Mechanistic Insights into Photodegradation of Organic Dyes Using Heterostructure Photocatalysts





Abstract
1. Introduction
2. Classification of Organic Dyes
3. Kinetics Study for Photodegradation Reactions
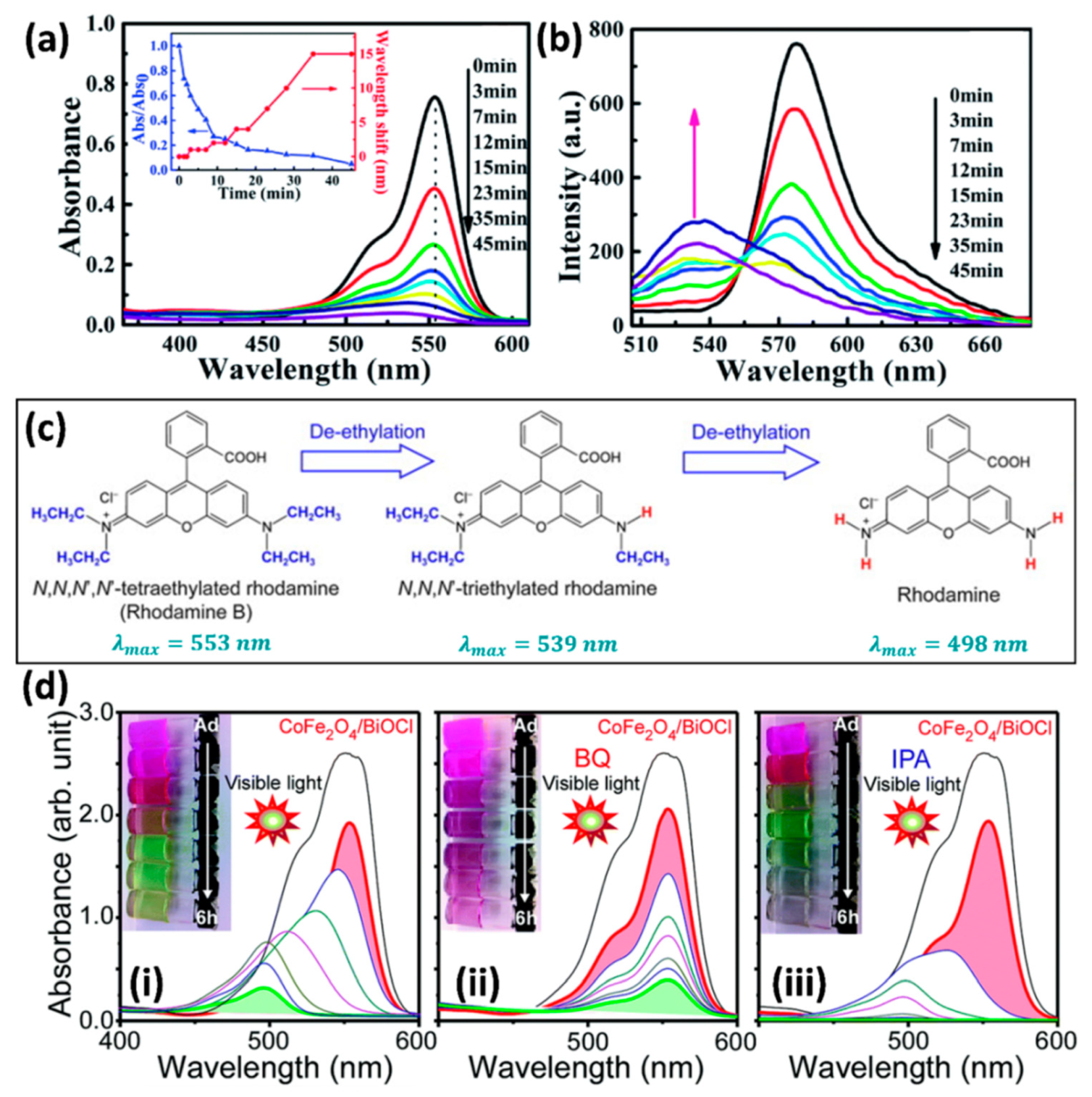
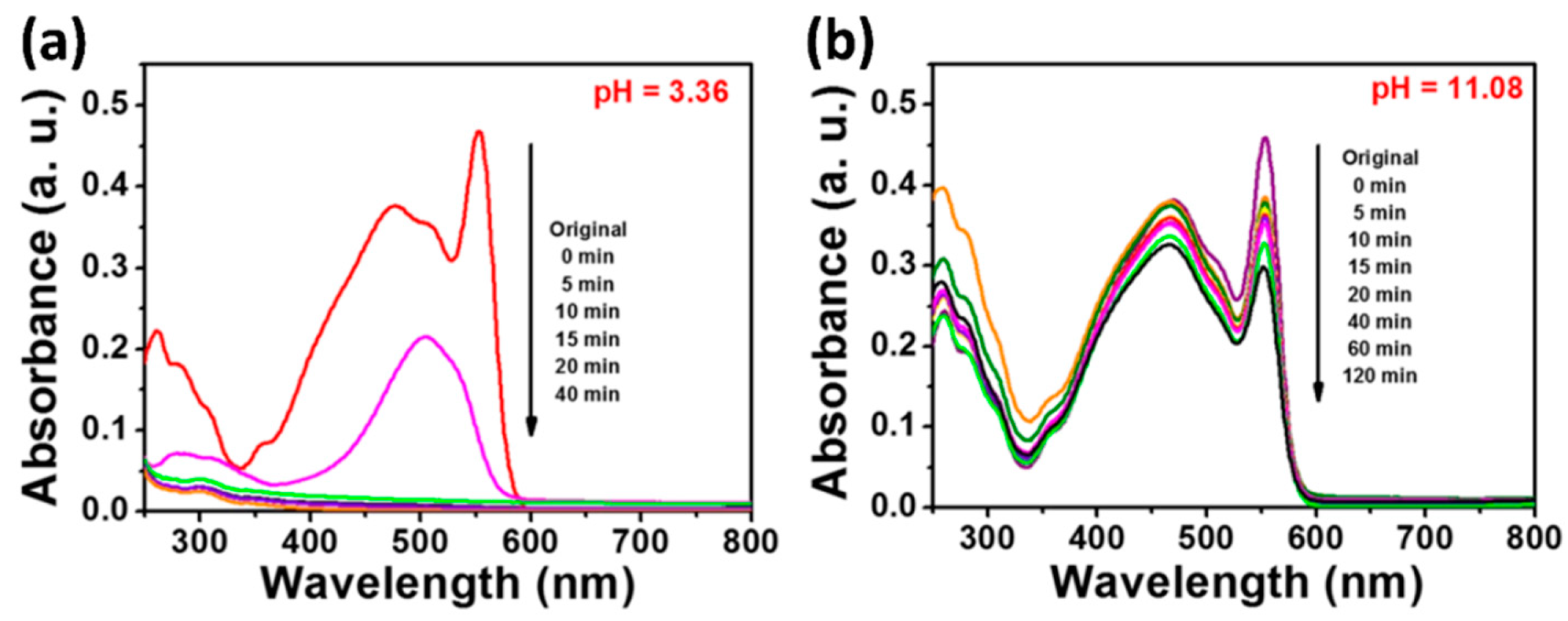
3.1. Absorption Peak Shift of Dye Molecules
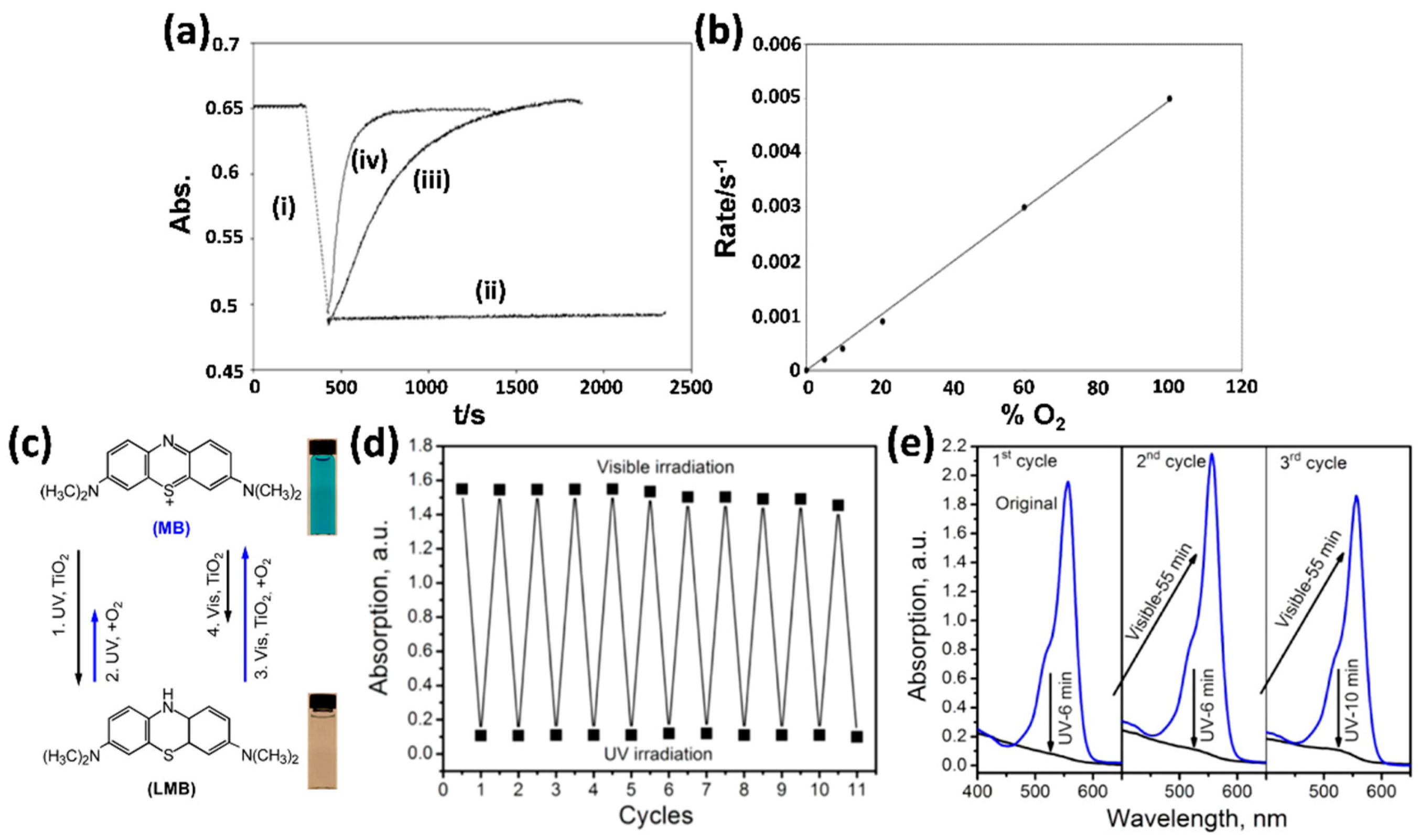
3.2. Photobleaching of Dye Molecules
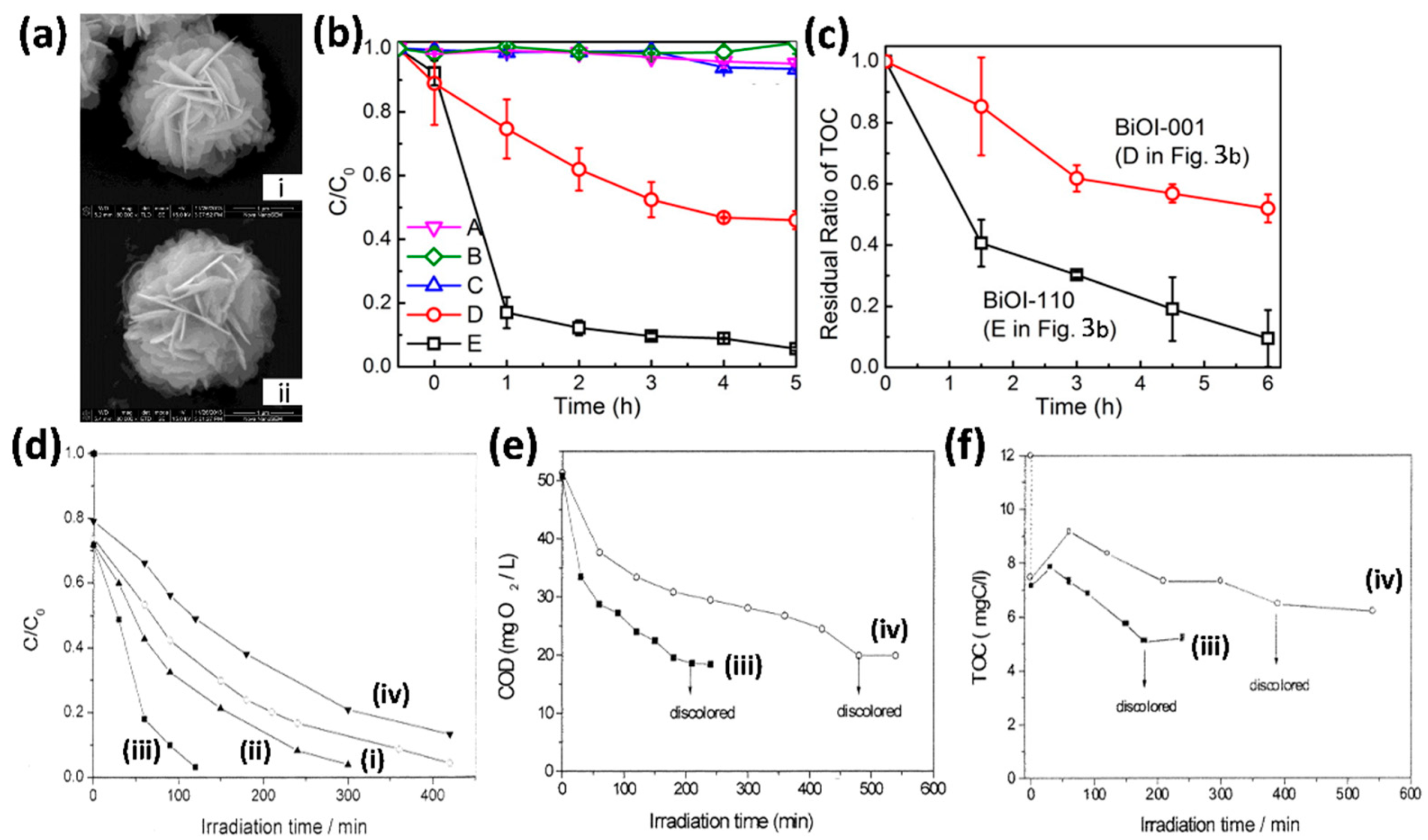
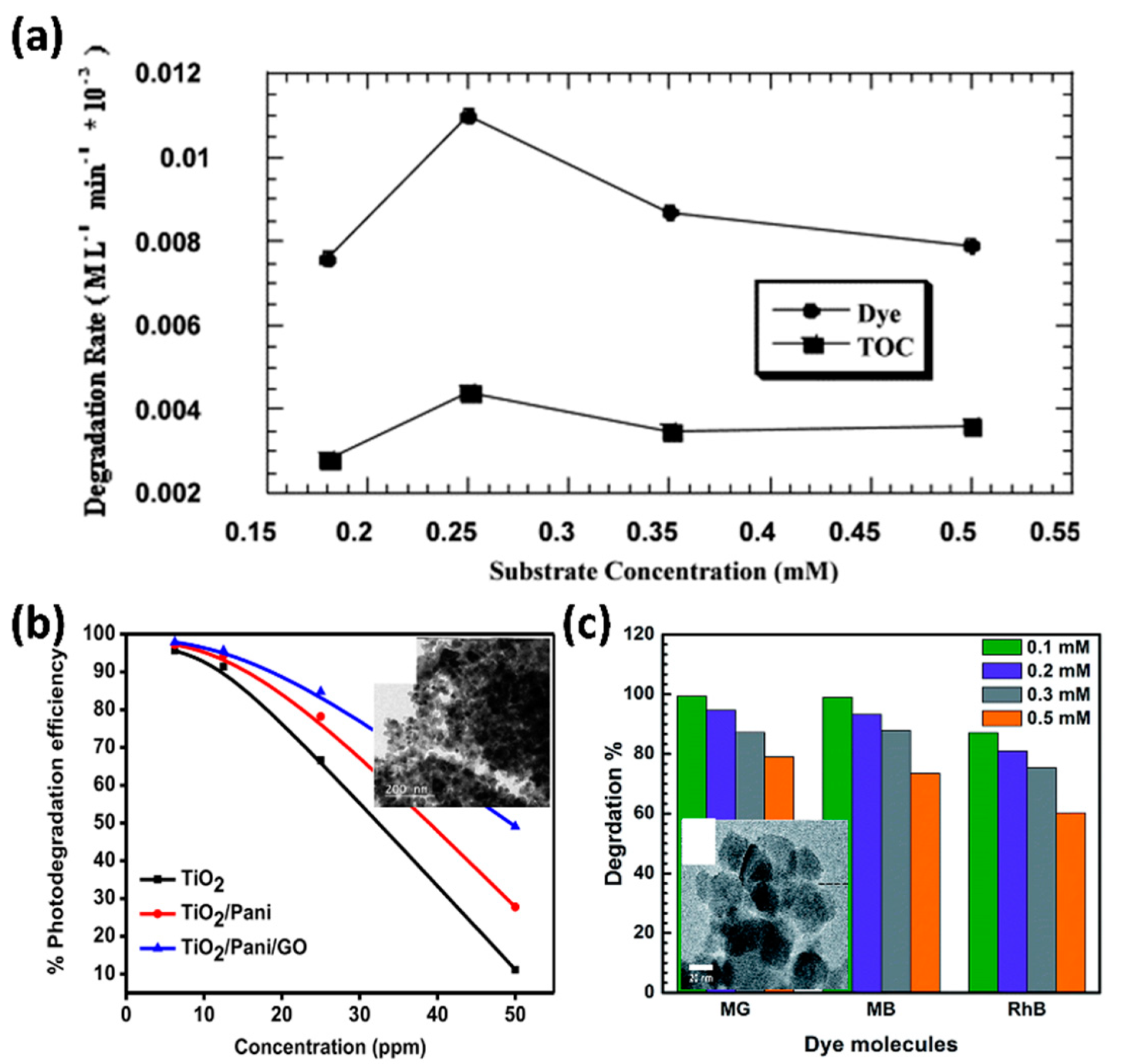
3.3. Chemical Oxygen Demand (COD) and Total Organic Carbon (TOC) Analysis
3.4. Pseudo Kinetics
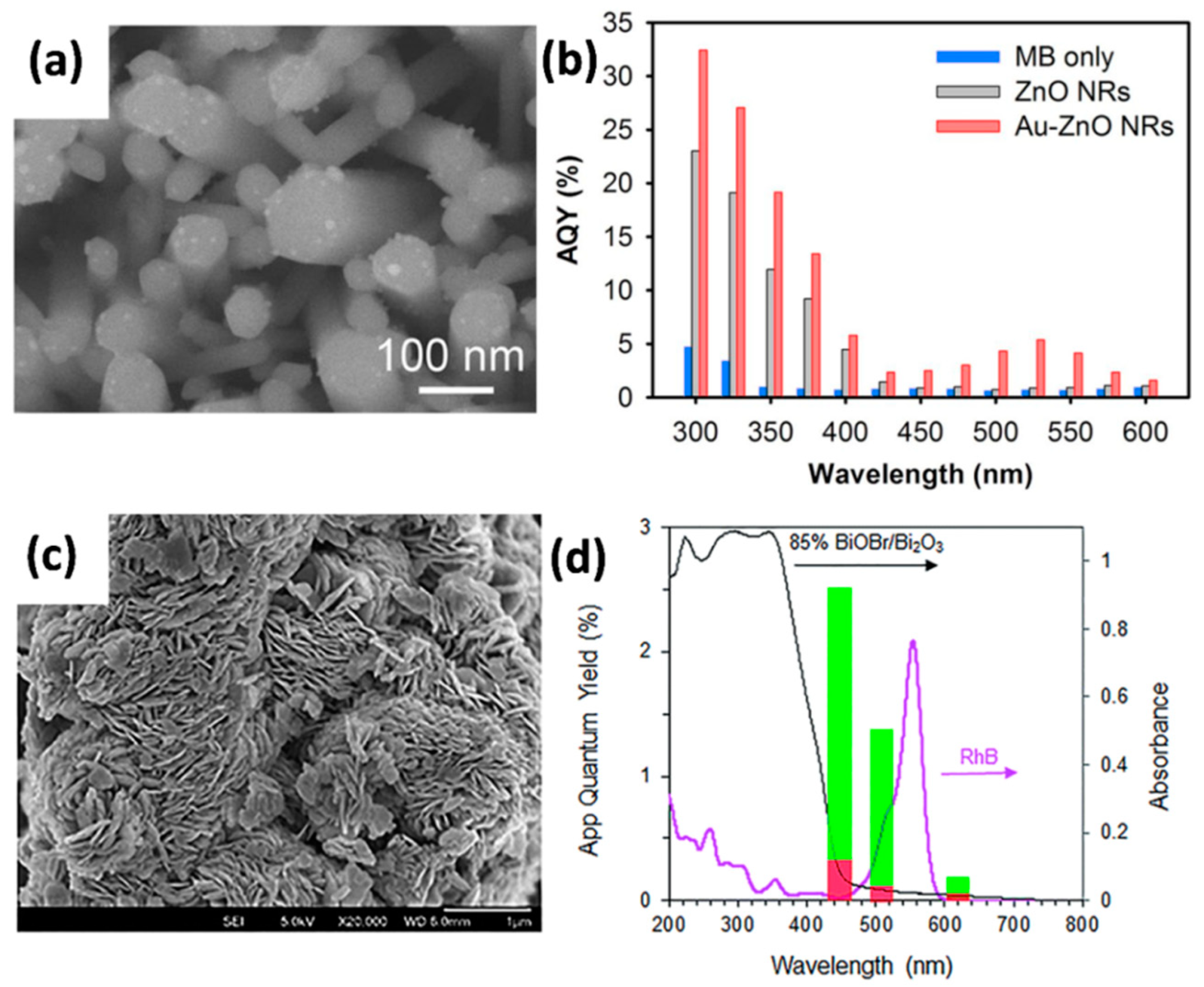
3.5. Quantum Yield of Photodegradation
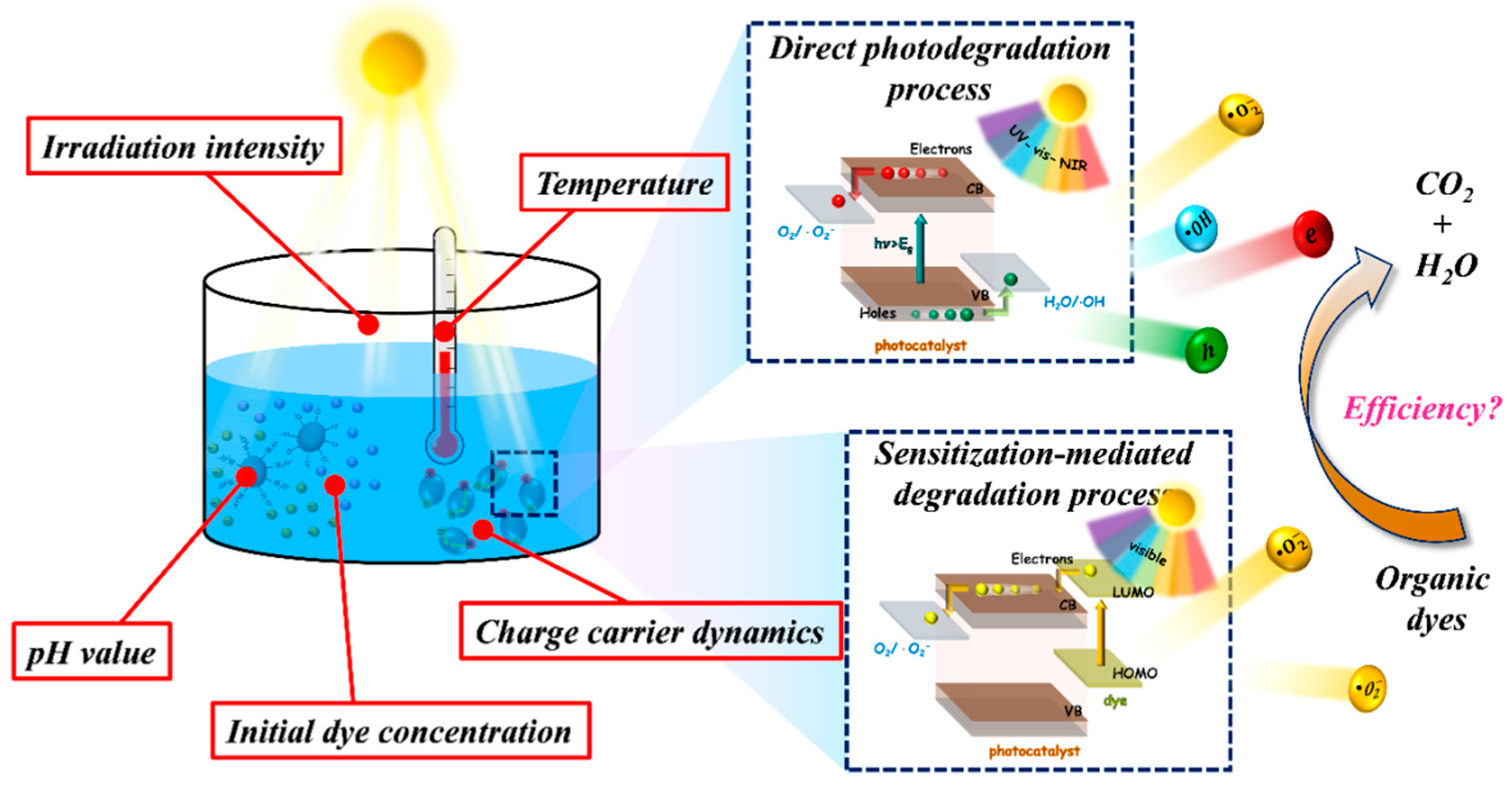
4. Factors Influencing the Photodegradation Reaction
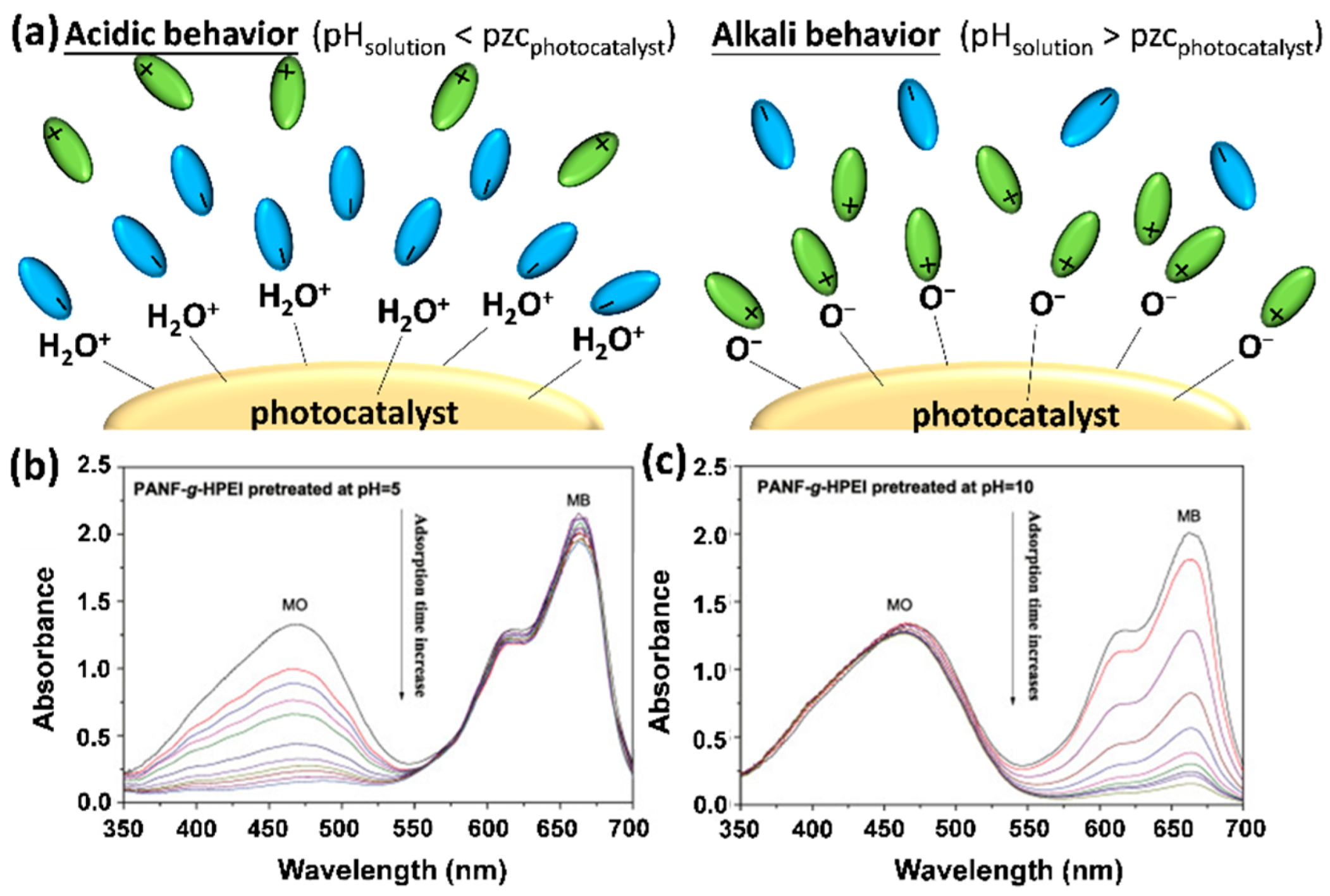
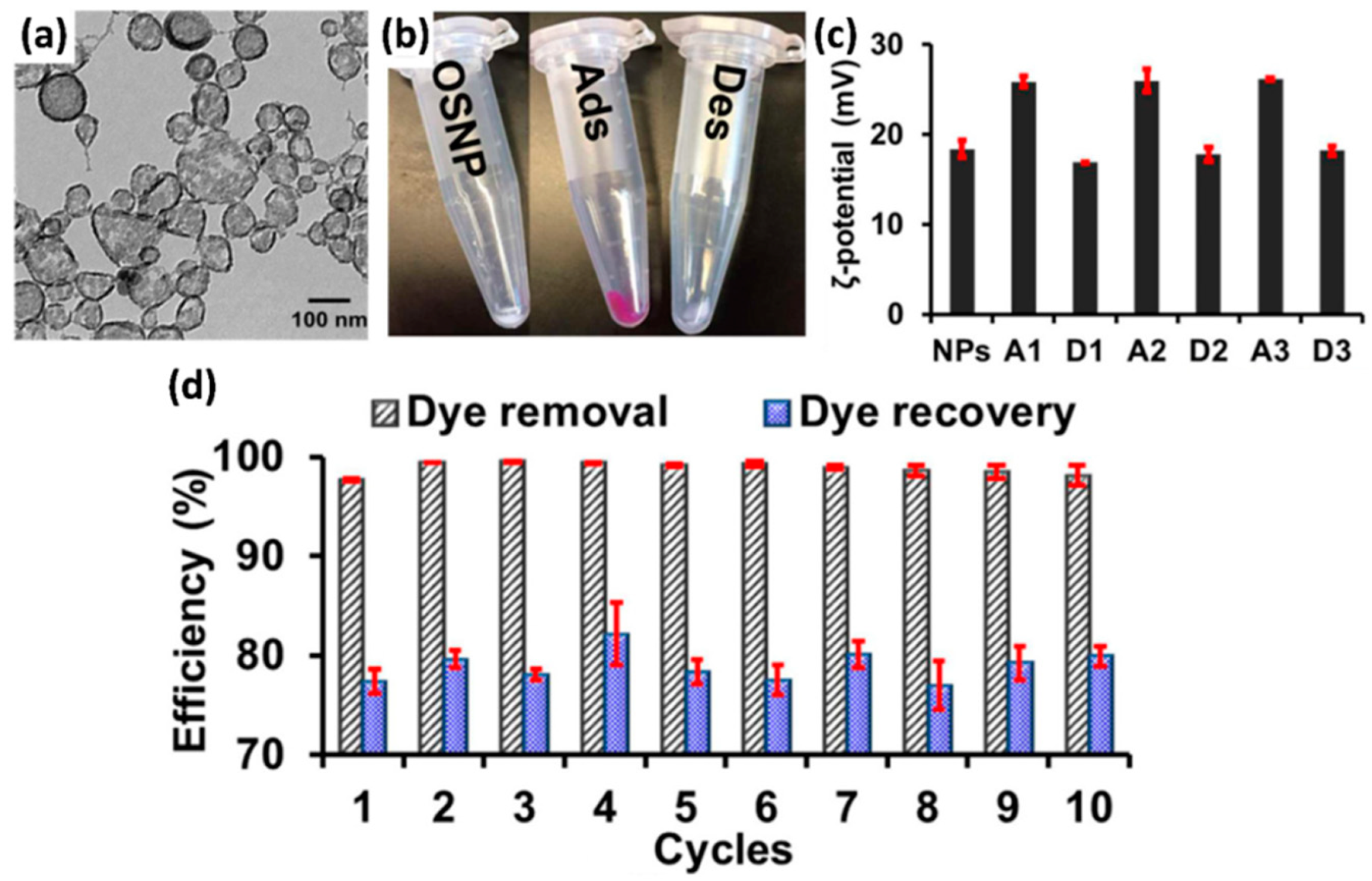
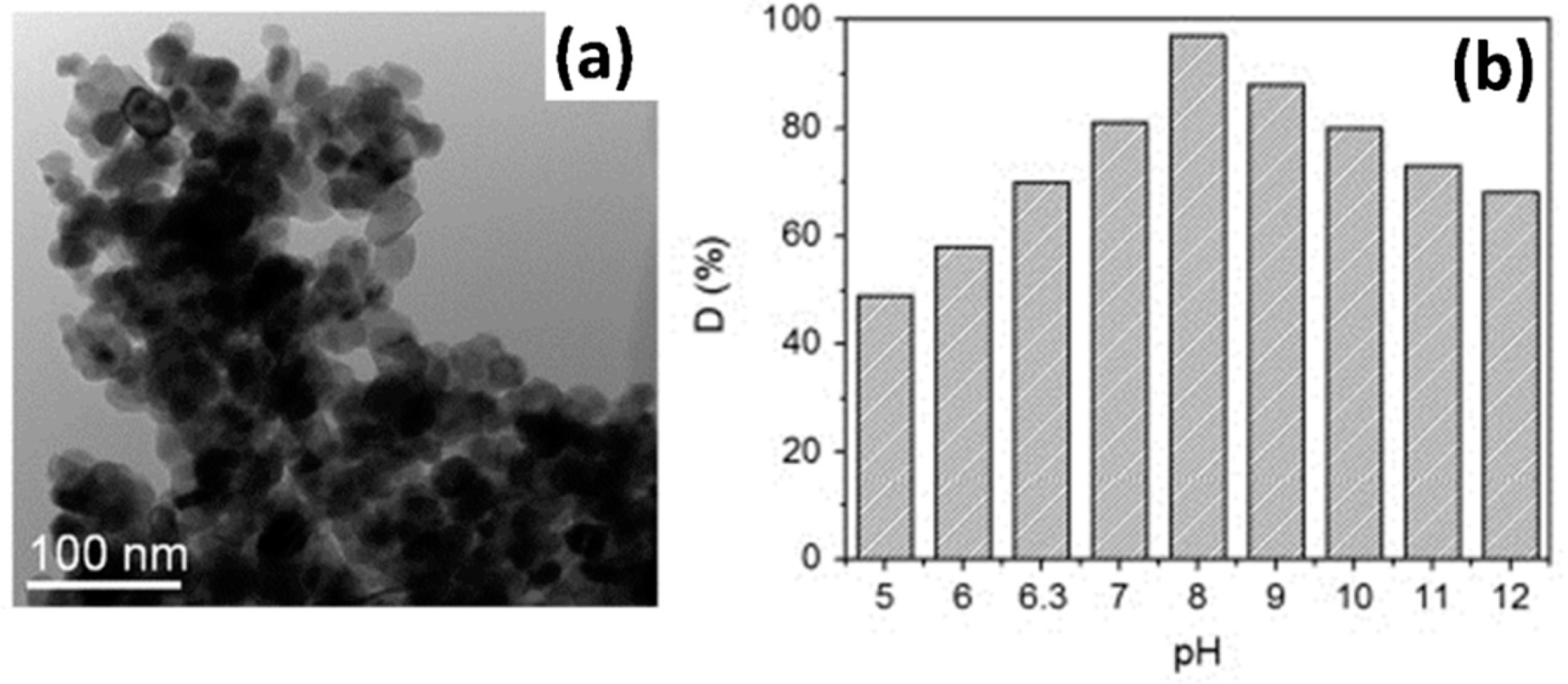
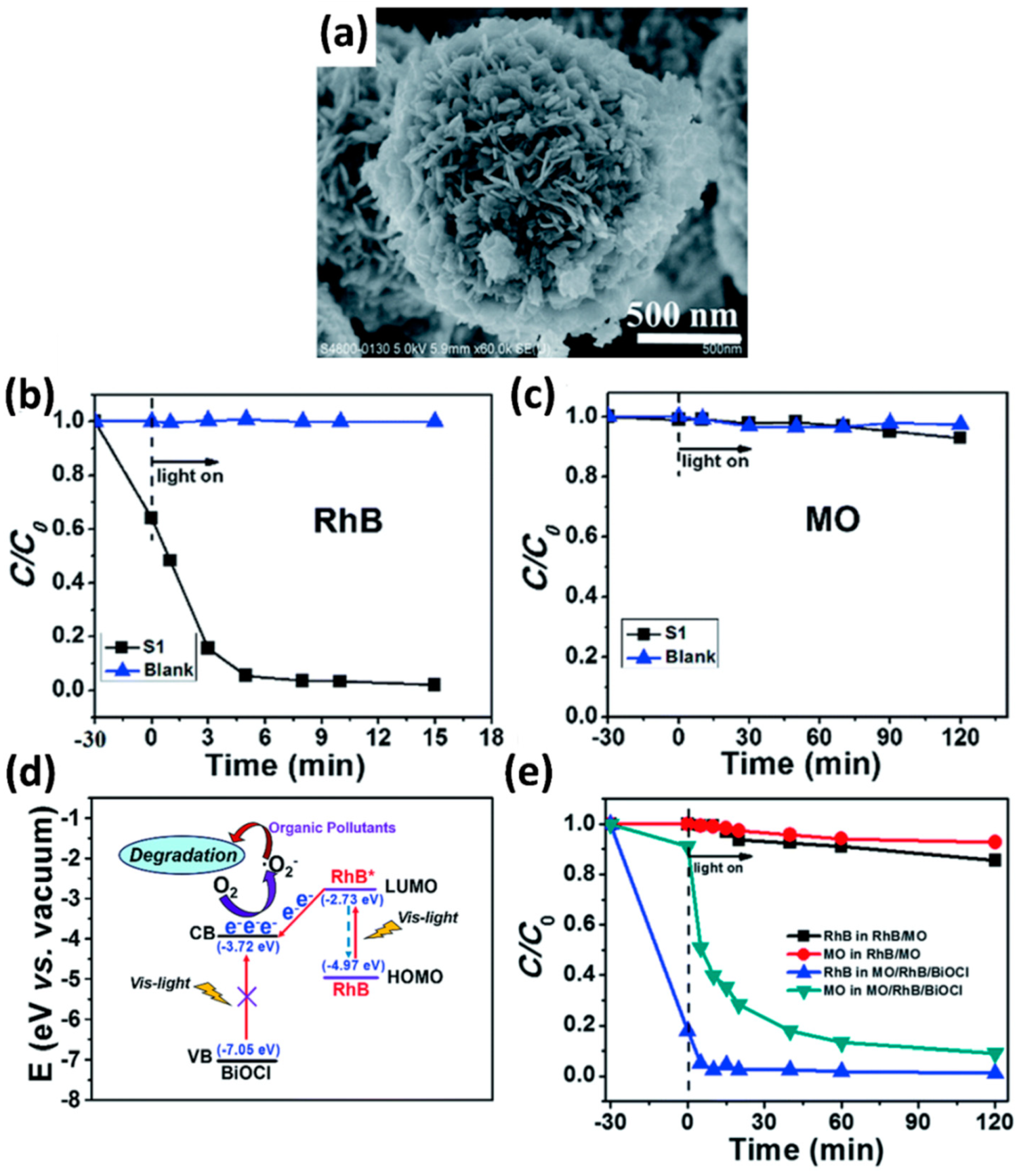
4.1. Interaction between Dye Molecules and Photocatalysts
4.2. Operational Parameters
4.2.1. Initial Dye Concentration
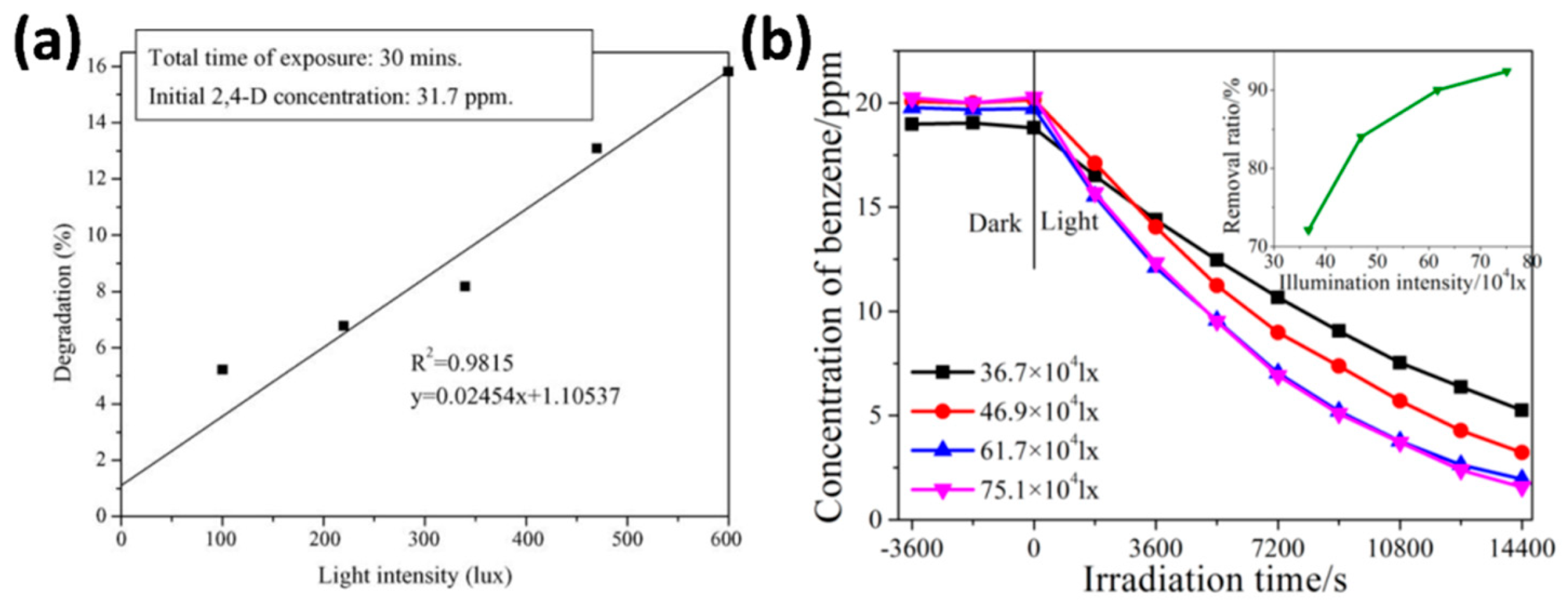
4.2.2. Light Intensity
4.2.3. Reaction Temperature
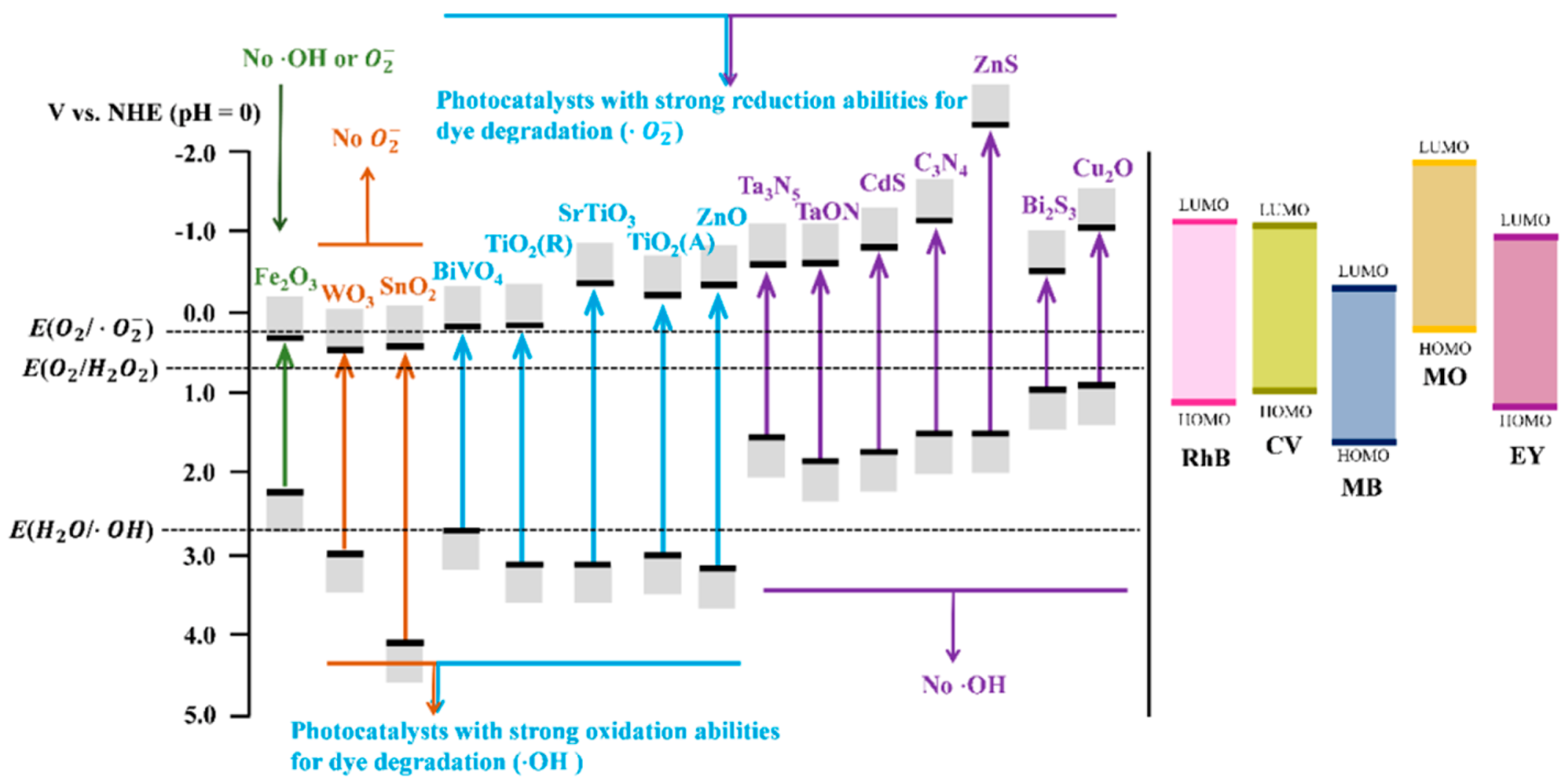
4.3. Intrinsic Properties of Photocatalysts
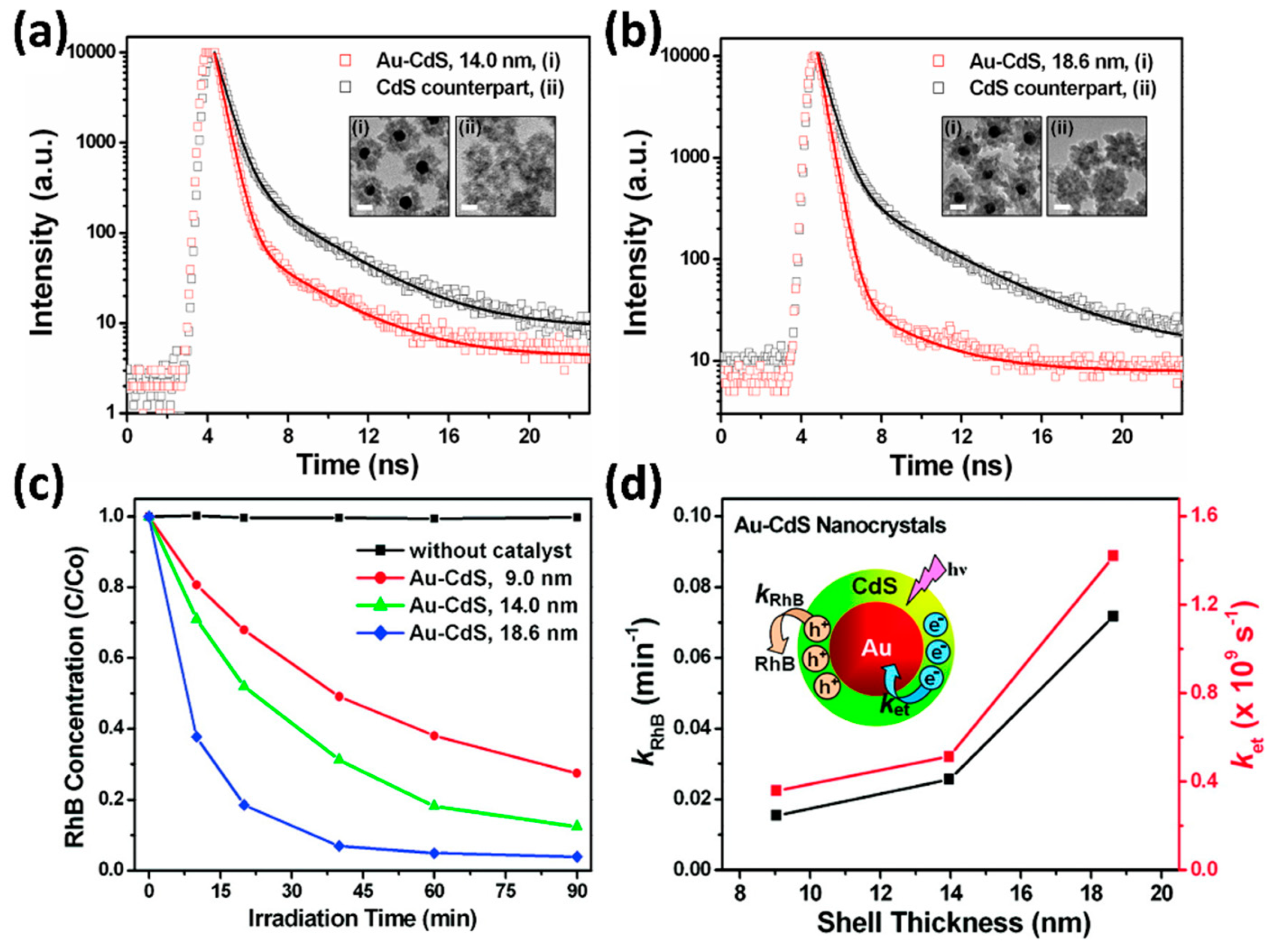
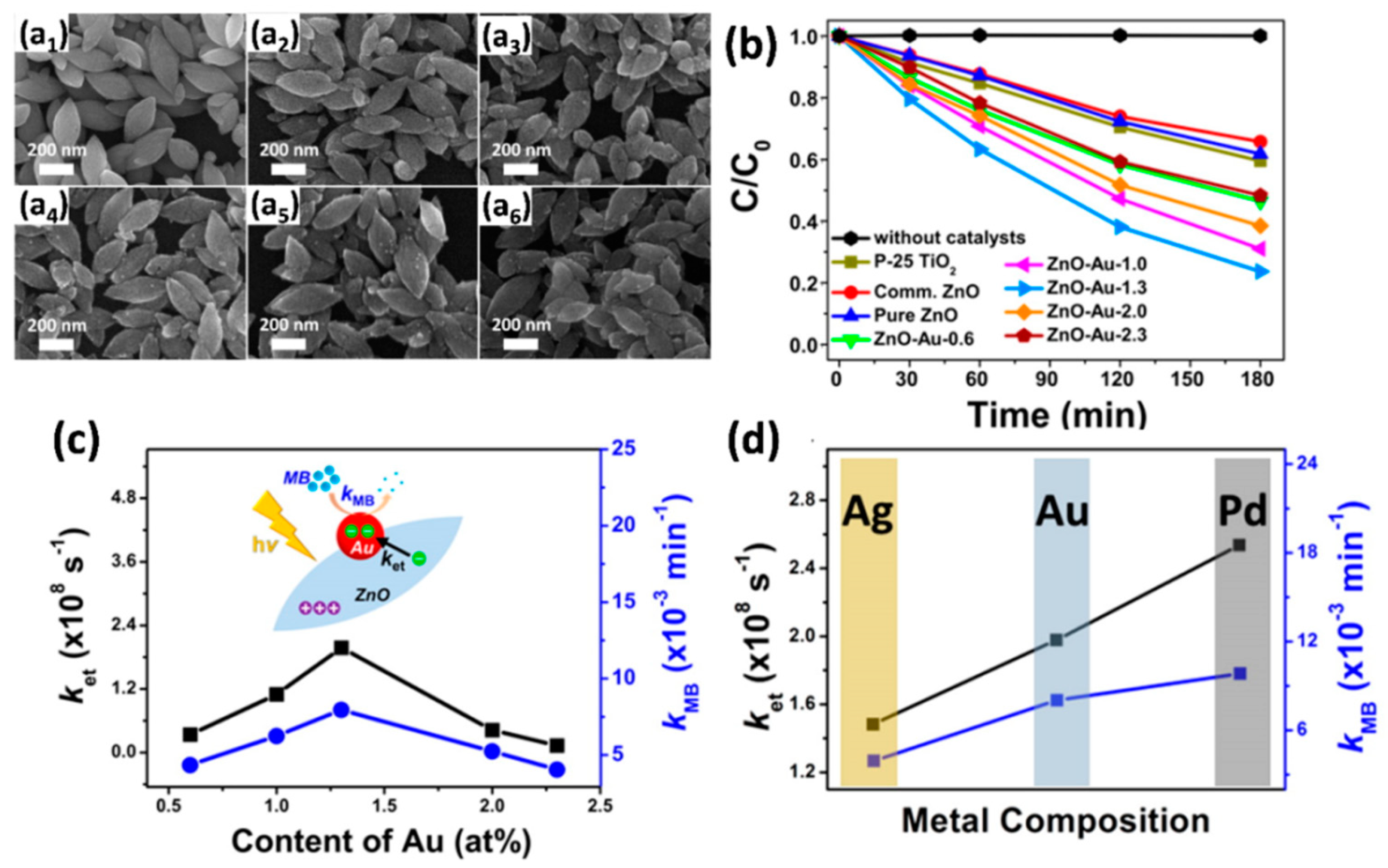
4.3.1. Modification with Metals
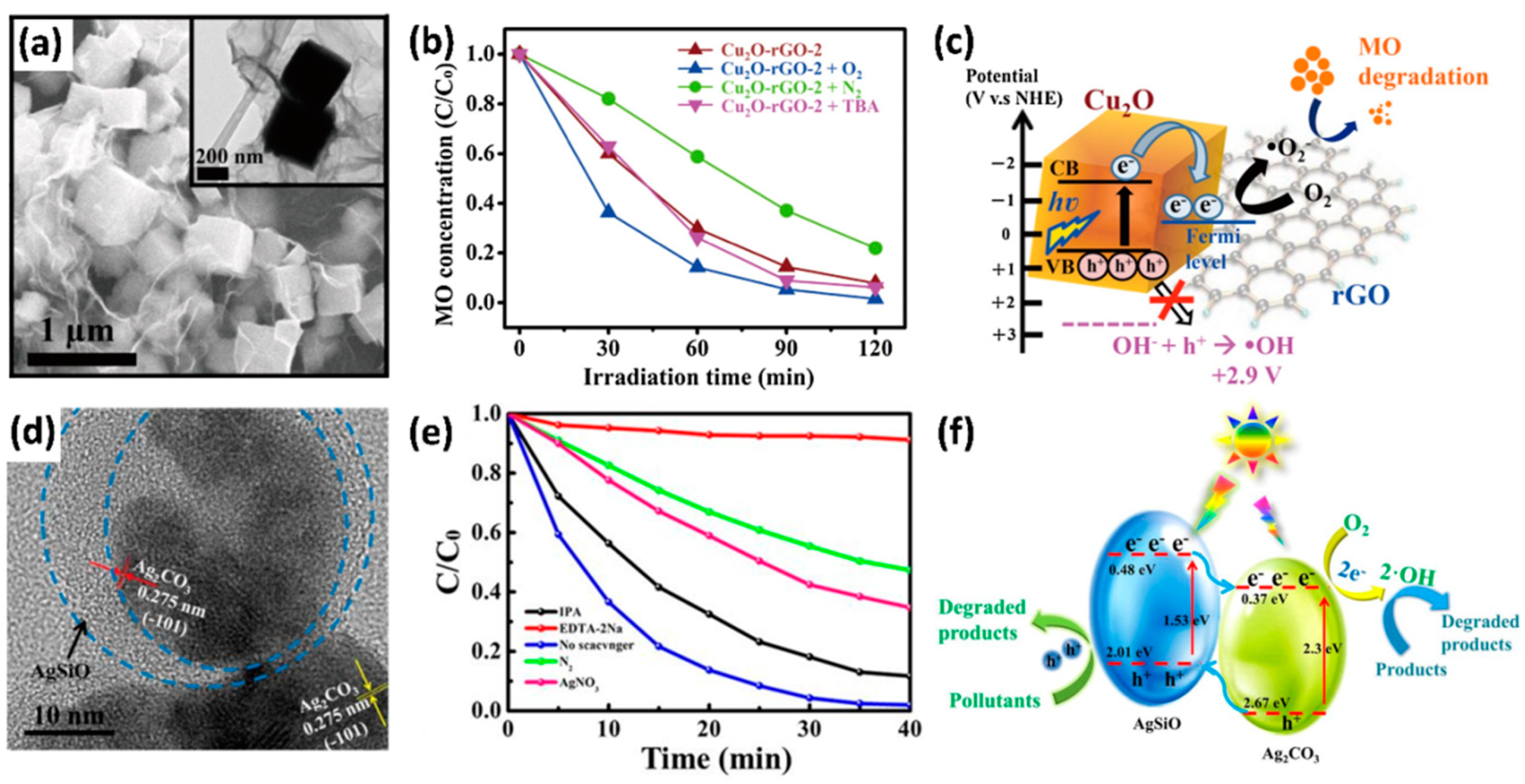
4.3.2. Modification with Semiconductors
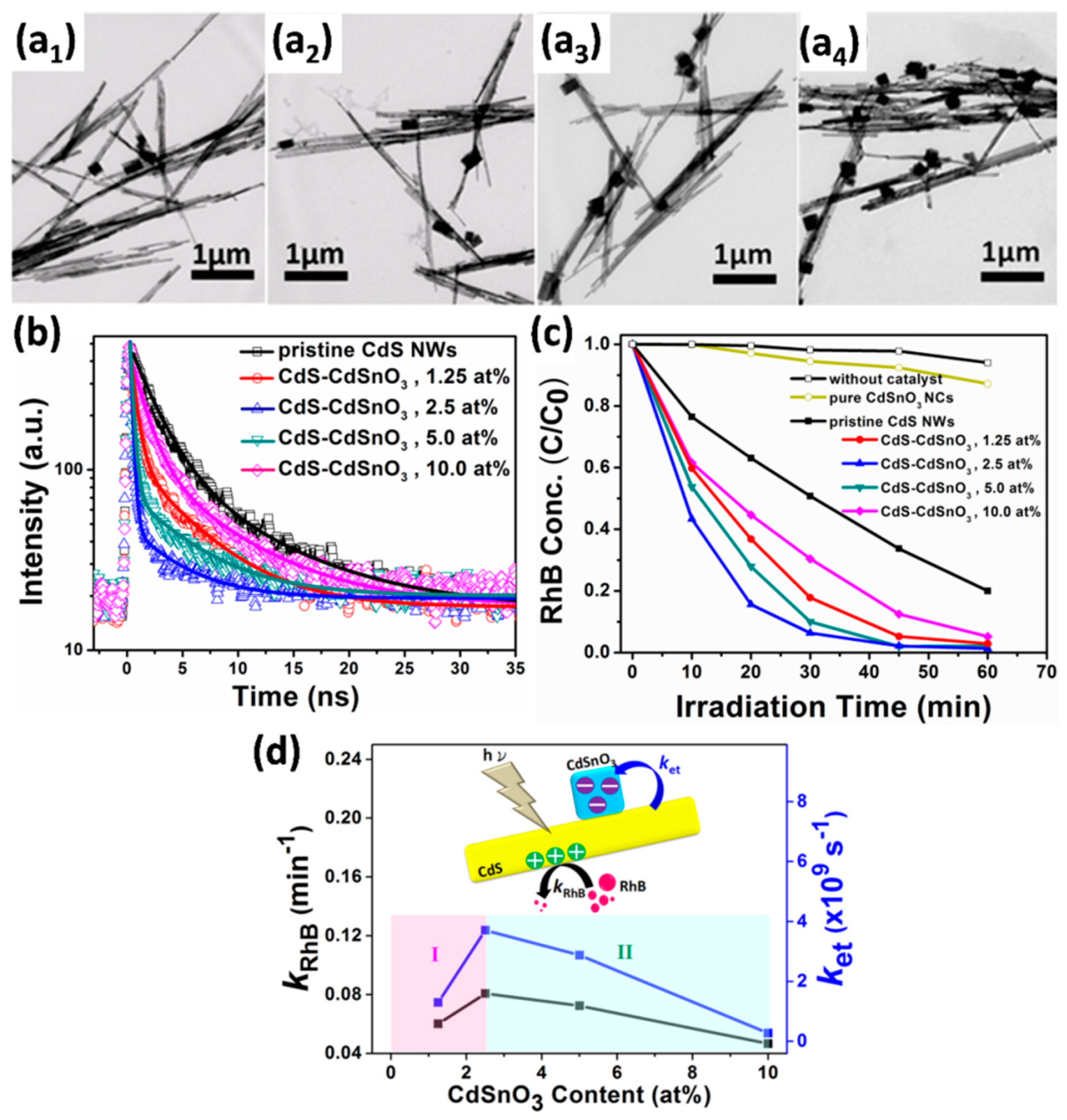
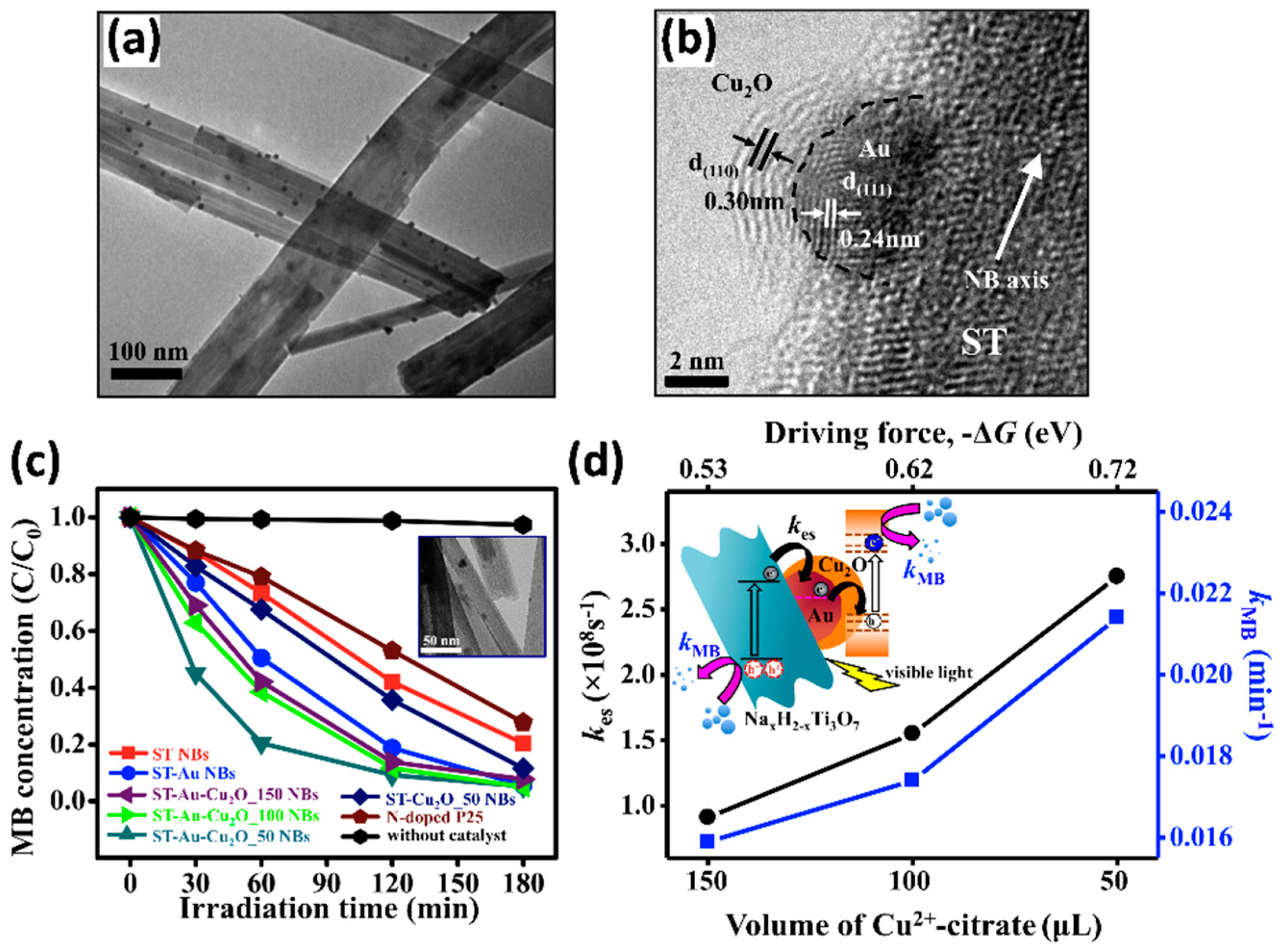
4.3.3. Modification with Metals and Semiconductors
5. Mechanism for Photodegradation of Dye
5.1. Direct Photodegradation Process
5.2. Sensitization-Mediated Degradation Process
6. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Brown, M.A.; De Vito, S.C. Predicting azo dye toxicity. Crit. Rev. Environ. Sci. Technol. 1993, 23, 249–324. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, J.; Zhang, C.; Yue, Q.; Li, Y.; Li, C. Equilibrium and kinetic studies of methyl orange and methyl violet adsorption on activated carbon derived from Phragmites australis. Desalination 2010, 252, 149–156. [Google Scholar] [CrossRef]
- Daneshvar, N.; Salari, D.; Khataee, A.R. Photocatalytic degradation of azo dye acid red 14 in water: Investigation of the effect of operational parameters. J. Photochem. Photobiol. A Chem. 2003, 157, 111–116. [Google Scholar] [CrossRef]
- Ledakowicz, S.; Gonera, M. Optimisation of oxidants dose for combined chemical and biological treatment of textile wastewater. Water Res. 1999, 33, 2511–2516. [Google Scholar] [CrossRef]
- Grzechulska, J.; Morawski, A.W. Photocatalytic decomposition of azo-dye acid black 1 in water over modified titanium dioxide. Appl. Catal. B Environ. 2002, 36, 45–51. [Google Scholar] [CrossRef]
- Haque, E.; Jun, J.W.; Jhung, S.H. Adsorptive removal of methyl orange and methylene blue from aqueous solution with a metal-organic framework material, iron terephthalate (MOF-235). J. Hazard. Mater. 2011, 185, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Li, G.; Wang, D.; Feng, C.; Tang, H. Removal of direct dyes by coagulation: The performance of preformed polymeric aluminum species. J. Hazard. Mater. 2007, 143, 567–574. [Google Scholar] [CrossRef]
- Galindo, C.; Jacques, P.; Kalt, A. Photooxidation of the phenylazonaphthol AO20 on TiO2: Kinetic and mechanistic investigations. Chemosphere 2001, 45, 997–1005. [Google Scholar] [CrossRef]
- Aleboyeh, A.; Aleboyeh, H.; Moussa, Y. “Critical” effect of hydrogen peroxide in photochemical oxidative decolorization of dyes: Acid Orange 8, Acid Blue 74 and Methyl Orange. Dyes Pigments 2003, 57, 67–75. [Google Scholar] [CrossRef]
- Ayodhya, D.; Veerabhadram, G. A review on recent advances in photodegradation of dyes using doped and heterojunction based semiconductor metal sulfide nanostructures for environmental protection. Mater. Today Energy 2018, 9, 83–113. [Google Scholar] [CrossRef]
