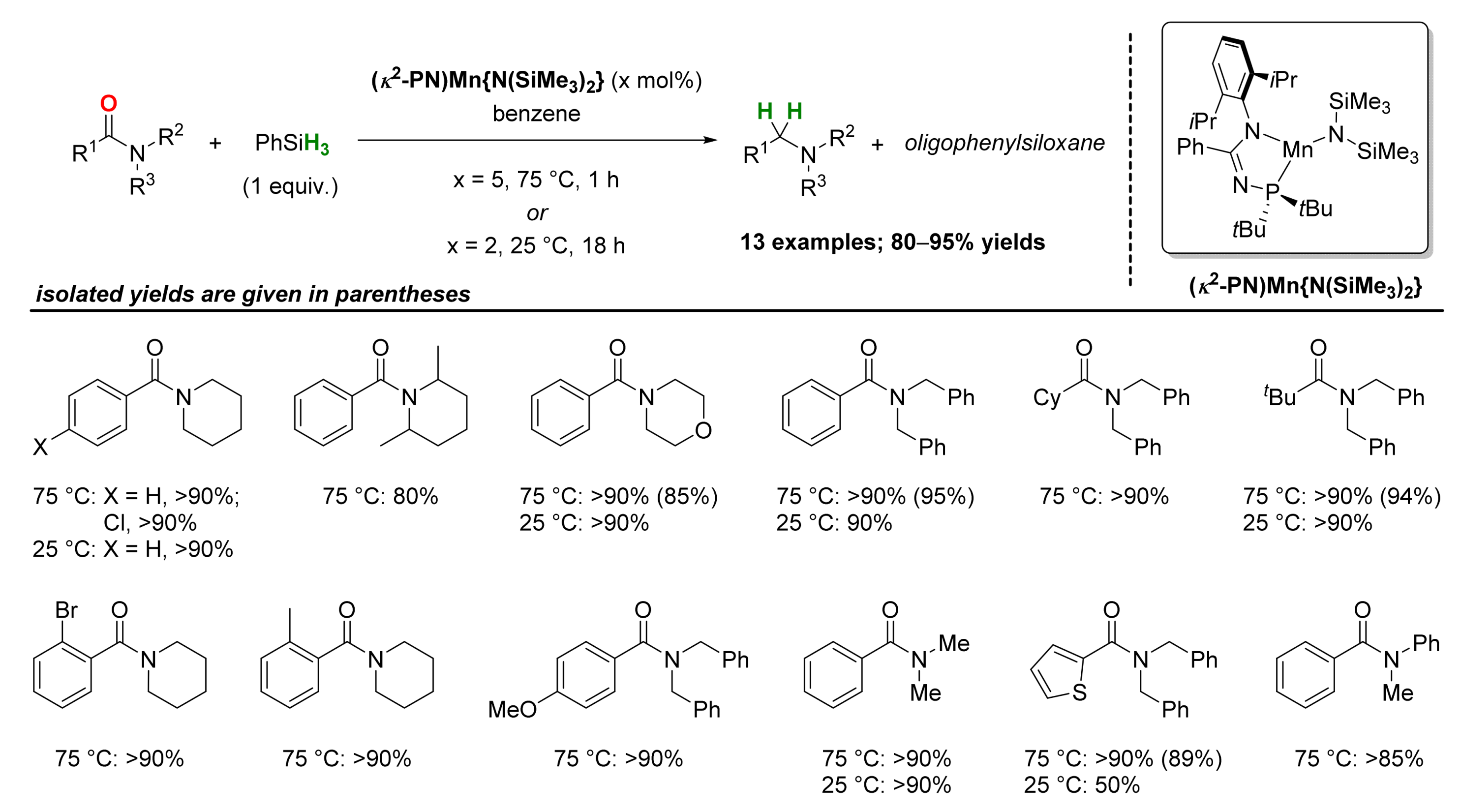
2.2. Manganese-Catalyzed Reactions
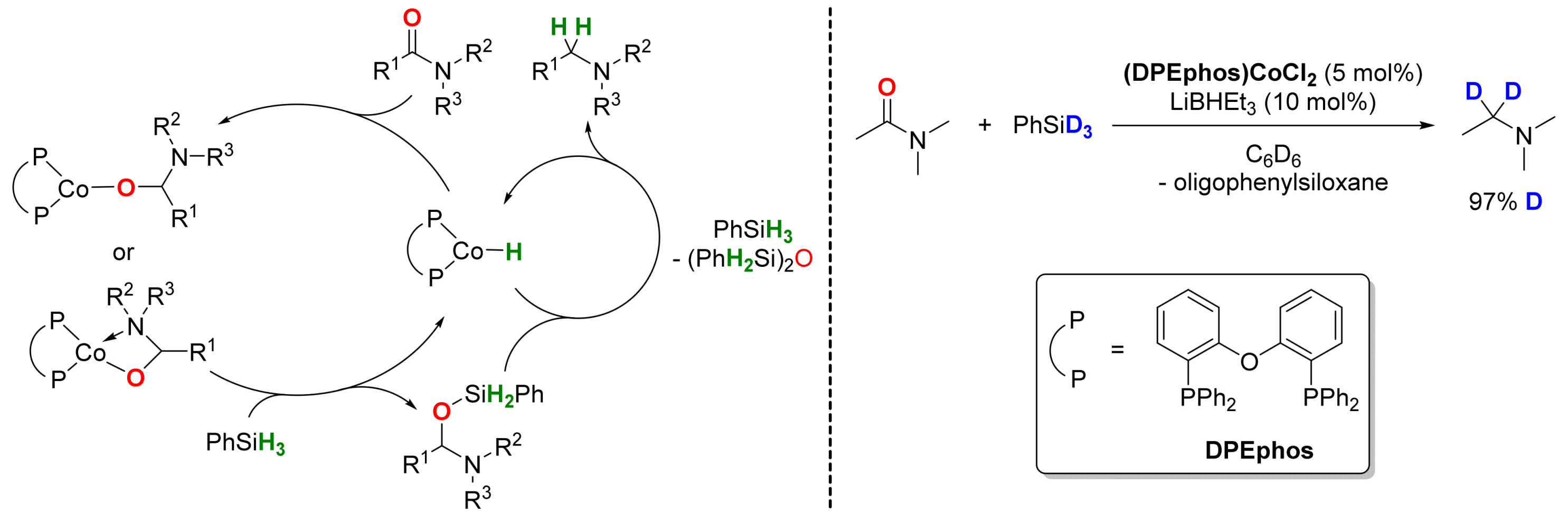
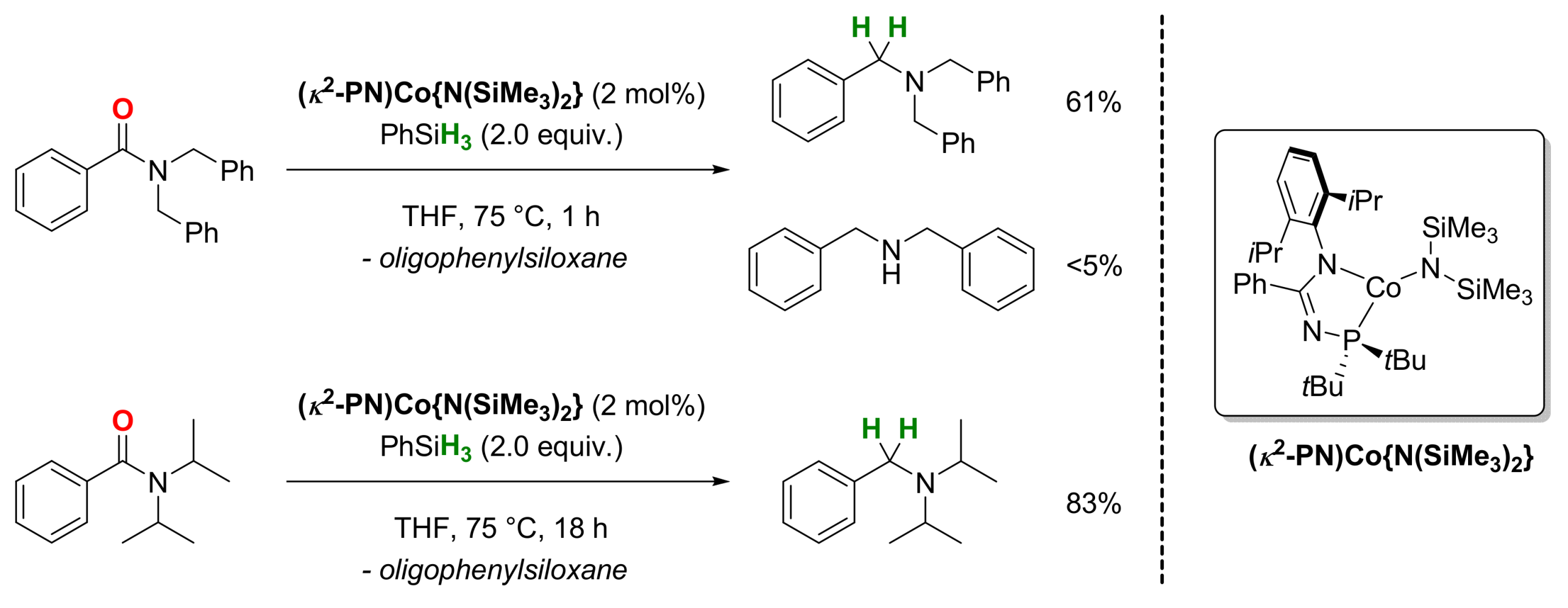
2.3. Cobalt-Catalyzed Reactions
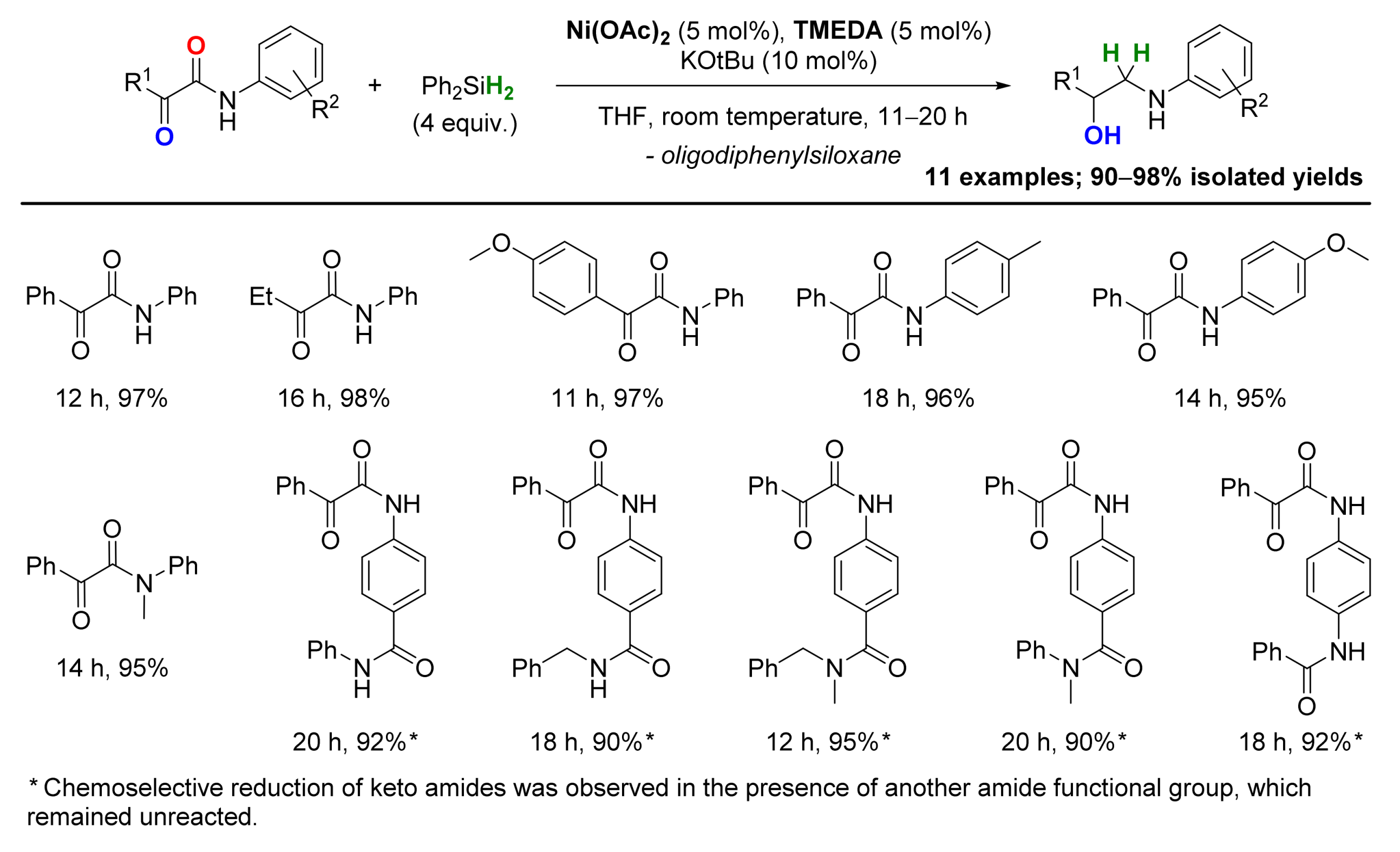
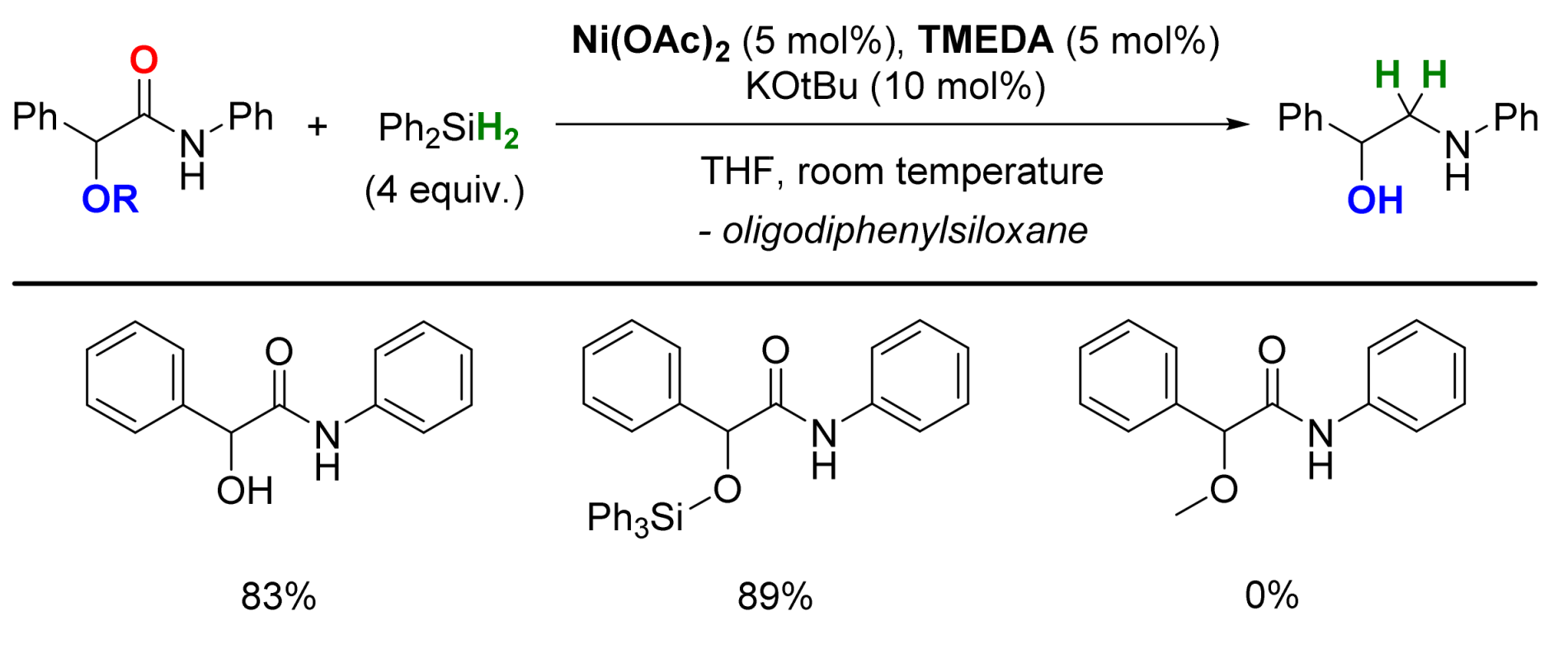
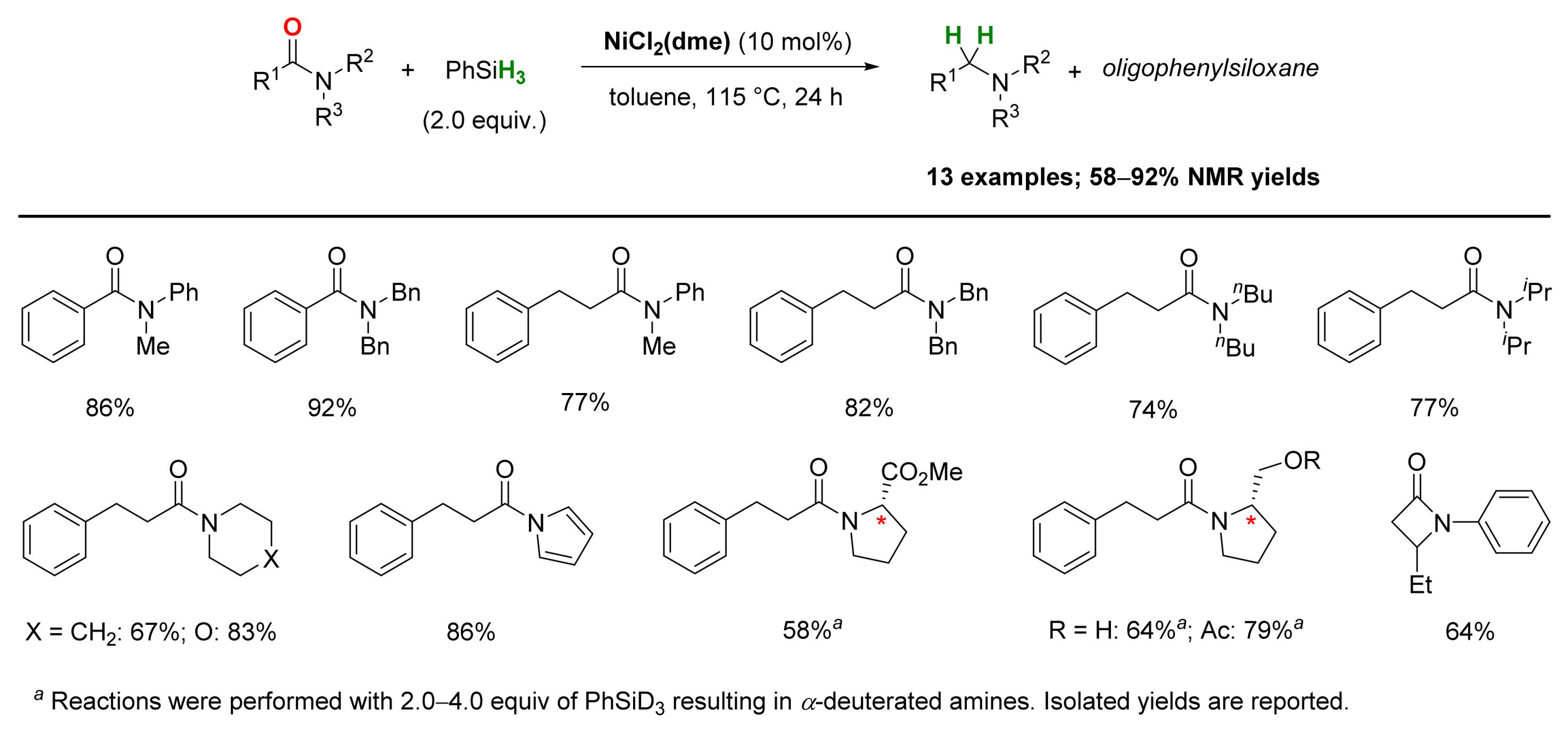
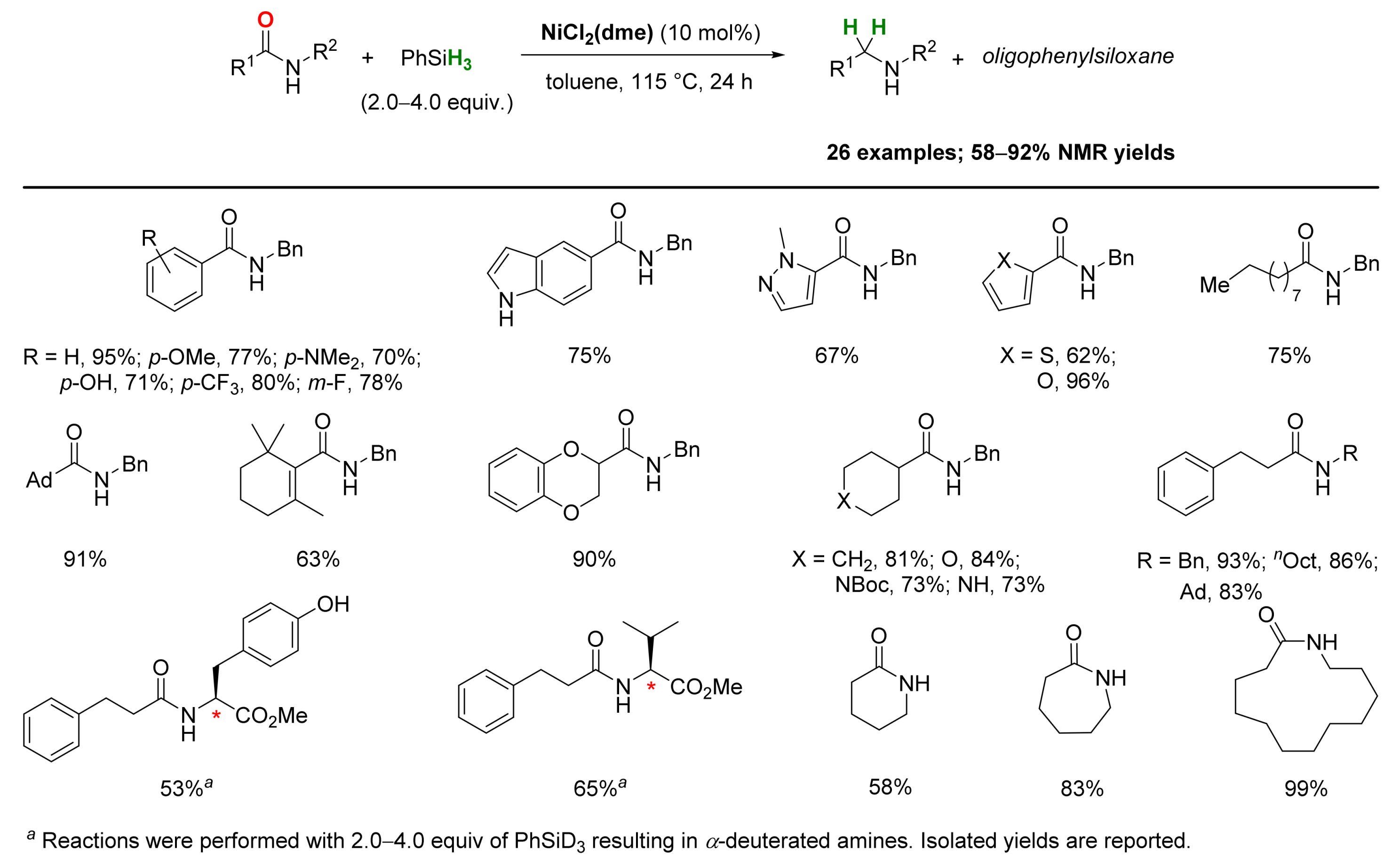
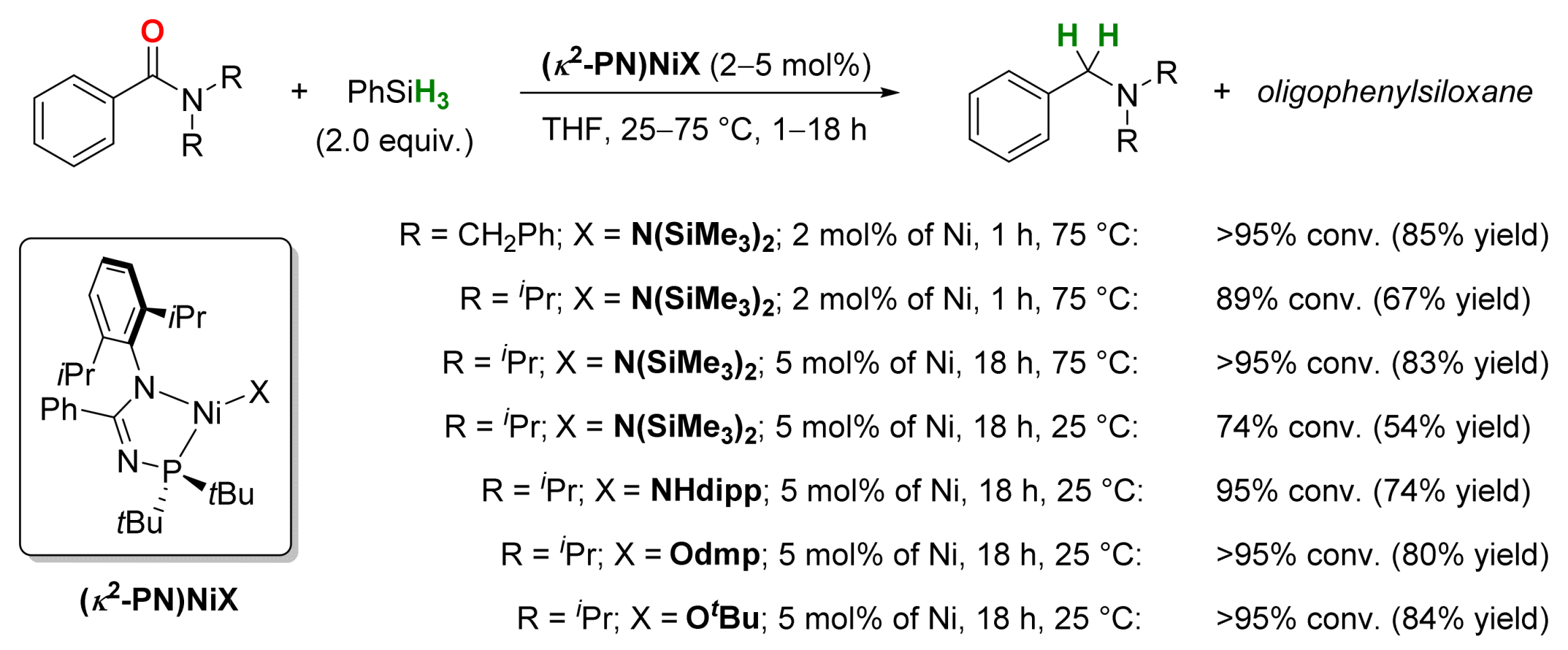
2.4. Nickel-Catalyzed Reactions
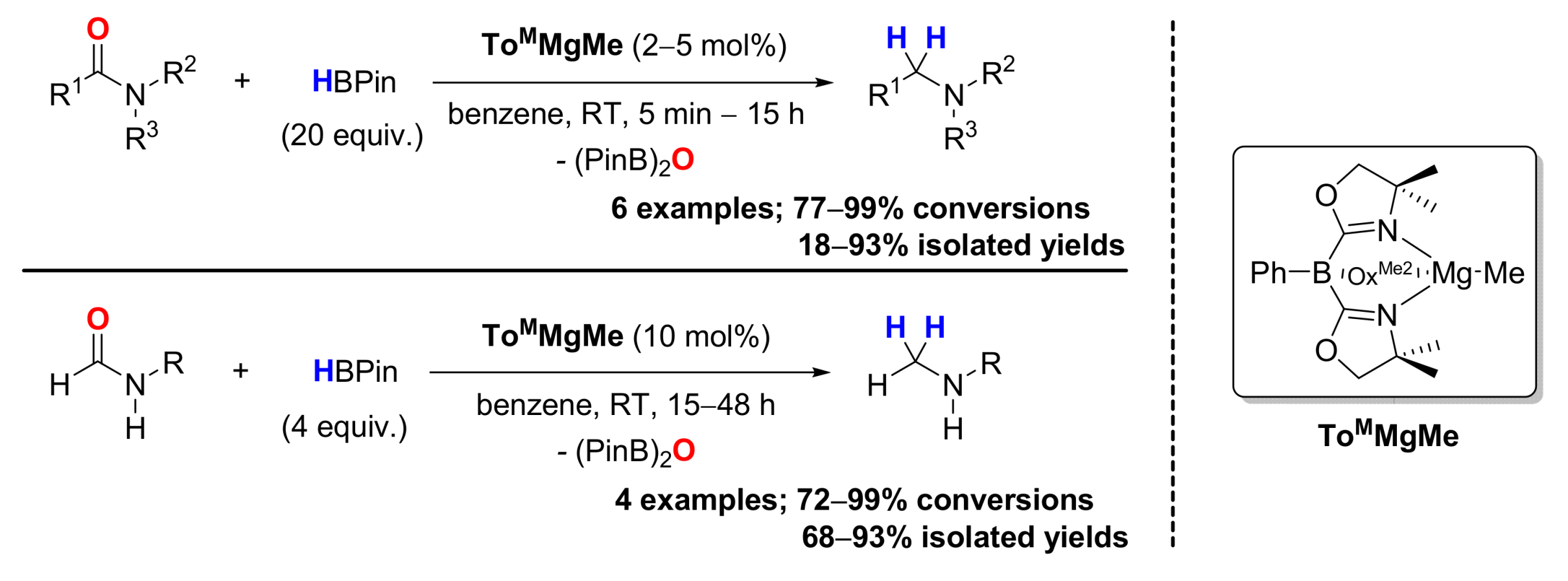
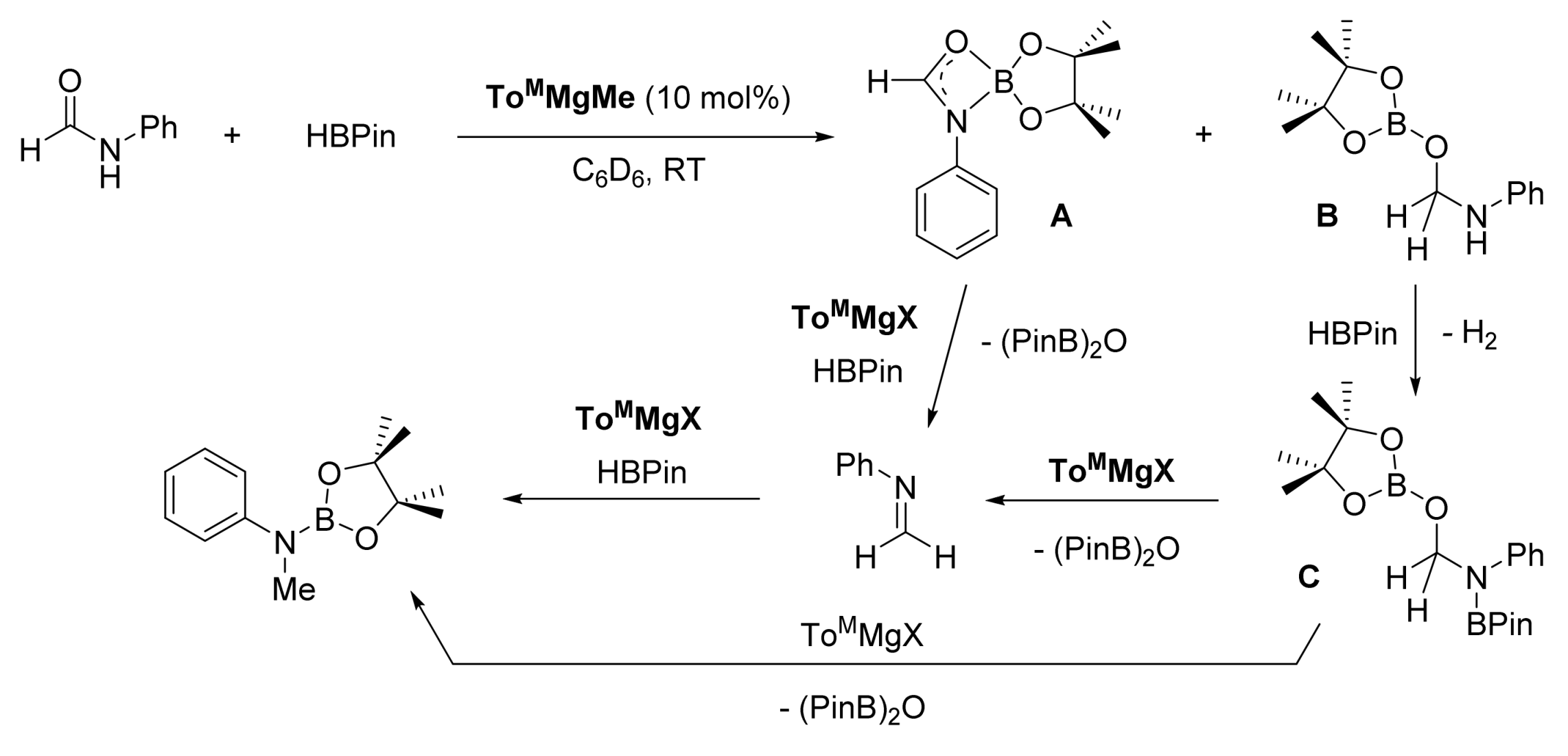
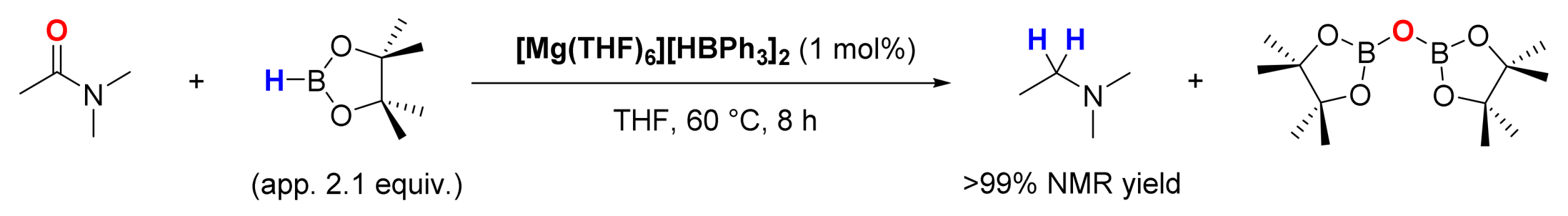
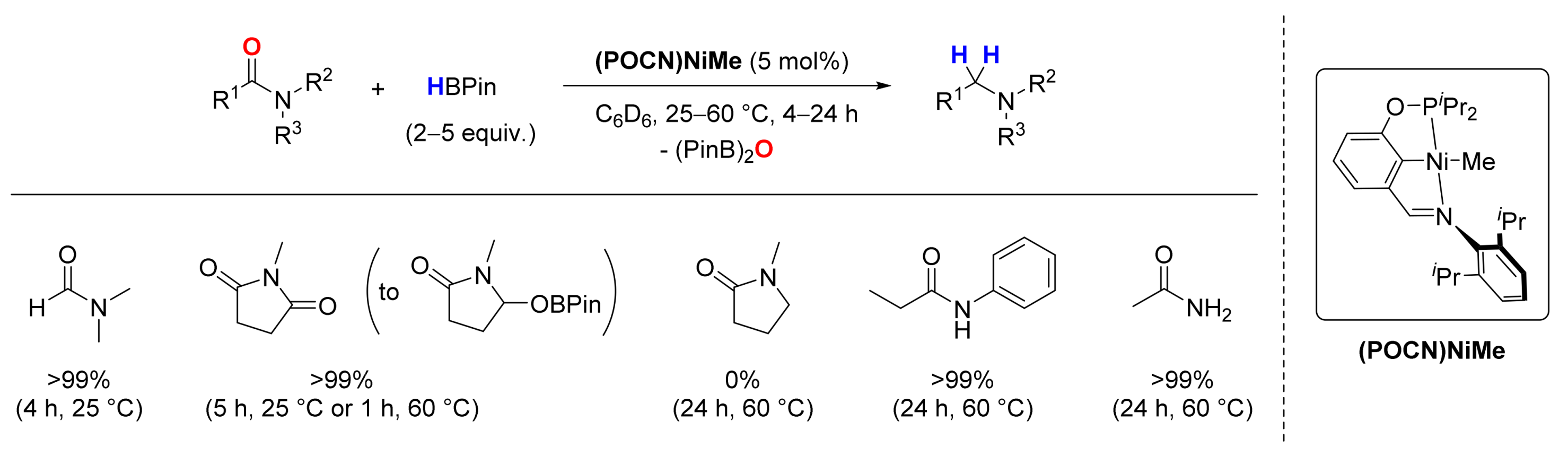
3. Deoxygenative Hydroboration of Amides
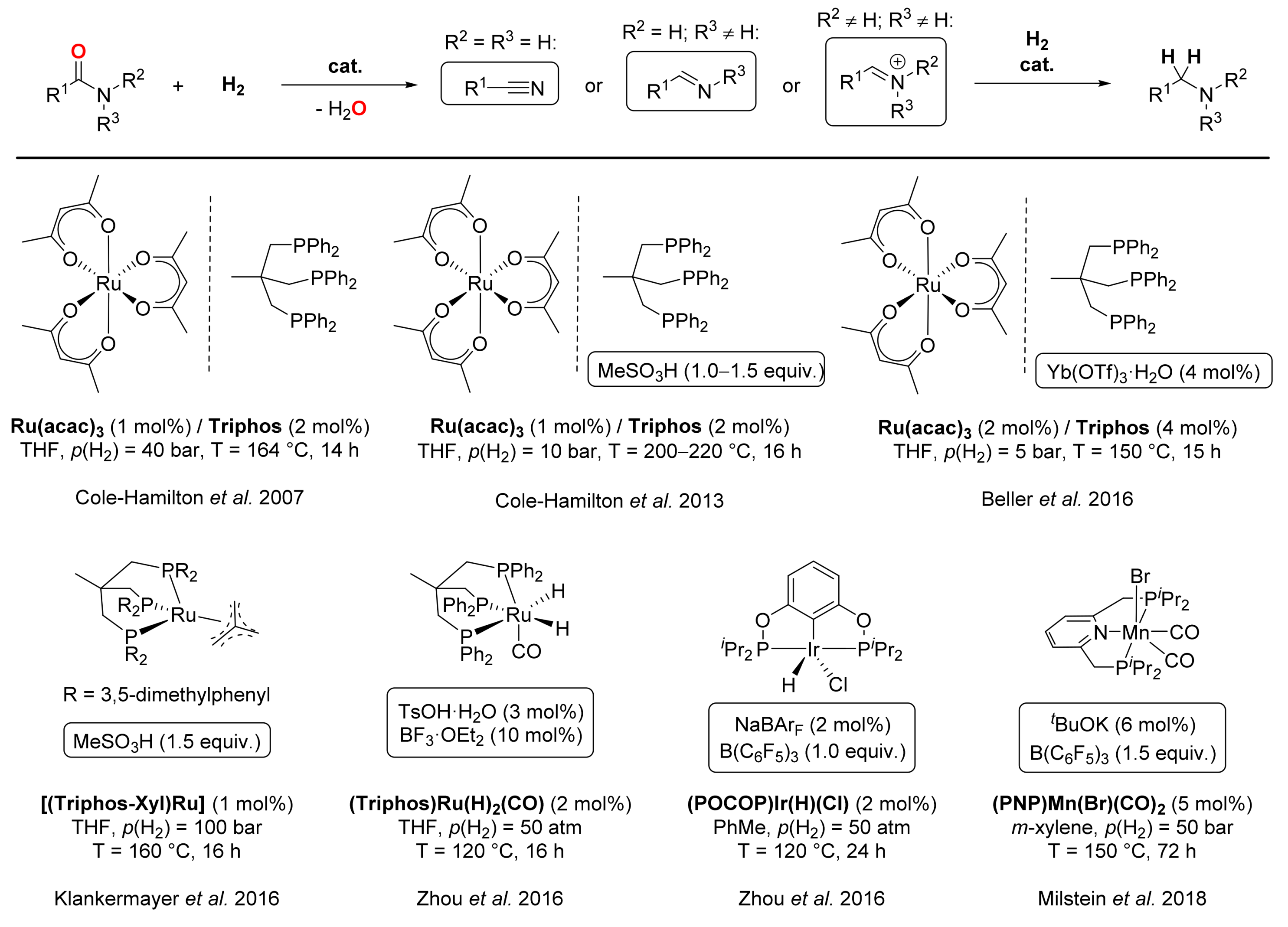
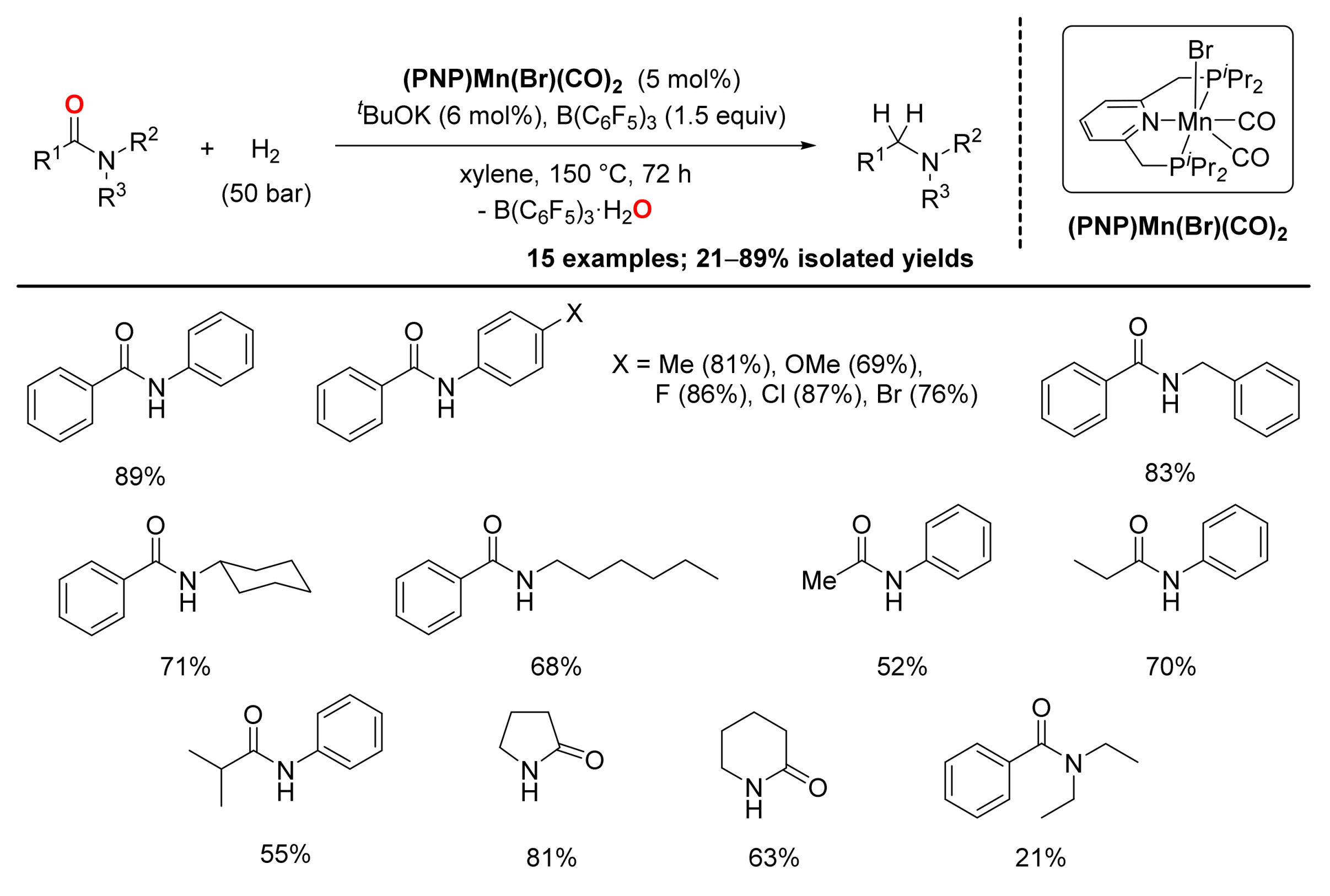
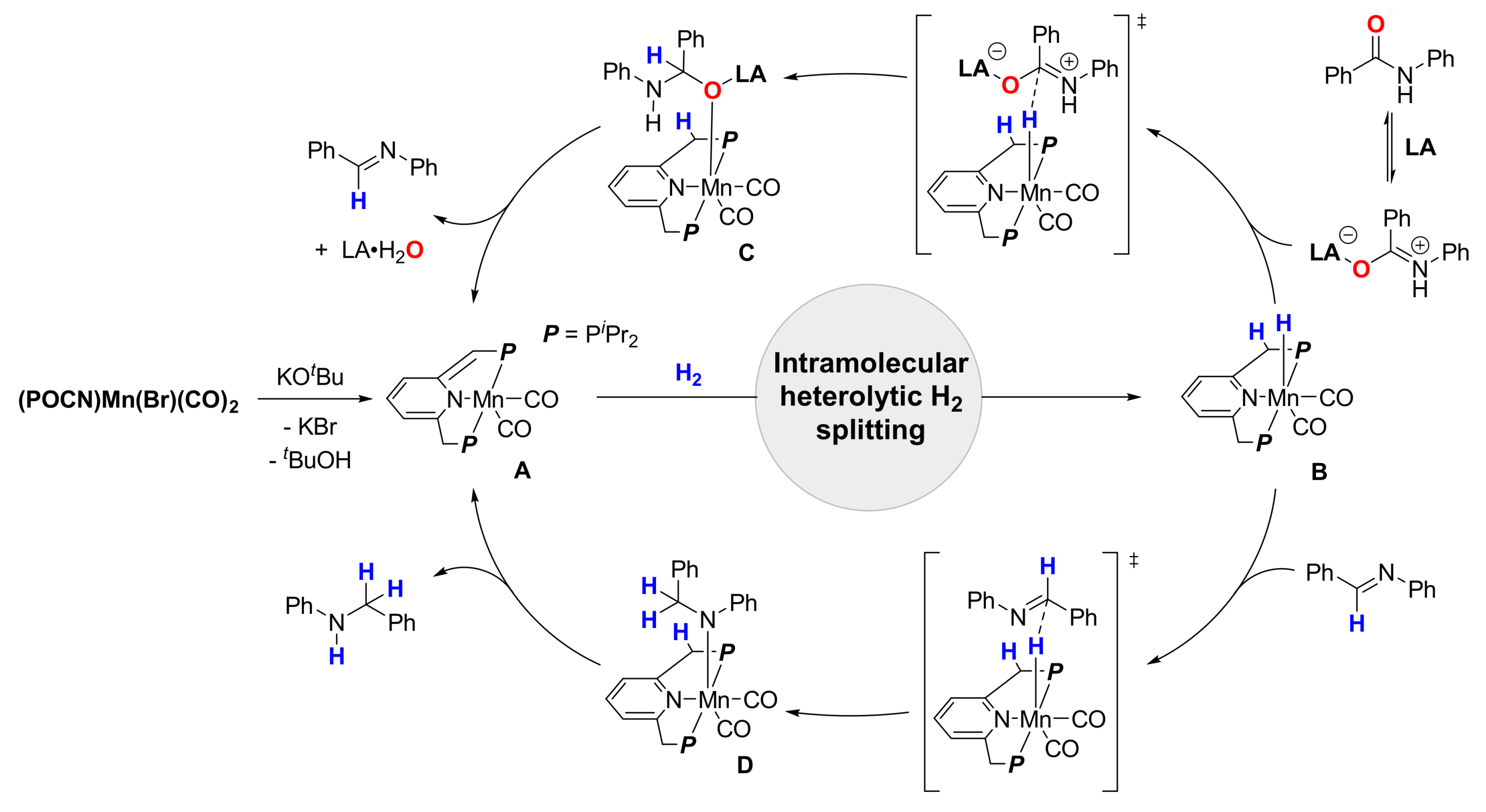
4. Deoxygenative Hydrogenation of Amides
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Smith, M. Organic Synthesis, 4th ed.; Academic Press: London, UK, 2017. [Google Scholar]
- Lawrence, S.A. Amines: Synthesis, Properties and Applications, 1st ed.; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. Amines, Aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Larock, R.C. Comprehensive Organic Transformations: A Guide to Functional Group Preparation; Wiley-VCH: New York, NY, USA, 1989. [Google Scholar]
- Ricci, A. Amino Group Chemistry. From Synthesis to the Life Sciences; Ricci, A., Ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Seyden-Penne, J. Reductions by the Alumino- and Borohydrides in Organic Synthesis, 2nd ed.; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Magro, A.A.N.; Eastham, G.R.; Cole-Hamilton, D.J. The Synthesis of Amines by the Homogeneous Hydrogenation of Secondary and Primary Amides. Chem. Commun. 2007, 3154–3156. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, J.; Dodds, D.L.; Klankermayer, J.; Brosinski, S.; Leitner, W.; Slawin, A.M.Z.; Cole-Hamilton, D.J. Homogeneous Catalytic Hydrogenation of Amides to Amines. Chem. Eur. J. 2013, 19, 11039–11050. [Google Scholar] [CrossRef] [PubMed]
- Meuresch, M.; Westhues, S.; Leitner, W.; Klankermayer, J. Tailor−Made Ruthenium−Triphos Catalysts for the Selective Homogeneous Hydrogenation of Lactams. Angew. Chem. Int. Ed. 2016, 55, 1392–1395. [Google Scholar] [CrossRef]
- Cabrero-Antonino, J.R.; Alberico, E.; Junge, K.; Junge, H.; Beller, M. Towards a General Ruthenium-Catalyzed Hydrogenation of Secondary and Tertiary Amides to Amines. Chem. Sci. 2016, 7, 3432–3442. [Google Scholar] [CrossRef]
- Yuan, M.-L.; Xie, J.-H.; Zhou, Q.-L. Boron Lewis Acid Promoted Ruthenium−Catalyzed Hydrogenation of Amides: An Efficient Approach to Secondary Amines. ChemCatChem 2016, 8, 3036–3040. [Google Scholar] [CrossRef]
