Combining Planar Laser-Induced Fluorescence with Stagnation Point Flows for Small Single-Crystal Model Catalysts: CO Oxidation on a Pd(100)

 , ,
, , 
Abstract
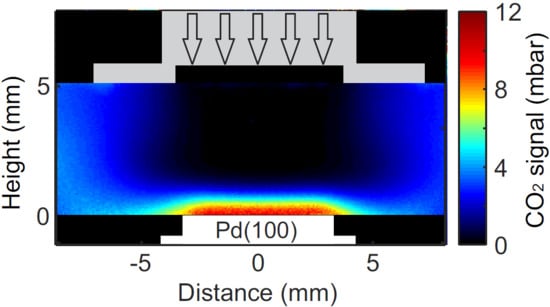
{kind=link}
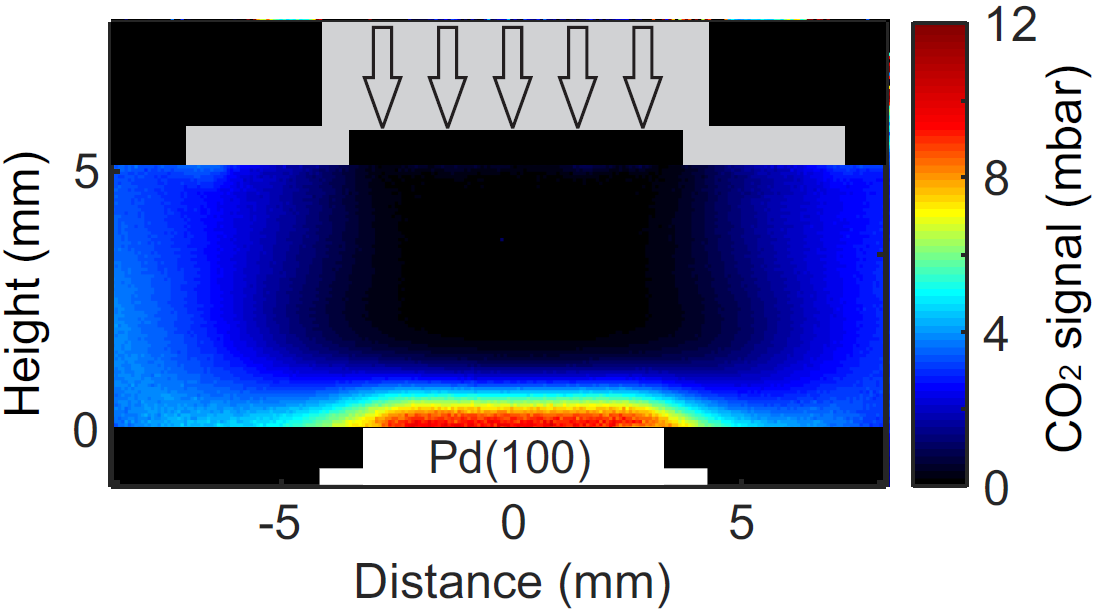
{kind=link}
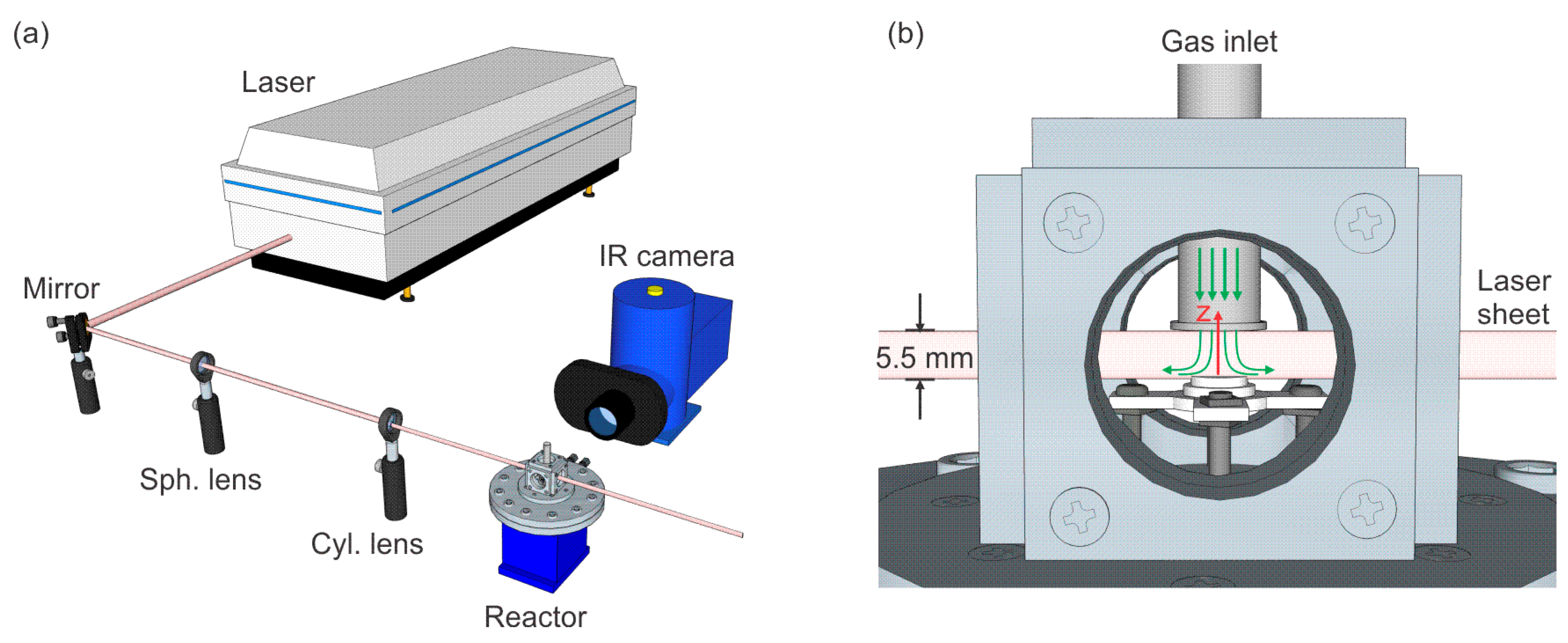
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Study
2.1. Reactor Chamber
2.2. PLIF
2.3. CO2 Visualization
3. Modeling
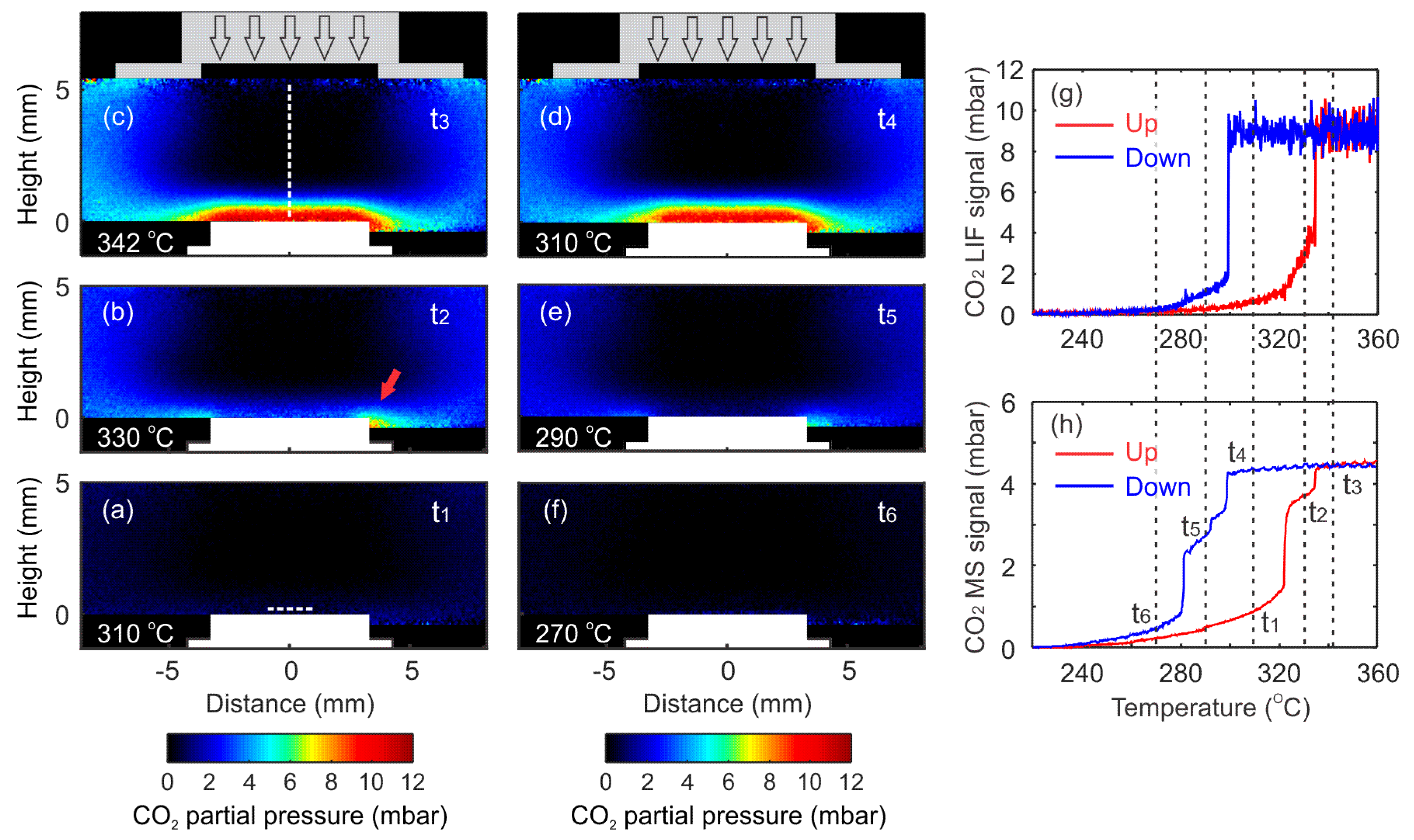
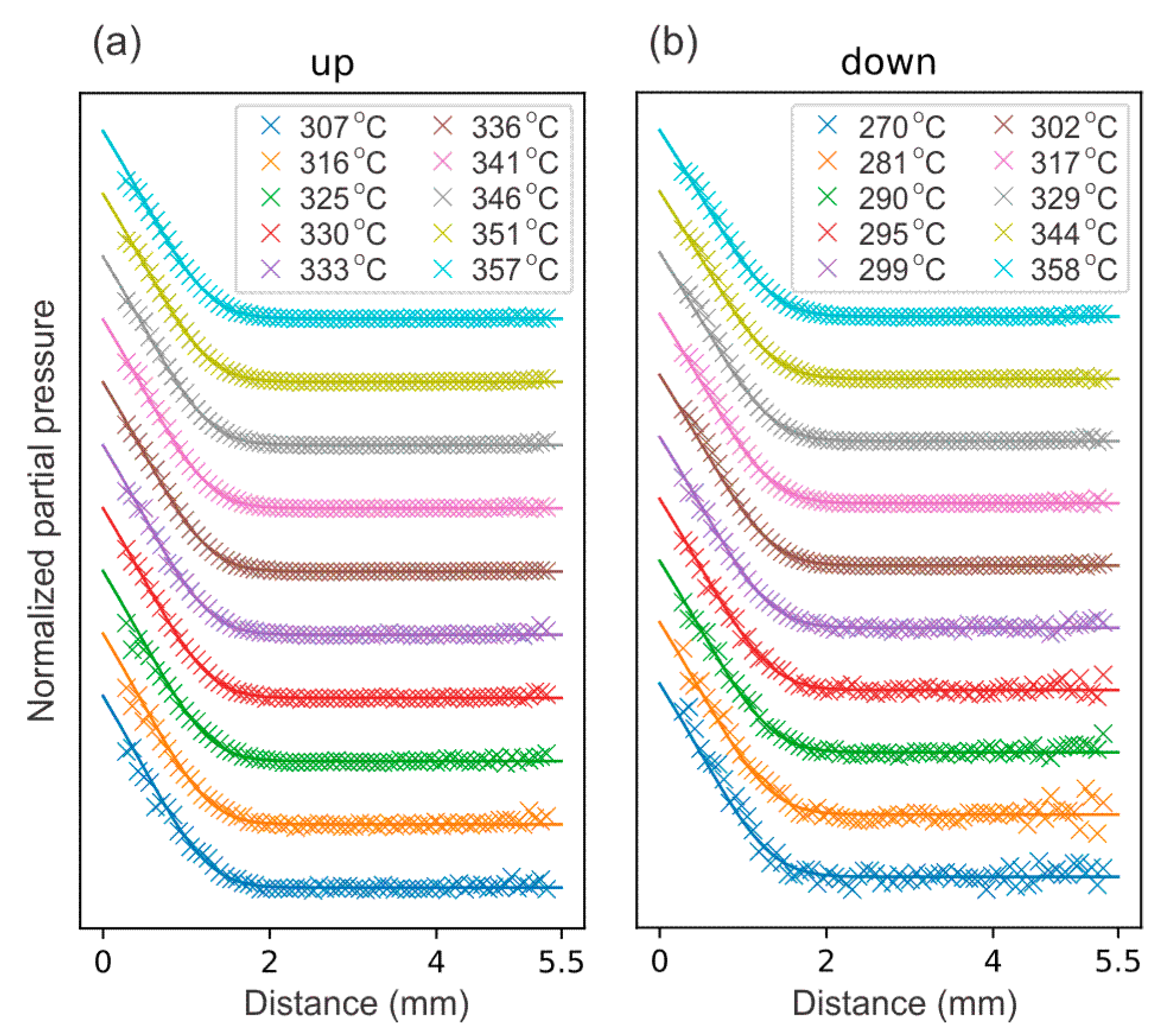
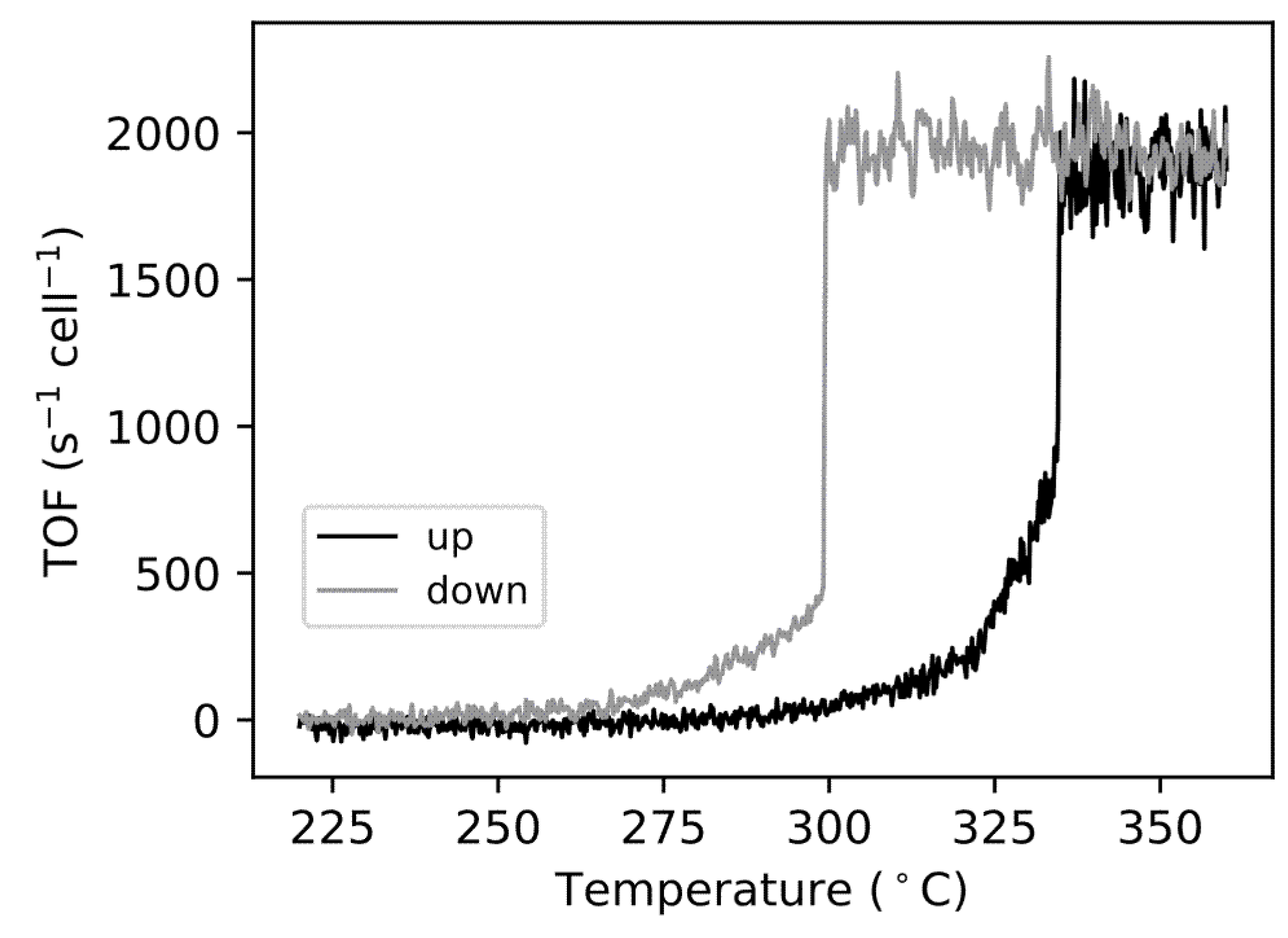
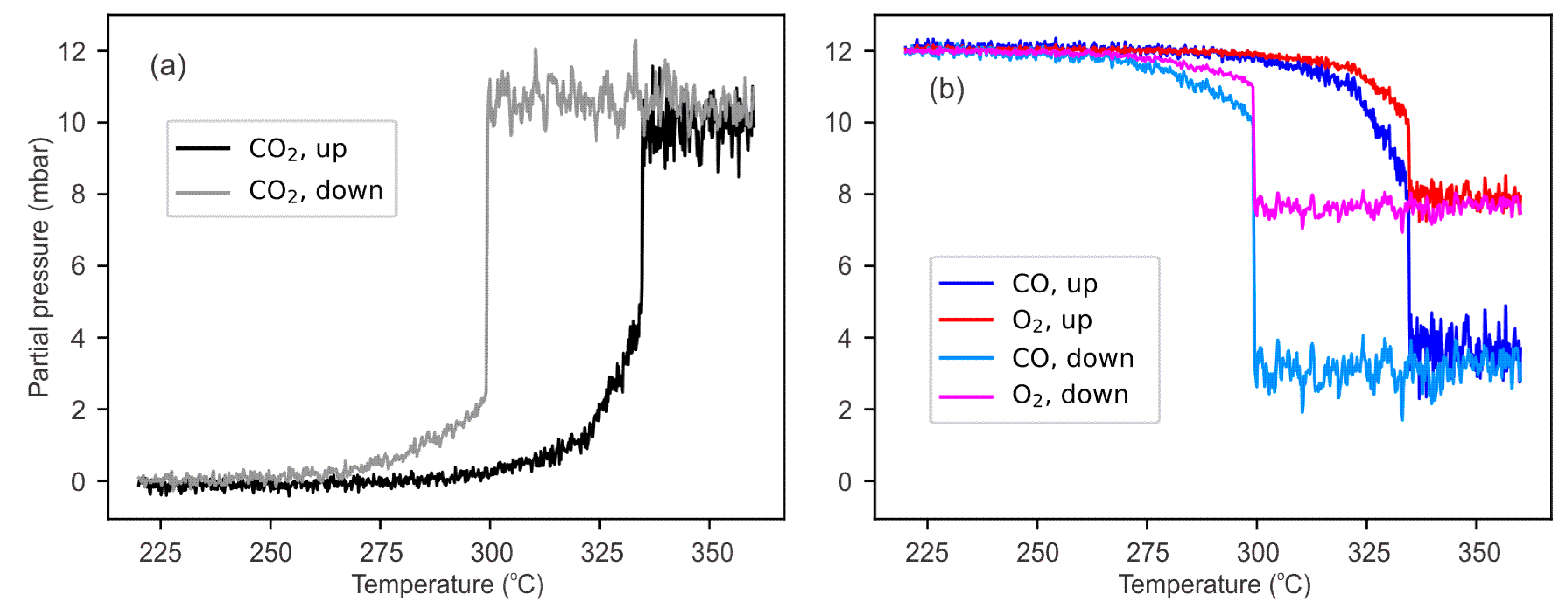
4. Results and Discussion
5. Summary and Outlook
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rupprechter, G.; Weilach, C. Mind the gap! Spectroscopy of catalytically active phases. Nano Today 2007, 2, 20–29. [Google Scholar] [CrossRef]
- Frenken, J.; Groot, I. (Eds.) Operando Research in Heterogeneous Catalysis; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Blomberg, S.; Hoffmann, M.J.; Gustafson, J.; Martin, N.M.; Fernandes, V.R.; Borg, A.; Liu, Z.; Chang, R.; Matera, S.; Reuter, K.; et al. In-situ X-ray photoelectron spectroscopy of model catalysts: At the edge of the gap. Phys. Rev. Lett. 2013, 110, 117601. [Google Scholar] [CrossRef]