- Aramendía, M.A.; Marinas, A.; Marinas, J.M.; Moreno, J.M.; Urbano, F.J. Photocatalytic degradation of herbicide fluroxypyr in aqueous suspension of TiO2. Catal. Today 2005, 101, 187–193. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Moon, J.H.; Choi, Y.S.; Park, G.O.; Jin, M.; Jin, L.Y.; Li, D.; Lee, J.Y.; Son, S.U.; Kim, J.M. Visible-Light Driven Photocatalytic Degradation of Organic Dyes over Ordered Mesoporous CdxZn1–xS Materials. J. Phys. Chem. C 2017, 121, 5137–5144. [Google Scholar] [CrossRef]
- Shahwan, T.; Abu Sirriah, S.; Nairat, M.; Boyacı, E.; Eroğlu, A.E.; Scott, T.B.; Hallam, K.R. Green synthesis of iron nanoparticles and their application as a Fenton-like catalyst for the degradation of aqueous cationic and anionic dyes. Chem. Eng. J. 2011, 172, 258–266. [Google Scholar] [CrossRef]
- Neppolian, B.; Choi, H.C.; Sakthivel, S.; Arabindoo, B.; Murugesan, V. Solar/UV-induced photocatalytic degradation of three commercial textile dyes. J. Hazard. Mater. 2002, 89, 303–317. [Google Scholar] [CrossRef]
- Shams-Ghahfarokhi, Z.; Nezamzadeh-Ejhieh, A. As-synthesized ZSM-5 zeolite as a suitable support for increasing the photoactivity of semiconductors in a typical photodegradation process. Mater. Sci. Semicond. Process. 2015, 39, 265–275. [Google Scholar] [CrossRef]
- Guillard, C.; Lachheb, H.; Houas, A.; Ksibi, M.; Elaloui, E.; Herrmann, J.-M. Influence of chemical structure of dyes, of pH and of inorganic salts on their photocatalytic degradation by TiO2 comparison of the efficiency of powder and supported TiO2. J. Photochem. Photobiol. A Chem. 2003, 158, 27–36. [Google Scholar] [CrossRef]
- Akbal, F. Photocatalytic degradation of organic dyes in the presence of titanium dioxide under UV and solar light: Effect of operational parameters. Environ. Prog. 2005, 24, 317–322. [Google Scholar] [CrossRef]
- Harish, S.; Archana, J.; Navaneethan, M.; Ponnusamy, S.; Singh, A.; Gupta, V.; Aswal, D.K.; Ikeda, H.; Hayakawa, Y. Synergetic effect of CuS@ZnS nanostructures on photocatalytic degradation of organic pollutant under visible light irradiation. RSC Adv. 2017, 7, 34366–34375. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Y.; Xiao, Z.; Li, W.; Li, B.; Huang, X.; Liu, X.; Hu, J. Heterostructures of CuS nanoparticle/ZnO nanorod arrays on carbon fibers with improved visible and solar light photocatalytic properties. J. Mater. Chem. A 2015, 3, 7304–7313. [Google Scholar] [CrossRef]
- Shafi, P.M.; Dhanabal, R.; Chithambararaj, A.; Velmathi, S.; Bose, A.C. α-MnO2/h-MoO3 Hybrid Material for High Performance Supercapacitor Electrode and Photocatalyst. ACS Sustain. Chem. Eng. 2017, 5, 4757–4770. [Google Scholar] [CrossRef]
- Jing, Q.; Feng, X.; Zhao, X.; Duan, Z.; Pan, J.; Chen, L.; Liu, Y. Bi/BiVO4 Chainlike Hollow Microstructures: Synthesis, Characterization, and Application as Visible-Light-Active Photocatalysts. ACS Appl. Nano Mater. 2018, 1, 2653–2661. [Google Scholar] [CrossRef]
- Cho, I.-S.; Kim, D.W.; Lee, S.; Kwak, C.H.; Bae, S.-T.; Noh, J.H.; Yoon, S.H.; Jung, H.S.; Kim, D.-W.; Hong, K.S. Synthesis of Cu2PO4OH Hierarchical Superstructures with Photocatalytic Activity in Visible Light. Adv. Funct. Mater. 2008, 18, 2154–2162. [Google Scholar] [CrossRef]
- Bi, Y.; Ouyang, S.; Umezawa, N.; Cao, J.; Ye, J. Facet Effect of Single-Crystalline Ag3PO4 Sub-microcrystals on Photocatalytic Properties. J. Am. Chem. Soc. 2011, 133, 6490–6492. [Google Scholar] [CrossRef]
- Cho, I.-S.; Kwak, C.H.; Kim, D.W.; Lee, S.; Hong, K.S. Photophysical, Photoelectrochemical, and Photocatalytic Properties of Novel SnWO4 Oxide Semiconductors with Narrow Band Gaps. J. Phys. Chem. C 2009, 113, 10647–10653. [Google Scholar] [CrossRef]
- Yang, X.; Cui, H.; Li, Y.; Qin, J.; Zhang, R.; Tang, H. Fabrication of Ag3PO4-Graphene Composites with Highly Efficient and Stable Visible Light Photocatalytic Performance. ACS Catal. 2013, 3, 363–369. [Google Scholar] [CrossRef]
- Clarke, E.A.; Anliker, R. Organic Dyes and Pigments. In Anthropogenic Compounds. The Handbook of Environmental Chemistry; Springer: Berlin/Heidelberg, Germany, 1980; Volume 3/3A, pp. 181–215. [Google Scholar]
- Demirbas, A. Agricultural based activated carbons for the removal of dyes from aqueous solutions: A review. J. Hazard. Mater. 2009, 167, 1–9. [Google Scholar] [CrossRef]