- Yuan, M.-L.; Xie, J.-H.; Zhu, S.-F.; Zhou, Q.-L. Deoxygenative Hydrogenation of Amides Catalyzed by a Well−Defined Iridium Pincer Complex. ACS Catal. 2016, 6, 3665–3669. [Google Scholar] [CrossRef]
- Zou, Y.-Q.; Chkraborty, S.; Nerush, A.; Oren, D.; Diskin-Posner, Y.; David-Ben, Y.; Milstein, D. Highly Selective, Efficient Deoxygenative Hydrogenation of Amides Catalyzed by a Manganese Pincer Complex via Metal-Ligand Cooperation. ACS Catal. 2018, 8, 8014–8019. [Google Scholar] [CrossRef]
- Li, B.; Sortais, J.-B.; Darcel, C. Amines synthesis via transition metal homogeneous catalyzed hydrosilylation. RSC Adv. 2016, 6, 57603–57625. [Google Scholar] [CrossRef]
- Gudun, K.A.; Segizbayev, M.; Adamov, A.; Plessow, P.N.; Lyssenko, K.A.; Balanay, M.P.; Khalimon, A.Y. POCN Ni(II) pincer complexes: synthesis, characterization and evaluation of catalytic hydrosilylation and hydroboration activities. Dalton Trans. 2019, 48, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Modern Reduction Methods; Andersson, P.G., Munslow, I.J., Eds.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Igarashi, M.; Fuchikami, T. Transition-metal complex-catalyzed reduction of amides with hydrosilanes: a facile transformation of amides to amines. Tetrahedron Lett. 2001, 42, 1945–1947. [Google Scholar] [CrossRef]
- Matsubara, K.; Iura, T.; Maki, T.; Nagashima, H. A Triruthenium Carbonyl Cluster Bearing a Bridging Acenaphthylene Ligand: An Efficient Catalyst for Reduction of Esters, Carboxylic Acids, and Amides by Trialkylsilanes. J. Org. Chem. 2001, 67, 4985–4988. [Google Scholar] [CrossRef]
- Motoyama, Y.; Mitsui, K.; Ishida, T.; Nagashima, H. Self-Encapsulation of Homogeneous Catalyst Species into Polymer Gel Leading to a Facile and Efficient Separation System of Amine Products in the Ru-Catalyzed Reduction of Carboxamides with Polymethylhydrosiloxane (PMHS). J. Am. Chem. Soc. 2005, 127, 13150–13151. [Google Scholar] [CrossRef] [PubMed]
- Sasakuma, H.; Motoyama, Y.; Nagashima, H. Functional group-selective poisoning of molecular catalysts: a ruthenium cluster-catalysed highly amide-selective silane reduction that does not affect ketones or esters. Chem. Commun. 2007, 4916–4918. [Google Scholar] [CrossRef]
- Hanada, S.; Ishida, T.; Motoyama, Y.; Nagashima, H. The Ruthenium-Catalyzed Reduction and Reductive N-Alkylation of Secondary Amides with Hydrosilanes: Practical Synthesis of Secondary and Tertiary Amines by Judicious Choice of Hydrosilanes. J. Org. Chem. 2007, 72, 7551–7559. [Google Scholar] [CrossRef]