- Karslıoğlu, O.; Bluhm, H. Ambient-pressure X-ray photoelectron spectroscopy (APXPS). In Operando Research in Heterogeneous Catalysis; Frenken, J., Groot, I., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 31–57. [Google Scholar]
- Toyoshima, R.; Yoshida, M.; Monya, Y.; Suzuki, K.; Mun, B.S.; Amemiya, K.; Mase, K.; Kondoh, H. Active surface oxygen for catalytic CO oxidation on Pd(100) proceeding under near ambient pressure conditions. J. Phys. Chem. Lett. 2012, 3, 3182–3187. [Google Scholar] [CrossRef] [PubMed]
- Frenken, J.; Groot, I. Live observations of catalysts using high-pressure scanning probe microscopy. In Operando Research in Heterogeneous Catalysis; Frenken, J., Groot, I., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–30. [Google Scholar]
- Hendriksen, B.L.M.; Bobaru, S.C.; Frenken, J.W.M. Looking at heterogeneous catalysis at atmospheric pressure using tunnel vision. Top. Catal. 2005, 36, 43–54. [Google Scholar] [CrossRef]
- Gao, F.; McClure, S.M.; Cai, Y.; Gath, K.K.; Wang, Y.; Chen, M.S.; Guo, Q.L.; Goodman, D.W. CO oxidation trends on Pt-group metals from ultrahigh vacuum to near atmospheric pressures: A combined in situ PM-IRAS and reaction kinetics study. Surf. Sci. 2009, 603, 65–70. [Google Scholar] [CrossRef]