- Ramesha, G.K.; Vijaya Kumara, A.; Muralidhara, H.B.; Sampath, S. Graphene and graphene oxide as effective adsorbents toward anionic and cationic dyes. J. Colloid Interface Sci. 2011, 361, 270–277. [Google Scholar] [CrossRef]
- Mishra, G.; Tripathy, M. A Critical Review of the Treatments for Decolourization of Textile Effluent. Colourage 1993, 40, 35–38. [Google Scholar]
- Singhal, G.S.; Rabinowitch, E. Changes in the absorption spectrum of methylene blue with pH. J. Phys. Chem. 1967, 71, 3347–3349. [Google Scholar] [CrossRef]
- Xu, B.; Wang, X.; Zhu, C.; Ran, X.; Li, T.; Guo, L. Probing the inhomogeneity and intermediates in the photosensitized degradation of rhodamine B by Ag3PO4 nanoparticles from an ensemble to a single molecule approach. RSC Adv. 2017, 7, 40896–40904. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, H.; Li, Q. Visible-light-driven photocatalytic properties of ZnO/ZnFe2O4 core/shell nanocable arrays. Appl. Catal. B Environ. 2014, 160, 408–414. [Google Scholar] [CrossRef]
- Xing, Y.; Que, W.; Liu, X.; Javed, H.A.; He, Z.; He, Y.; Zhou, T. Bi2Sn2O7–TiO2 nanocomposites for enhancing visible light photocatalytic activity. RSC Adv. 2014, 4, 49900–49907. [Google Scholar] [CrossRef]
- Choi, Y.I.; Kim, Y.-I.; Cho, D.W.; Kang, J.-S.; Leung, K.; Sohn, Y. Recyclable magnetic CoFe2O4/BiOX (X = Cl, Br and I) microflowers for photocatalytic treatment of water contaminated with methyl orange, rhodamine B, methylene blue, and a mixed dye. RSC Adv. 2015, 5, 79624–79634. [Google Scholar] [CrossRef]
- Chen, C.; Lu, C.; Chung, Y.; Jan, J. UV light induced photodegradation of malachite green on TiO2 nanoparticles. J. Hazard. Mater. 2007, 141, 520–528. [Google Scholar] [CrossRef]
- Lee, S.-K.; Mills, A.; Lepre, A. An intelligence ink for oxygen. Chem. Commun. 2004, 1912–1913. [Google Scholar] [CrossRef]
- Lee, S.-K.; Sheridan, M.; Mills, A. Novel UV-activated colorimetric oxygen indicator. Chem. Mater. 2005, 17, 2744–2751. [Google Scholar] [CrossRef]
- Wang, W.; Ye, M.; He, L.; Yin, Y. Nanocrystalline TiO2-catalyzed photoreversible color switching. Nano Lett. 2014, 14, 1681–1686. [Google Scholar] [CrossRef]
- Pan, M.; Zhang, H.; Gao, G.; Liu, L.; Chen, W. Facet-Dependent Catalytic Activity of Nanosheet-Assembled Bismuth Oxyiodide Microspheres in Degradation of Bisphenol A. Environ. Sci. Technol. 2015, 49, 6240–6248. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, C.; Li, X.; Zhao, J.; Hidaka, H.; Serpone, N. Photodegradation of Sulforhodamine-B Dye in Platinized Titania Dispersions under Visible Light Irradiation: Influence of Platinum as a Functional Co-catalyst. J. Phys. Chem. B 2002, 106, 5022–5028. [Google Scholar] [CrossRef]
- Mahdiani, M.; Soofivand, F.; Ansari, F.; Salavati-Niasari, M. Grafting of CuFe12O19 nanoparticles on CNT and graphene: Eco-friendly synthesis, characterization and photocatalytic activity. J. Clean. Prod. 2018, 176, 1185–1197. [Google Scholar] [CrossRef]
- Khorshidi, N.; Khorrami, S.; Olya, M.; Mottiee, F. Photodegradation of Basic Dyes using Nanocomposite (Silver Zinc Oxide-Copper Oxide) and Kinetic Studies. Orient. J. Chem. 2016, 32, 1205–1214. [Google Scholar] [CrossRef]
- Vasanth Kumar, K.; Porkodi, K.; Selvaganapathi, A. Constrain in solving Langmuir–Hinshelwood kinetic expression for the photocatalytic degradation of Auramine O aqueous solutions by ZnO catalyst. Dyes Pigments 2007, 75, 246–249. [Google Scholar] [CrossRef]
- Bora, T.; Zoepfl, D.; Dutta, J. Importance of Plasmonic Heating on Visible Light Driven Photocatalysis of Gold Nanoparticle Decorated Zinc Oxide Nanorods. Sci. Rep. 2016, 6, 26913. [Google Scholar] [CrossRef]
- Han, A.; Sun, J.; Chuah, G.K.; Jaenicke, S. Enhanced p-cresol photodegradation over BiOBr/Bi2O3 in the presence of rhodamine B. RSC Adv. 2017, 7, 145–152. [Google Scholar] [CrossRef]
- Tsui, S.M.; Chu, W. Quantum yield study of the photodegradation of hydrophobic dyes in the presence of acetone sensitizer. Chemosphere 2001, 44, 17–22. [Google Scholar] [CrossRef]