- Reeves, J.T.; Tan, Z.; Marsini, M.A.; Han, Z.S.; Xu, Y.; Reeves, D.C.; Lee, H.; Lu, B.Z.; Senanayake, C.H. A Practical Procedure for Reduction of Primary, Secondary and Tertiary Amides to Amines. Adv. Synth. Catal. 2013, 355, 47–52. [Google Scholar] [CrossRef]
- Li, B.; Sortais, J.-B.; Darcel, C. Unexpected selectivity in ruthenium-catalyzed hydrosilylation of primary amides: synthesis of secondary amines. Chem. Commun. 2013, 49, 3691–3693. [Google Scholar] [CrossRef]
- Kuwano, R.; Takahashi, M.; Ito, Y. Reduction of Amides to Amines via Catalytic Hydrosilylation by a Rhodium Complex. Tetrahedron Lett. 1998, 39, 1017–1020. [Google Scholar] [CrossRef]
- Bornschein, C.; Lennox, A.J.J.; Werkmeister, S.; Junge, K.; Beller, M. A Mild and Selective Reduction of β-Lactams: Rh-Catalyzed Hydrosilylation towards Important Pharmacological Building Blocks. Eur. J. Org. Chem. 2015, 1915–1919. [Google Scholar] [CrossRef]
- Das, S.; Li, Y.; Bornschein, C.; Pisiewicz, S.; Kiersch, K.; Michalik, D.; Gallou, F.; Junge, K.; Beller, M. Selective Rhodium-Catalyzed Reduction of Tertiary Amides in Amino Acid Esters and Peptides. Angew. Chem. Int. Ed. 2015, 54, 12389–12393. [Google Scholar] [CrossRef]
- Park, S.; Brookhart, M. Development and Mechanistic Investigation of a Highly Efficient Iridium(V) Silyl Complex for the Reduction of Tertiary Amides to Amines. J. Am. Chem. Soc. 2012, 134, 640–653. [Google Scholar] [CrossRef]
- Cheng, C.; Brookhart, M. Iridium-Catalyzed Reduction of Secondary Amides to Secondary Amines and Imines by Diethylsilane. J. Am. Chem. Soc. 2012, 134, 11304–11307. [Google Scholar] [CrossRef]
- Motoyama, Y.; Aoki, M.; Takaoka, N.; Aoto, R.; Nagashima, H. Highly efficient synthesis of aldenamines from carboxamides by iridium-catalyzed silane-reduction/dehydration under mild conditions. Chem. Commun. 2009, 1574–1576. [Google Scholar] [CrossRef]
- Tahara, A.; Miyamoto, Y.; Aoto, R.; Shigeta, K.; Une, Y.; Sunada, Y.; Motoyama, Y.; Nagashima, H. Catalyst Design of Vaska-Type Iridium Complexes for Highly Efficient Synthesis of π–Conjugated Enamines. Organometallics 2015, 34, 4895–4907. [Google Scholar] [CrossRef]
- Guzmán, J.; Bernal, A.M.; Garcia-Orduna, P.; Lahoz, F.J.; Oro, L.A.; Fernández-Alvarez, F.J. Selective reduction of formamides to O-silylated hemiaminals or methylamines with HSiMe2Ph catalyzed by iridium complexes. Dalton Trans. 2019, 48, 4255–4262. [Google Scholar] [CrossRef]
- Hanada, S.; Motoyama, Y.; Nagashima, H. Dual Si–H effects in platinum-catalyzed silane reduction of carboxamides leading to a practical synthetic process of tertiary-amines involving self-encapsulation of the catalyst species into the insoluble silicone resin formed. Tetrahedron Lett. 2006, 47, 6173–6177. [Google Scholar] [CrossRef]