- Ryczkowski, J. IR spectroscopy in catalysis. Catal. Today 2001, 68, 263–381. [Google Scholar] [CrossRef]
- Gustafson, J.; Shipilin, M.; Zhang, C.; Stierle, A.; Hejral, U.; Ruett, U.; Gutowski, O.; Carlsson, P.A.; Skoglundh, M.; Lundgren, E. High-energy surface X-ray diffraction for fast surface structure determination. Science 2014, 343, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Stierle, A.; Gustafson, J.; Lundgren, E. Surface-sensitive X-ray diffraction across the pressure gap. In Operando Research in Heterogeneous Catalysis; Frenken, J., Groot, I., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 59–87. [Google Scholar]
- Lundgren, E.; Zhang, C.; Merte, L.R.; Shipilin, M.; Blomberg, S.; Hejral, U.; Zhou, J.; Zetterberg, J.; Gustafson, J. Novel in situ techniques for studies of model catalysts. Acc. Chem. Res. 2017, 50, 2326–2333. [Google Scholar] [CrossRef]
- Zhou, J.; Blomberg, S.; Gustafson, J.; Lundgren, E.; Zetterberg, J. Visualization of gas distribution in a model AP-XPS reactor by PLIF: CO oxidation over a Pd(100) catalyst. Catalysts 2017, 7, 29. [Google Scholar] [CrossRef]
- Zellner, A.; Suntz, R.; Deutschmann, O. Two-dimensional spatial resolution of concentration profiles in catalytic reactors by planar laser-induced fluorescence: NO reduction over diesel oxidation catalysts. Angew. Chem. Int. Ed. 2015, 54, 2653–2655. [Google Scholar] [CrossRef] [PubMed]