- Nawrocka, A.; Krawczyk, S. Electronic Excited State of Alizarin Dye Adsorbed on TiO2 Nanoparticles: A Study by Electroabsorption (Stark Effect) Spectroscopy. J. Phys. Chem. C 2008, 112, 10233–10241. [Google Scholar] [CrossRef]
- Zhao, D.; Chen, C.; Wang, Y.; Ma, W.; Zhao, J.; Rajh, T.; Zang, L. Enhanced Photocatalytic Degradation of Dye Pollutants under Visible Irradiation on Al(III)-Modified TiO2: Structure, Interaction, and Interfacial Electron Transfer. Environ. Sci. Technol. 2008, 42, 308–314. [Google Scholar] [CrossRef]
- Chen, F.; Zhao, E.; Kim, T.; Wang, J.; Hableel, G.; Reardon, P.J.T.; Ananthakrishna, S.J.; Wang, T.; Arconada-Alvarez, S.; Knowles, J.C.; et al. Organosilica Nanoparticles with an Intrinsic Secondary Amine: An Efficient and Reusable Adsorbent for Dyes. ACS Appl. Mater. Interfaces 2017, 9, 15566–15576. [Google Scholar] [CrossRef]
- Li, L.; Chen, W.; Li, X. Efficient adsorption of Norfloxacin by Fe-MCM-41 molecular sieves: Kinetic, isotherm and thermodynamic studies. Chem. Eng. J. 2015, 281, 397–403. [Google Scholar]
- Dai, Y.; Zhang, K.; Li, J.; Jiang, Y.; Chen, Y.; Tanaka, S. Adsorption of copper and zinc onto carbon material in an aqueous solution oxidized by ammonium peroxydisulphate. Sep. Purif. Technol. 2017, 186, 255–263. [Google Scholar] [CrossRef]
- Hu, Y.; Guo, T.; Ye, X.; Li, Q.; Guo, M.; Liu, H.; Wu, Z. Dye adsorption by resins: Effect of ionic strength on hydrophobic and electrostatic interactions. Chem. Eng. J. 2013, 228, 392–397. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, H.; Dong, X. The amphoteric properties of g-C3N4 nanosheets and fabrication of their relevant heterostructure photocatalysts by an electrostatic re-assembly route. Chem. Commun. 2015, 51, 7176–7179. [Google Scholar] [CrossRef]
- Poulios, I.; Avranas, A.; Rekliti, E.; Zouboulis, A. Photocatalytic oxidation of Auramine O in the presence of semiconducting oxides. J. Chem. Technol. Biotechnol. 2000, 75, 205–212. [Google Scholar] [CrossRef]
- Faria, P.C.C.; Órfão, J.J.M.; Pereira, M.F.R. Adsorption of anionic and cationic dyes on activated carbons with different surface chemistries. Water Res. 2004, 38, 2043–2052. [Google Scholar] [CrossRef]
- Chun, H.; Yizhong, W.; Hongxiao, T. Influence of adsorption on the photodegradation of various dyes using surface bond-conjugated TiO2/SiO2 photocatalyst. Appl. Catal. B Environ. 2001, 35, 95–105. [Google Scholar] [CrossRef]
- Bourikas, K.; Stylidi, M.; Kondarides, D.I.; Verykios, X.E. Adsorption of Acid Orange 7 on the Surface of Titanium Dioxide. Langmuir 2005, 21, 9222–9230. [Google Scholar] [CrossRef]
- Kansal, S.K.; Kaur, N.; Singh, S. Photocatalytic Degradation of Two Commercial Reactive Dyes in Aqueous Phase Using Nanophotocatalysts. Nanoscale Res. Lett. 2009, 4, 709. [Google Scholar] [CrossRef]
- Baran, W.; Makowski, A.; Wardas, W. The effect of UV radiation absorption of cationic and anionic dye solutions on their photocatalytic degradation in the presence TiO2. Dyes Pigments 2008, 76, 226–230. [Google Scholar] [CrossRef]
- Devi, L.G.; Kumar, S.G. Exploring the critical dependence of adsorption of various dyes on the degradation rate using Ln3+-TiO2 surface under UV/solar light. Appl. Surf. Sci. 2012, 261, 137–146. [Google Scholar] [CrossRef]
- Zając, K.; Choina, J.; Dolat, D.; Morawski, A. Methylene Blue and Phenol Photocatalytic Degradation on Nanoparticles of Anatase TiO2. Pol. J. Environ. Stud. 2010, 19, 685–691. [Google Scholar]
- Fan, Y.; Liu, H.-J.; Zhang, Y.; Chen, Y. Adsorption of anionic MO or cationic MB from MO/MB mixture using polyacrylonitrile fiber hydrothermally treated with hyperbranched polyethylenimine. J. Hazard. Mater. 2015, 283, 321–328. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, Y.; Li, G.; Tian, F.; Tang, H.; Chen, R. Rhodamine B-sensitized BiOCl hierarchical nanostructure for methyl orange photodegradation. RSC Adv. 2016, 6, 7772–7779. [Google Scholar] [CrossRef]
- Kong, J.-Z.; Li, A.-D.; Li, X.-Y.; Zhai, H.-F.; Zhang, W.-Q.; Gong, Y.-P.; Li, H.; Wu, D. Photo-degradation of methylene blue using Ta-doped ZnO nanoparticle. J. Solid State Chem. 2010, 183, 1359–1364. [Google Scholar] [CrossRef]