- Hanada, S.; Tsutsumi, E.; Motoyama, Y.; Nagashima, H. Practical Access to Amines by Platinum-Catalyzed Reduction of Carboxamides with Hydrosilanes: Synergy of Dual Si-H Groups Leads to High Efficiency and Selectivity. J. Am. Chem. Soc. 2009, 131, 15032–15040. [Google Scholar] [CrossRef] [PubMed]
- Pisiewicz, S.; Junge, K.; Beller, M. Mild Hydrosilylation of Amides by Platinum N-Heterocyclic Carbene Catalysts. Eur. J. Inorg. Chem. 2014, 2345–2349. [Google Scholar] [CrossRef]
- Selvakumar, K.; Rangareddy, K.; Harrod, J.F. The titanocene-catalyzed reduction of acetamides to tertiary amines by PhMeSiH2. Can. J. Chem. 2004, 82, 1244–1248. [Google Scholar] [CrossRef]
- Laval, S.; Dayoub, W.; Pehlivan, L.; Métay, E.; Favre-Réguillon, A.; Delbrayelle, D.; Mignani, G.; Lemaire, M. Hydrosiloxane–Ti(OiPr)4: an efficient system for the reduction of primary amides into primary amines as their hydrochloride salts. Tetrahedron Lett. 2011, 52, 4072–4075. [Google Scholar] [CrossRef]
- Fernandes, A.C.; Romao, C.C. Reduction of amides with silanes catalyzed by MoO2Cl2. J. Mol. Catal. A Chem. 2007, 272, 60–63. [Google Scholar] [CrossRef]
- Volkov, A.; Tinnis, F.; Slagbrand, T.; Pershagen, I.; Adolfsson, H. Mo(CO)6 catalysed chemoselective hydrosilylation of α,β-unsaturated amides for the formation of allylamines. Chem. Commun. 2014, 50, 14508–14511. [Google Scholar] [CrossRef]
- Das, S.; Join, B.; Junge, K.; Beller, M. A general and selective copper-catalyzed reduction of secondary amides. Chem. Commun. 2012, 48, 2683–2685. [Google Scholar] [CrossRef]
- Calas, R. Thirty years in organosilicon chemistry. J. Organomet. Chem. 1980, 200, 11–36. [Google Scholar] [CrossRef]
- Das, S.; Addis, D.; Zhou, S.; Junge, K.; Beller, M. Zinc-Catalyzed Reduction of Amides: Unprecedented Selectivity and Functional Group Tolerance. J. Am. Chem. Soc. 2010, 132, 1770–1771. [Google Scholar] [CrossRef]
- Das, S.; Addis, D.; Junge, K.; Beller, M. Zinc-Catalyzed Chemoselective Reduction of Tertiary and Secondary Amides to Amines. Chem. Eur. J. 2011, 17, 12186–12192. [Google Scholar] [CrossRef]
- Kovalenko, O.O.; Volkov, A.; Adolfsson, H. Mild and Selective Et2Zn-Catalyzed Reduction of Tertiary Amides under Hydrosilylation Conditions. Org. Lett. 2015, 17, 446–449. [Google Scholar] [CrossRef]
- Chardon, A.; El Dine, T.M.; Legay, R.; De Paolis, M.; Rouden, J.; Blanchet, J. Borinic Acid Catalysed Reduction of Tertiary Amides with Hydrosilanes: A Mild and Chemoselective Synthesis of Amines. Chem. Eur. J. 2017, 23, 2005–2009. [Google Scholar] [CrossRef]