- Matera, S.; Reuter, K. First-principles approach to heat and mass transfer effects in model catalyst studies. Catal. Lett. 2009, 133, 156–159. [Google Scholar] [CrossRef]
- Matera, S.; Reuter, K. When atomic-scale resolution is not enough: Spatial effects on in situ model catalyst studies. J. Catal. 2012, 295, 261–268. [Google Scholar] [CrossRef]
- Zhou, J.; Blomberg, S.; Gustafson, J.; Lundgren, E.; Zetterberg, J. Simultaneous imaging of gas phase over and surface reflectance of a Pd(100) single crystal during CO oxidation. J. Phys. Chem. C 2017, 121, 23511–23519. [Google Scholar] [CrossRef]
- Onderwaater, W.G.; Taranovskyy, A.; van Baarle, G.C.; Frenken, J.W.M.; Groot, I.M.N. In situ optical reflectance difference observations of CO oxidation over Pd(100). J. Phys. Chem. C 2017, 121, 11407–11415. [Google Scholar] [CrossRef] [PubMed]
- Matera, S.; Reuter, K. Transport limitations and bistability for in situ CO oxidation at RuO2(110): First-principles based multiscale modeling. Phys. Rev. B 2010, 82. [Google Scholar] [CrossRef]
- Roos, M.; Kielbassa, S.; Schirling, C.; Häring, T.; Bansmann, J.; Behm, R.J. Scanning mass spectrometer for quantitative reaction studies on catalytically active microstructures. Rev. Sci. Instrum. 2007, 78. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.C.; Delgass, W.N.; Baertsch, C.D. Spatially resolved in situ FTIR analysis of CO adsorption and reaction on Pt/SiO2 in a silicon microreactor. Appl. Catal. B Environ. 2009, 93, 66–74. [Google Scholar] [CrossRef]
- Zetterberg, J.; Blomberg, S.; Gustafson, J.; Evertsson, J.; Zhou, J.; Adams, E.C.; Carlsson, P.A.; Aldén, M.; Lundgren, E. Spatially and temporally resolved gas distributions around heterogeneous catalysts using infrared planar laser-induced fluorescence. Nat. Commun. 2015, 6, 7076. [Google Scholar] [CrossRef]
- Blomberg, S.; Zhou, J.; Gustafson, J.; Zetterberg, J.; Lundgren, E. 2D and 3D imaging of the gas phase close to an operating model catalyst by planar laser induced fluorescence. J. Phys. Condens. Matter 2016, 28. [Google Scholar] [CrossRef]
- Mantzaras, J. Progress in non-intrusive laser-based measurements of gas-phase thermoscalars and supporting modeling near catalytic interfaces. Prog. Energy Combust. Sci. 2019, 70, 169–211. [Google Scholar] [CrossRef]