- Gomathi Devi, L.; Narasimha Murthy, B.; Girish Kumar, S. Heterogeneous photo catalytic degradation of anionic and cationic dyes over TiO2 and TiO2 doped with Mo6+ ions under solar light: Correlation of dye structure and its adsorptive tendency on the degradation rate. Chemosphere 2009, 76, 1163–1166. [Google Scholar] [CrossRef]
- Saquib, M.; Muneer, M. TiO2-mediated photocatalytic degradation of a triphenylmethane dye (gentian violet), in aqueous suspensions. Dyes Pigments 2003, 56, 37–49. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, R.; Pandey, G. Synthesis, Characterization of Titania/Polyaniline/GO Nanocomposites, and Its Photocatalytic Activity Under UV-Visible Light. Macromol. Symp. 2018, 379, 1600192. [Google Scholar] [CrossRef]
- Giwa, A.; Nkeonye, P.O.; Bello, K.A.; Kolawole, E.G.; Campos, A.O. Solar Photocatalytic Degradation of Reactive Yellow 81 and Reactive Violet 1 in Aqueous Solution Containing Semiconductor Oxides. Int. J. Appl. Sci. Technol. 2012, 2, 90–105. [Google Scholar]
- Saggioro, E.; Oliveira, A.; Pavesi, T.; Maia, C.; Vieira Ferreira, L.F.; Moreira, J. Use of Titanium Dioxide Photocatalysis on the Remediation of Model Textile Wastewaters Containing Azo Dyes. Molecules 2011, 16, 10370–10386. [Google Scholar] [CrossRef]
- Boruah, P.K.; Borthakur, P.; Darabdhara, G.; Kamaja, C.K.; Karbhal, I.; Shelke, M.V.; Phukan, P.; Saikia, D.; Das, M.R. Sunlight assisted degradation of dye molecules and reduction of toxic Cr(vi) in aqueous medium using magnetically recoverable Fe3O4/reduced graphene oxide nanocomposite. RSC Adv. 2016, 6, 11049–11063. [Google Scholar] [CrossRef]
- Kiriakidou, F.; Kondarides, D.I.; Verykios, X.E. The effect of operational parameters and TiO2-doping on the photocatalytic degradation of azo-dyes. Catal. Today 1999, 54, 119–130. [Google Scholar] [CrossRef]
- Nam, W.; Kim, J.; Han, G. Photocatalytic oxidation of methyl orange in a three-phase fluidized bed reactor. Chemosphere 2002, 47, 1019–1024. [Google Scholar] [CrossRef]
- Kundu, S.; Pal, A.; Dikshit, A.K. UV induced degradation of herbicide 2,4-D: Kinetics, mechanism and effect of various conditions on the degradation. Sep. Purif. Technol. 2005, 44, 121–129. [Google Scholar] [CrossRef]
- Sun, P.; Zhang, J.; Liu, W.; Wang, Q.; Cao, W. Modification to L-H Kinetics Model and Its Application in the Investigation on Photodegradation of Gaseous Benzene by Nitrogen-Doped TiO2. Catalysts 2018, 8, 326. [Google Scholar] [CrossRef]
- Ollis, D.F.; Pelizzetti, E.; Serpone, N. Photocatalyzed destruction of water contaminants. Environ. Sci. Technol. 1991, 25, 1522–1529. [Google Scholar] [CrossRef]
- Mehrotra, K.; Yablonsky, G.S.; Ray, A.K. Macro kinetic studies for photocatalytic degradation of benzoic acid in immobilized systems. Chemosphere 2005, 60, 1427–1436. [Google Scholar] [CrossRef]
- Hashimoto, K.; Irie, H.; Fujishima, A. TiO2 Photocatalysis: A Historical Overview and Future Prospects. Jpn. J. Appl. Phys. 2005, 44, 8269–8285. [Google Scholar] [CrossRef]
- Barakat, N.A.M.; Kanjwal, M.A.; Chronakis, I.S.; Kim, H.Y. Influence of temperature on the photodegradation process using Ag-doped TiO2 nanostructures: Negative impact with the nanofibers. J. Mol. Catal. A Chem. 2013, 366, 333–340. [Google Scholar] [CrossRef]
- Yang, T.-T.; Chen, W.-T.; Hsu, Y.-J.; Wei, K.-H.; Lin, T.-Y.; Lin, T.-W. Interfacial Charge Carrier Dynamics in Core−Shell Au-CdS Nanocrystals. J. Phys. Chem. C 2010, 114, 11414–11420. [Google Scholar] [CrossRef]
- Lin, W.-H.; Chiu, Y.-H.; Shao, P.-W.; Hsu, Y.-J. Metal-Particle-Decorated ZnO Nanocrystals: Photocatalysis and Charge Dynamics. ACS Appl. Mater. Interfaces 2016, 8, 32754–32763. [Google Scholar] [CrossRef]
- Lin, Y.-F.; Hsu, Y.-J. Interfacial charge carrier dynamics of type-II semiconductor nanoheterostructures. Appl. Catal. B Environ. 2013, 130–131, 93–98. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Hsu, Y.-J. Type-II nanorod heterostructure formation through one-step cation exchange. Nanoscale 2013, 5, 363–368. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Pu, Y.-C.; Hsu, Y.-J. Interfacial Charge Carrier Dynamics of the Three-Component In2O3–TiO2–Pt Heterojunction System. J. Phys. Chem. C 2012, 116, 2967–2975. [Google Scholar] [CrossRef]