- Peruzzi, M.T.; Mei, Q.Q.; Lee, S.J.; Gagne, M.R. Chemoselective amide reductions by heteroleptic fluoroaryl boron Lewis acids. Chem. Commun. 2018, 54, 5855–5858. [Google Scholar] [CrossRef]
- Bower, S.; Kreutzer, K.A.; Buchwald, S.L. A Mild General Procedure for the One-Pot Conversion of Amides to Aldehydes. Angew. Chem. Int. Ed. 1996, 35, 1515–1516. [Google Scholar] [CrossRef]
- Krackl, S.; Someya, C.I.; Enthaler, S. Reductive Cleavage of Amides to Alcohols and Amines Catalyzed by Well-Defined Bimetallic Molybdenum Complexes. Chem. Eur. J. 2012, 18, 15267–15271. [Google Scholar] [CrossRef]
- Chirik, P.J. Catalysis without Precious Metals; Ballock, R.M., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp. 83–110. [Google Scholar]
- Nurseiit, A.; Janabel, J.; Gudun, K.A.; Kassymbek, A.; Segizbayev, M.; Seilkhanov, T.M.; Khalimon, A.Y. Bench-Stable Cobalt Pre-Catalysts for Mild Hydrosilative Reduction of Tertiary Amides and Beyond. ChemCatChem 2019, 11, 790–798. [Google Scholar] [CrossRef]
- Kelly, C.M.; McDonald, R.; Sydora, O.L.; Stradiotto, M.; Turculet, L. A Manganese Pre-Catalyst: Mild Reduction of Amides, Ketones, Aldehydes, and Esters. Angew. Chem. Int. Ed. 2017, 56, 15901–15904. [Google Scholar] [CrossRef]
- Macaulay, C.M.; Ogawa, T.; McDonald, R.; Sydora, O.L.; Stradiotto, M.; Turculet, L. A comparative analysis of hydrosilative amide reduction catalyzed by first-row transition metal (Mn, Fe, Co and Ni) N-phosphinoamidinate complexes. Dalton Trans. 2019. [Google Scholar] [CrossRef]
- Calas, R. Sur Quelques Aspects De La Reactivitie Des Hydrogenosilanes En Chimie Organique. Pure Appl. Chem. 1966, 13, 61–79. [Google Scholar] [CrossRef]
- Zhou, S.; Junge, K.; Addis, D.; Das, S.; Beller, M. A Convenient and General Iron-Catalyzed Reduction of Amides to Amines. Angew. Chem. Int. Ed. 2009, 48, 9507–9510. [Google Scholar] [CrossRef]
- Sunada, Y.; Kawakami, H.; Imaoka, T.; Motoyama, Y.; Nagashima, H. Hydrosilane Reduction of Tertiary Carboxamides by Iron Carbonyl Catalysts. Angew. Chem. Int. Ed. 2009, 48, 9511–9514. [Google Scholar] [CrossRef]
- Zhou, S.; Addis, D.; Das, S.; Junge, K.; Beller, M. New catalytic properties of iron complexes: dehydration of amides to nitriles. Chem. Commun. 2009, 4883–4885. [Google Scholar] [CrossRef]
- Das, S.; Wendt, B.; Möller, K.; Junge, K.; Beller, M. Two Iron Catalysts are Better than One: A General and Convenient Reduction of Aromatic and Aliphatic Primary Amides. Angew. Chem. Int. Ed. 2012, 51, 1662–1666. [Google Scholar] [CrossRef]
- Itazaki, M.; Nakazawa, H. Selective Double Addition Reaction of an E-H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles. Molecules 2018, 23, 2769–2787. [Google Scholar] [CrossRef]