- Matera, S.; Maestri, M.; Cuoci, A.; Reuter, K. Predictive-quality surface reaction chemistry in real reactor models: Integrating first-principles kinetic monte carlo simulations into computational fluid dynamics. ACS Catal. 2014, 4, 4081–4092. [Google Scholar] [CrossRef]
- Matera, S.; Blomberg, S.; Hoffmann, M.J.; Zetterberg, J.; Gustafson, J.; Lundgren, E.; Reuter, K. Evidence for the active phase of heterogeneous catalysts through in situ reaction product imaging and multiscale modeling. ACS Catal. 2015, 5, 4514–4518. [Google Scholar] [CrossRef]
- Kee, R.J.; Coltrin, M.E.; Glarborg, P.; Zhu, H. Chemically Reacting Flow: Theory, Modeling, and Simulation; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Zanier, F.; Michelon, N.; Canu, P. Design and characterization of a stagnation flow reactor for heterogeneous microkinetic studies. Chem. Eng. J. 2017, 315, 67–82. [Google Scholar] [CrossRef]
- Zetterberg, J.; Blomberg, S.; Zhou, J.; Gustafson, J.; Lundgren, E. Planar laser induced fluorescence applied to catalysis. In Operando Research in Heterogeneous Catalysis; Springer International Publishing: Cham, Switzerland, 2017; pp. 131–149. [Google Scholar]
- Kohsehoinghaus, K. Laser techniques for the quantitative detection of reactive intermediates in combustion systems. Prog. Energy Combust. Sci. 1994, 20, 203–279. [Google Scholar] [CrossRef]