- Pu, Y.-C.; Chou, H.-Y.; Kuo, W.-S.; Wei, K.-H.; Hsu, Y.-J. Interfacial charge carrier dynamics of cuprous oxide-reduced graphene oxide (Cu2O-rGO) nanoheterostructures and their related visible-light-driven photocatalysis. Appl. Catal. B Environ. 2017, 204, 21–32. [Google Scholar] [CrossRef]
- Chiu, Y.-H.; Hsu, Y.-J. Au@Cu7S4 yolk@shell nanocrystal-decorated TiO2 nanowires as an all-day-active photocatalyst for environmental purification. Nano Energy 2017, 31, 286–295. [Google Scholar] [CrossRef]
- Naresh, G.; Hsieh, P.-L.; Meena, V.; Lee, S.-K.; Chiu, Y.-H.; Madasu, M.; Lee, A.-T.; Tsai, H.-Y.; Lai, T.-H.; Hsu, Y.-J.; et al. Facet-Dependent Photocatalytic Behaviors of ZnS-Decorated Cu2O Polyhedra Arising from Tunable Interfacial Band Alignment. ACS Appl. Mater. Interfaces 2019, 11, 3582–3589. [Google Scholar] [CrossRef]
- Pu, Y.-C.; Lin, W.-H.; Hsu, Y.-J. Modulation of charge carrier dynamics of NaxH2−xTi3O7-Au-Cu2O Z-scheme nanoheterostructures through size effect. Appl. Catal. B Environ. 2015, 163, 343–351. [Google Scholar] [CrossRef]
- Chen, F.; Deng, Z.; Li, X.; Zhang, J.; Zhao, J. Visible light detoxification by 2,9,16,23-tetracarboxyl phthalocyanine copper modified amorphous titania. Chem. Phys. Lett. 2005, 415, 85–88. [Google Scholar] [CrossRef]
- Yan, S.C.; Li, Z.S.; Zou, Z.G. Photodegradation of Rhodamine B and Methyl Orange over Boron-Doped g-C3N4 under Visible Light Irradiation. Langmuir 2010, 26, 3894–3901. [Google Scholar] [CrossRef]
- Cao, R.; Yang, H.; Deng, X.; Zhang, S.; Xu, X. In-situ synthesis of amorphous silver silicate/carbonate composites for selective visible-light photocatalytic decomposition. Sci. Rep. 2017, 7, 15001. [Google Scholar] [CrossRef]
- Qiu, P.; Chen, H.; Xu, C.; Zhou, N.; Jiang, F.; Wang, X.; Fu, Y. Fabrication of an exfoliated graphitic carbon nitride as a highly active visible light photocatalyst. J. Mater. Chem. A 2015, 3, 24237–24244. [Google Scholar] [CrossRef]
- Xu, T.; Zhang, L.; Cheng, H.; Zhu, Y. Significantly enhanced photocatalytic performance of ZnO via graphene hybridization and the mechanism study. Appl. Catal. B Environ. 2011, 101, 382–387. [Google Scholar] [CrossRef]
- Tian, Y.; Chang, B.; Yang, Z.; Zhou, B.; Xi, F.; Dong, X. Graphitic carbon nitride–BiVO4 heterojunctions: Simple hydrothermal synthesis and high photocatalytic performances. RSC Adv. 2014, 4, 4187–4193. [Google Scholar] [CrossRef]
- Teng, W.; Li, X.; Zhao, Q.; Zhao, J.; Zhang, D. In situ capture of active species and oxidation mechanism of RhB and MB dyes over sunlight-driven Ag/Ag3PO4 plasmonic nanocatalyst. Appl. Catal. B Environ. 2012, 125, 538–545. [Google Scholar] [CrossRef]
- Hu, Y.; Li, D.; Wang, H.; Zeng, G.; Li, X.; Shao, Y. Role of active oxygen species in the liquid-phase photocatalytic degradation of RhB using BiVO4/TiO2 heterostructure under visible light irradiation. J. Mol. Catal. A Chem. 2015, 408, 172–178. [Google Scholar] [CrossRef]
- Hou, J.; Jiang, K.; Shen, M.; Wei, R.; Wu, X.; Idrees, F.; Cao, C. Micro and nano hierachical structures of BiOI/activated carbon for efficient visible-light-photocatalytic reactions. Sci. Rep. 2017, 7, 11665. [Google Scholar] [CrossRef]
- Jiang, Y.-R.; Lin, H.-P.; Chung, W.-H.; Dai, Y.-M.; Lin, W.-Y.; Chen, C.-C. Controlled hydrothermal synthesis of BiOxCly/BiOmIn composites exhibiting visible-light photocatalytic degradation of crystal violet. J. Hazard. Mater. 2015, 283, 787–805. [Google Scholar] [CrossRef]
- Chou, S.-Y.; Chung, W.-H.; Chen, L.-W.; Dai, Y.-M.; Lin, W.-Y.; Lin, J.-H.; Chen, C.-C. A series of BiOxIy/GO photocatalysts: Synthesis, characterization, activity, and mechanism. RSC Adv. 2016, 6, 82743–82758. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, T.; Xu, D.; Zhang, L. Microwave-Assisted Catalytic Degradation of Crystal Violet with Barium Ferrite Nanomaterial. Ind. Eng. Chem. Res. 2016, 55, 11869–11877. [Google Scholar] [CrossRef]