- Khalimon, A.Y.; Simionescu, R.; Kuzmina, L.G.; Howard, J.A.K.; Nikonov, G.I. Agostic NSiH···Mo Complexes: From Curiosity to Catalysis. Angew. Chem. Int. Ed. 2008, 47, 7701–7704. [Google Scholar] [CrossRef]
- Peterson, E.; Khalimon, A.Y.; Simionescu, R.; Kuzmina, L.G.; Howard, J.A.K.; Nikonov, G.I. Diversity of Catalysis by an Imido-Hydrido Complex of Molybdenum. Mechanism of Carbonyl Hydrosilylation and Silane Alcoholysis. J. Am. Chem. Soc. 2009, 131, 908–909. [Google Scholar] [CrossRef]
- Khalimon, A.Y.; Simionescu, R.; Nikonov, G.I. Catalytic and Stoichiometric Reactivity of β-Silylamido Agostic Complex of Mo: Intermediacy of a Silanimine Complex and Applications to Multicomponent Coupling. J. Am. Chem. Soc. 2011, 133, 7033–7053. [Google Scholar] [CrossRef]
- Khalimon, A.Y.; Ignatov, S.K.; Okhapkin, A.I.; Simionescu, R.; Kuzmina, L.G.; Howard, J.A.K.; Nikonov, G.I. Unusual Structure, Fluxionality, and Reaction Mechanism of Carbonyl Hydrosilylation by Silyl Hydride Complex [(ArN=)Mo(H)(SiH2Ph)(PMe3)3]. Chem. Eur. J. 2013, 19, 8573–8590. [Google Scholar] [CrossRef]
- Marciniec, B.; Gulinski, J.; Urbaniak, W.; Kornetka, Z.W. Comprehensive Handbook on Hydrosilylation; Marciniec, B., Ed.; Pergamon Press: Oxford, UK, 1992. [Google Scholar]
- Tsutsumi, H.; Sunada, Y.; Nagashima, H. New catalyst systems for iron-catalyzed hydrosilane reduction of carboxamides. Chem. Commun. 2011, 47, 6581–6583. [Google Scholar] [CrossRef]
- Bézier, D.; Venkanna, G.T.; Sortais, J.-B.; Darcel, C. Well-Defined Cyclopentadienyl NHC Iron Complex as the Catalyst for Efficient Hydrosilylation of Amides to Amines and Nitriles. ChemCatChem 2011, 3, 1747–1750. [Google Scholar] [CrossRef]
- Volkov, A.; Buitrago, E.; Adolfsson, H. Direct Hydrosilylation of Tertiary Amides to Amines by an In Situ Formed Iron/N-Heterocyclic Carbene Catalyst. Eur. J. Org. Chem. 2013, 2066–2070. [Google Scholar] [CrossRef]
- Blom, B.; Tan, G.; Enthaler, S.; Inoue, S.; Epping, J.D.; Driess, M. Bis-N-Heterocyclic Carbene (NHC) Stabilized η6-Arene Iron(0) Complexes: Synthesis, Structure, Reactivity, and Catalytic Activity. J. Am. Chem. Soc. 2013, 135, 18108–18120. [Google Scholar] [CrossRef]
- Macaulay, C.M.; Gustafson, S.J.; Fuller, J.T., III; Kwon, D.-H.; Ogawa, T.; Ferguson, M.J.; McDonald, R.; Lumsden, M.D.; Bischof, S.M.; Sydora, O.L.; et al. Alkene Isomerization–Hydroboration Catalyzed by First-Row Transition-Metal (Mn, Fe, Co, and Ni) N-Phosphinoamidinate Complexes: Origin of Reactivity and Selectivity. ACS Catal. 2018, 8, 9907–9925. [Google Scholar] [CrossRef]