- Aldén, M.; Bood, J.; Li, Z.; Richter, M. Visualization and understanding of combustion processes using spatially and temporally resolved laser diagnostic techniques. Proc. Combust. Inst. 2011, 33, 69–97. [Google Scholar] [CrossRef]
- Hanson, R.K. Planar laser-induced fluorescence imaging. J. Quant. Spectrosc. Radiat. Transf. 1988, 40, 343–362. [Google Scholar] [CrossRef]
- Zhou, J.; Pfaff, S.; Lundgren, E.; Zetterberg, J. A convenient setup for laser-induced fluorescence imaging of both CO and CO2 during catalytic CO oxidation. Appl. Phys. B 2017, 123, 87. [Google Scholar] [CrossRef]
- Gudmundson, F.; Persson, J.L.; Försth, M.; Behrendt, F.; Kasemo, B.; Rosen, A. OH gas phase chemistry outside a Pt catalyst. J. Catal. 1998, 179, 420–430. [Google Scholar] [CrossRef]
- Fernandes, V.R.; Van den Bossche, M.; Knudsen, J.; Farstad, M.H.; Gustafson, J.; Venvik, H.J.; Grönbeck, H.; Borg, A. Reversed hysteresis during CO oxidation over Pd75Ag25(100). ACS Catal. 2016, 6, 4154–4161. [Google Scholar] [CrossRef]
- Rosell, J.; Bai, X.S.; Sjoholm, J.; Zhou, B.; Li, Z.; Wang, Z.; Pettersson, P.; Li, Z.; Richter, M.; Aldén, M. Multi-species PLIF study of the structures of turbulent premixed methane/air jet flames in the flamelet and thin-reaction zones regimes. Combust. Flame 2017, 182, 324–338. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Matera, S.; Pfaff, S.; Blomberg, S.; Lundgren, E.; Zetterberg, J. Combining Planar Laser-Induced Fluorescence with Stagnation Point Flows for Small Single-Crystal Model Catalysts: CO Oxidation on a Pd(100). Catalysts 2019, 9, 484. https://doi.org/10.3390/catal9050484
Zhou J, Matera S, Pfaff S, Blomberg S, Lundgren E, Zetterberg J. Combining Planar Laser-Induced Fluorescence with Stagnation Point Flows for Small Single-Crystal Model Catalysts: CO Oxidation on a Pd(100). Catalysts. 2019; 9(5):484. https://doi.org/10.3390/catal9050484
Chicago/Turabian StyleZhou, Jianfeng, Sebastian Matera, Sebastian Pfaff, Sara Blomberg, Edvin Lundgren, and Johan Zetterberg. 2019. "Combining Planar Laser-Induced Fluorescence with Stagnation Point Flows for Small Single-Crystal Model Catalysts: CO Oxidation on a Pd(100)" Catalysts 9, no. 5: 484. https://doi.org/10.3390/catal9050484
APA StyleZhou, J., Matera, S., Pfaff, S., Blomberg, S., Lundgren, E., & Zetterberg, J. (2019). Combining Planar Laser-Induced Fluorescence with Stagnation Point Flows for Small Single-Crystal Model Catalysts: CO Oxidation on a Pd(100). Catalysts, 9(5), 484. https://doi.org/10.3390/catal9050484