- Liu, X.; An, S.; Shi, W.; Yang, Q.; Zhang, L. Microwave-induced catalytic oxidation of malachite green under magnetic Cu-ferrites: New insight into the degradation mechanism and pathway. J. Mol. Catal. A Chem. 2014, 395, 243–250. [Google Scholar] [CrossRef]
- Arifin, S.A.; Jalaludin, S.; Saleh, R. Photocatalytic Decolorization of Malachite Green in the Presence of Fe3O4/TiO2/CuO Nanocomposites. Mater. Sci. Forum 2015, 1123, 264–269. [Google Scholar]
- Kulsi, C.; Ghosh, A.; Mondal, A.; Kargupta, K.; Ganguly, S.; Banerjee, D. Remarkable photo-catalytic degradation of malachite green by nickel doped bismuth selenide under visible light irradiation. Appl. Surf. Sci. 2017, 392, 540–548. [Google Scholar] [CrossRef]
- Kushwaha, H.; Vaish, R. Enhanced Visible Light Photocatalytic Activity of Curcumin-Sensitized Perovskite Bi0.5Na0.5TiO3 for Rhodamine 6G Degradation. Int. J. Appl. Ceram. Technol. 2015, 13, 333–339. [Google Scholar] [CrossRef]
- Mohapatra, L.; Parida, K.; Satpathy, M. Molybdate/Tungstate Intercalated Oxo-Bridged Zn/Y LDH for Solar Light Induced Photodegradation of Organic Pollutants. J. Phys. Chem. C 2012, 116, 13063–13070. [Google Scholar] [CrossRef]
- Kaur, R.; Vellingiri, K.; Kim, K.-H.; Paul, A.K.; Deep, A. Efficient photocatalytic degradation of rhodamine 6G with a quantum dot-metal organic framework nanocomposite. Chemosphere 2016, 154, 620–627. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, S.; Wang, K.; Lou, L. Role of primary active species and TiO2 surface characteristic in UV-illuminated photodegradation of Acid Orange 7. J. Photochem. Photobiol. A Chem. 2005, 172, 47–54. [Google Scholar] [CrossRef]
- Li, G.; Wong, K.H.; Zhang, X.; Hu, C.; Yu, J.C.; Chan, R.C.Y.; Wong, P.K. Degradation of Acid Orange 7 using magnetic AgBr under visible light: The roles of oxidizing species. Chemosphere 2009, 76, 1185–1191. [Google Scholar] [CrossRef]
- Reza Pouretedal, H.; Hosseini, M. Bleaching Kinetic and Mechanism Study of Congo Red Catalyzed by ZrO2 Nanoparticles Prepared by Using a Simple Precipitation Method. Acta Chim. Slov. 2010, 57, 415–423. [Google Scholar]
- Bhoi, Y.P.; Pradhan, S.R.; Behera, C.; Mishra, B.G. Visible light driven efficient photocatalytic degradation of Congo red dye catalyzed by hierarchical CuS–Bi2CuxW1−xO6−2x nanocomposite system. RSC Adv. 2016, 6, 35589–35601. [Google Scholar] [CrossRef]
- Paramarta, V.; Taufik, A.; Munisa, L.; Saleh, R. Sono- and photocatalytic activities of SnO2 nanoparticles for degradation of cationic and anionic dyes. AIP Conf. Proc. 2017, 1788, 030125. [Google Scholar] [CrossRef]
- Kaur, S.; Sharma, S.; Kansal, S.K. Synthesis of ZnS/CQDs nanocomposite and its application as a photocatalyst for the degradation of an anionic dye, ARS. Superlattices Microstruct. 2016, 98, 86–95. [Google Scholar] [CrossRef]
- Ghugal, S.G.; Umare, S.S.; Sasikala, R. Photocatalytic mineralization of anionic dyes using bismuth doped CdS–Ta2O5 composite. RSC Adv. 2015, 5, 63393–63400. [Google Scholar] [CrossRef]
- Song, S.; Xu, L.; He, Z.; Chen, J.; Xiao, X.; Yan, B. Mechanism of the Photocatalytic Degradation of C.I. Reactive Black 5 at pH 12.0 Using SrTiO3/CeO2 as the Catalyst. Environ. Sci. Technol. 2007, 41, 5846–5853. [Google Scholar] [CrossRef]
- Roushani, M.; Mavaei, M.; Rajabi, H.R. Graphene quantum dots as novel and green nano-materials for the visible-light-driven photocatalytic degradation of cationic dye. J. Mol. Catal. A Chem. 2015, 409, 102–109. [Google Scholar] [CrossRef]
- Guo, J.; Liao, X.; Lee, M.-H.; Hyett, G.; Huang, C.-C.; Hewak, D.W.; Mailis, S.; Zhou, W.; Jiang, Z. Experimental and DFT insights of the Zn-doping effects on the visible-light photocatalytic water splitting and dye decomposition over Zn-doped BiOBr photocatalysts. Appl. Catal. B Environ. 2019, 243, 502–512. [Google Scholar] [CrossRef]
- Trandafilović, L.V.; Jovanović, D.J.; Zhang, X.; Ptasińska, S.; Dramićanin, M.D. Enhanced photocatalytic degradation of methylene blue and methyl orange by ZnO:Eu nanoparticles. Appl. Catal. B Environ. 2017, 203, 740–752. [Google Scholar] [CrossRef]