- Arias-Ugarte, R.; Sharma, H.K.; Morris, A.L.C.; Pannell, K.H. Metal-Catalyzed Reduction of HCONR’2, R’ = Me (DMF), Et (DEF), by Silanes to Produce R’2NMe and Disiloxanes: A Mechanism Unraveled. J. Am. Chem. Soc. 2012, 134, 848–851. [Google Scholar] [CrossRef]
- Weinmann, M.; Walter, G.; Huttner, G.; Heinrich, J. Reaction studies on hypervalent silicon hydride compounds. J. Organomet. Chem. 1998, 561, 131–141. [Google Scholar] [CrossRef]
- Chruściel, J.J. 29Si NMR studies on the mechanism of dehydrocoupling of hydrosilanes with hydroxylic reagents in DMF — The role of DMF. Can. J. Chem. 2005, 83, 508–516. [Google Scholar] [CrossRef]
- Dombray, T.; Helleu, C.; Darcel, C.; Sortais, J.-B. Cobalt Carbonyl-Based Catalyst for Hydrosilylation of Carboxamides. Adv. Synth. Catal. 2013, 355, 3358–3362. [Google Scholar] [CrossRef]
- Dierkes, P.; van Leeuwen, P.W.N.M. The bite angle makes the difference: a practical ligand parameter for diphosphine ligands. J. Chem. Soc. Dalton Trans. 1999, 1519–1530. [Google Scholar] [CrossRef]
- Mamillapalli, N.C.; Sekar, G. Chemoselective reduction of a-keto amides using nickel catalysts. Chem. Commun. 2014, 50, 7881–7884. [Google Scholar] [CrossRef]
- Simmons, B.J.; Hoffmann, M.; Hwang, J.; Jackl, M.K.; Garg, N.K. Nickel-Catalyzed Reduction of Secondary and Tertiary Amides. Org. Lett. 2017, 19, 1910–1913. [Google Scholar] [CrossRef]
- Mullard, A. Deuterated drugs draw heavier backing. Nat. Rev. Drug Discov. 2016, 15, 219–221. [Google Scholar] [CrossRef]
- Lampland, N.L.; Hovey, M.; Mukherjee, D.; Sadow, A.D. Magnesium-Catalyzed Mild Reduction of Tertiary and Secondary Amides to Amines. ACS Catal. 2015, 5, 4219–4226. [Google Scholar] [CrossRef]
- Mukherjee, D.; Shirase, S.; Spaniol, T.P.; Mashima, K.; Okuda, J. Magnesium hydridotriphenylborate [Mg(thf)6][HBPh3]2: A versatile hydroboration catalyst. Chem. Commun. 2016, 52, 13155–13158. [Google Scholar] [CrossRef]
- Adkins, H.; Wojcik, B. Hydrogenation of Amides to Amines. J. Am. Chem. Soc. 1934, 56, 247. [Google Scholar] [CrossRef]
- Beamson, G.; Papworth, A.J.; Philipps, C.; Smith, A.M.; Whyman, R. Selective Hydrogenation of Amides Using bimetallic Ru/Re and Rh/Re Catalysts. J. Catal. 2011, 278, 228–238. [Google Scholar] [CrossRef]
- Wojcik, B.; Adkins, H. Catalytic Hydrogenation of Amides to Amines. J. Am. Chem. Soc. 1934, 56, 2419–2424. [Google Scholar] [CrossRef]
- Stein, M.; Breit, B. Catalytic Hydrogenation of Amides to Amines under Mild Conditions. Angew. Chem. Int. Ed. 2013, 52, 2231–2234. [Google Scholar] [CrossRef]