- Chen, J.; Wang, H.; Huang, G.; Zhang, Z.; Han, L.; Song, W.; Li, M.; Zhang, Y. Facile synthesis of urchin-like hierarchical Nb2O5 nanospheres with enhanced visible light photocatalytic activity. J. Alloy. Compd. 2017, 728, 19–28. [Google Scholar] [CrossRef]
- Ohtani, B. Preparing Articles on Photocatalysis—Beyond the Illusions, Misconceptions, and Speculation. Chem. Lett. 2008, 37, 216–229. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cationic Dyes | Abbreviation | MW | Structure | (nm) |
| Methylene blue | MB | 799.81 |  | 664 |
| Rhodamine B | RhB | 479.02 |  | 553 |
| Rhodamine 6G | Rh6G | 479.02 |  | 534 |
| Malachite green | MG | 364.91 |  | 614 |
| Crystal violet | CV | 407.98 |  | 573 |
| Safranin O | SO | 350.85 |  | 520 |
| Auramine O | AO | 303.83 |  | 420 |
| Victoria blue B | VBB | 506.08 |  | 614 |
| Anionic Dyes | Abbreviation | Mw | Structure | (nm) |
| Methyl orange | MO | 327.33 |  | 464 |
| Eosin Y | EY | 691.85 |  | 518 |
| Acid orange 7 | AO7 | 350.32 |  | 484 |
| Acid red 14 | AR14 | 502.43 |  | 515 |
| Alizarin red S | ARS | 240.21 |  | 426 |
| Rose bengal | RB | 973.67 |  | 550 |
| Phenol red | PR | 354.38 |  | 560 |
| Congo red | CR | 696.67 |  | 497 |
| Acid violet 7 | AV7 | 566.47 |  | 522 |
| Reactive black 5 | RB5 | 991.82 |  | 602 |
| Type | Sacrifice Reagent | Abbreviation |
|---|---|---|
| Electron scavenger | AgNO3 | - |
| CCl4 | - | |
| K2Cr2O7 | - | |
| Hole scavenger | KI | - |
| Ethylenediaminetetraacetic acid | EDTA, EDTA-2Na | |
| Tri-ethanolamine | TEOA | |
| Ammonium oxalate | AO | |
| Sodium oxalate (Na2C2O4) | - | |
| Methanol | - | |
| Ascorbic acid | AA | |
| ·OH scavenger | tert-Butyl alcohol | TBA, t-BuOH |
| 2-Propanol | IPA | |
| ·O2− scavenger | Benzoquinone | BQ |
| Acrylamide | AC | |
| Superoxide dismutase | SOD |
| Class | Dye | Active Species | Photocatalysts |
|---|---|---|---|
| Cationic dye | RhB | H+ | g-C3N4 [89] |
| H+ | Ag/Ag3PO4 [94] | ||
| ·O2−/H+ | BiVO4/TiO2 [95] | ||
| ·O2−/H+ | BiOI/C [96] | ||
| H+ (major), ·O2− (minor) | CoFe2O4/BiO(Cl, Br, I) [34] | ||
| MB | ·O2−/H+ | g-C3N4 [91] | |
| ·O2−/ H+ | ZnO/graphene [92] | ||
| ·O2− | C3N4-BiVO4 [93] | ||
| ·O2−/H+ | AgSiO/Ag2CO3 [90] | ||
| CV | ·O2− (major), h+/·OH (minor) | BiOxCly/BiOmIn [97] | |
| ·O2− (major), h+/·OH (minor) | BiOxIy/GO [98] | ||
| ·O2−/h+ (major), ·OH (minor) | BaFe2O4 [99] | ||
| MG | H+/·OH /·O2− | CuFe2O4 [100] | |
| ·OH/e−/H+ | Fe3O4/TiO2/CuO [101] | ||
| ·OH/e−/H+ (major), ·O2− (minor) | Ni-Bi2Se3 [102] | ||
| Rh6G | H+ (major) ·OH/·O2− (minor) | Curcumin/Bi0.5Na0.5TiO3 [103] | |
| ·O2− | Zn/Y [104] | ||
| H+/·O2− | Quantum dot/Eu-metal organic framework [105] | ||
| Anionic dye | MO | ·O2− | Cu2O-rGO [84] |
| ·O2− | 2,9,16,23-tetracarboxyl phthalocyanine/amorphous TiO2 [88] | ||
| ·O2− (major) H+ (minor) | g-C3N4 [89] | ||
| AO7 | H+/·OH | TiO2 [106] | |
| h+ | Ag/AgBr/SiO2-coated Fe3O4 [107] | ||
| CR | ·OH | ZrO2 [108] | |
| H+/·OH | CuS-Bi2CuxW1−xO6−2x [109] | ||
| H+ (major), ·OH (minor) | SnO2 [110] | ||
| ARS | ·OH/e−/H+ | ZnS/carbon quantum dots [111] | |
| AV7 | ·O2−/H+/·OH | CdS/Ta2O5 [112] | |
| RB5 | H+ | SrTiO3/CeO2 [113] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, Y.-H.; Chang, T.-F.M.; Chen, C.-Y.; Sone, M.; Hsu, Y.-J. Mechanistic Insights into Photodegradation of Organic Dyes Using Heterostructure Photocatalysts. Catalysts 2019, 9, 430. https://doi.org/10.3390/catal9050430
Chiu Y-H, Chang T-FM, Chen C-Y, Sone M, Hsu Y-J. Mechanistic Insights into Photodegradation of Organic Dyes Using Heterostructure Photocatalysts. Catalysts. 2019; 9(5):430. https://doi.org/10.3390/catal9050430
Chicago/Turabian StyleChiu, Yi-Hsuan, Tso-Fu Mark Chang, Chun-Yi Chen, Masato Sone, and Yung-Jung Hsu. 2019. "Mechanistic Insights into Photodegradation of Organic Dyes Using Heterostructure Photocatalysts" Catalysts 9, no. 5: 430. https://doi.org/10.3390/catal9050430
APA StyleChiu, Y.-H., Chang, T.-F. M., Chen, C.-Y., Sone, M., & Hsu, Y.-J. (2019). Mechanistic Insights into Photodegradation of Organic Dyes Using Heterostructure Photocatalysts. Catalysts, 9(5), 430. https://doi.org/10.3390/catal9050430