- Toyao, T.; Siddiki, S.M.A.H.; Morita, Y.; Kamachi, T.; Touchy, A.S.; Onodera, W.; Kon, K.; Furukawa, S.; Ariga, H.; Asakura, K.; Yoshizawa, K.; Shimizu, K.-i. Rhenium-Loaded TiO2: A Highly Versatile and Chemoselective Catalyst for the Hydrogenation of Carboxylic Acid Derivatives and the N-Methylation of Amines Using H2 and CO2. Chem. Eur. J. 2017, 23, 14848–14859. [Google Scholar] [CrossRef]
- Beamson, G.; Papworth, A.J.; Philipps, C.; Smith, A.W.; Whyman, R. Selective Hydrogenation of Amides using Ruthenium/Molybdenum Catalysts. Adv. Synth. Catal. 2010, 352, 869–883. [Google Scholar] [CrossRef]
- Coetzee, J.; Manyar, H.G.; Hardacre, C.; Cole-Hamilton, D.J. The First Continuous Flow Hydrogenation of Amides to Amines. ChemCatChem 2013, 5, 2843–2847. [Google Scholar] [CrossRef]
- Fleury-Brégeot, N.; de la Fuente, V.; Castillón, S.; Claver, C. Highlights of Transition Metal-Catalyzed Asymmetric Hydrogenation of Imines. ChemCatChem 2010, 2, 1346–1371. [Google Scholar] [CrossRef]

































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| # | Amide | Pre-cat. | T, °C / t, h | Yield, % | # | Amide | Pre-cat. | T, °C / t, h | Yield, % |
|---|---|---|---|---|---|---|---|---|---|
| 1 |  | A–F A–C, F E D | 60/5 25/5 25/6.5 25/24 | >99 >99 >99 b 79 | 7 |  | A B E | 60/5 25/17 60/24 | >99 >99 61g |
| 2 |  | A B E E | 60/5 25/17 60/5 60/24 | >99 >99 82 94 | 8 |  | A A B E | 60/5 65/16 25/24 60/24 | >99 96 h 90 i 43 j |
| 3 |  | A B D E E | 60/5 25/3 25/24 25/32 60/5 | >99 >99 (95) 80 c 68 >99 | 9 |  | A A B | 60/5 65/16 100/24 | 28 44 k 15 |
| 4 |  | A B E E | 60/16 60/24 60/24 60/48 | 91 d >99 e 28 37 | 10 |  | B B B | 75/24 75/48 75/72 | 45 73 >99 |
| 5 |  | A B B E | 60/5 25/17 65/5 60/24 | >99 >99 (93) >99 72 | 11 |  | R=H: B R=Me: B R=Me: B | 100/24 75/24 75/48 | 83 l 89 m >99 m |
| 6 |  | A B B E | 60/5 25/17 60/5 60/24 | >99 87 85 87 f | 12 |  | B | 60/60 | >99 n |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalimon, A.Y.; Gudun, K.A.; Hayrapetyan, D. Base Metal Catalysts for Deoxygenative Reduction of Amides to Amines. Catalysts 2019, 9, 490. https://doi.org/10.3390/catal9060490
Khalimon AY, Gudun KA, Hayrapetyan D. Base Metal Catalysts for Deoxygenative Reduction of Amides to Amines. Catalysts. 2019; 9(6):490. https://doi.org/10.3390/catal9060490
Chicago/Turabian StyleKhalimon, Andrey Y., Kristina A. Gudun, and Davit Hayrapetyan. 2019. "Base Metal Catalysts for Deoxygenative Reduction of Amides to Amines" Catalysts 9, no. 6: 490. https://doi.org/10.3390/catal9060490
APA StyleKhalimon, A. Y., Gudun, K. A., & Hayrapetyan, D. (2019). Base Metal Catalysts for Deoxygenative Reduction of Amides to Amines. Catalysts, 9(6), 490. https://doi.org/10.3390/catal9060490