CaRMeN: An Improved Computer-Aided Method for Developing Catalytic Reaction Mechanisms



Abstract
:1. Introduction
2. Combustion over Rh-Based Catalysts
2.1. Overview of Mechanisms in the Literature
2.2. Overview of Experimental Setups
2.3. Overview of Simulation Tools
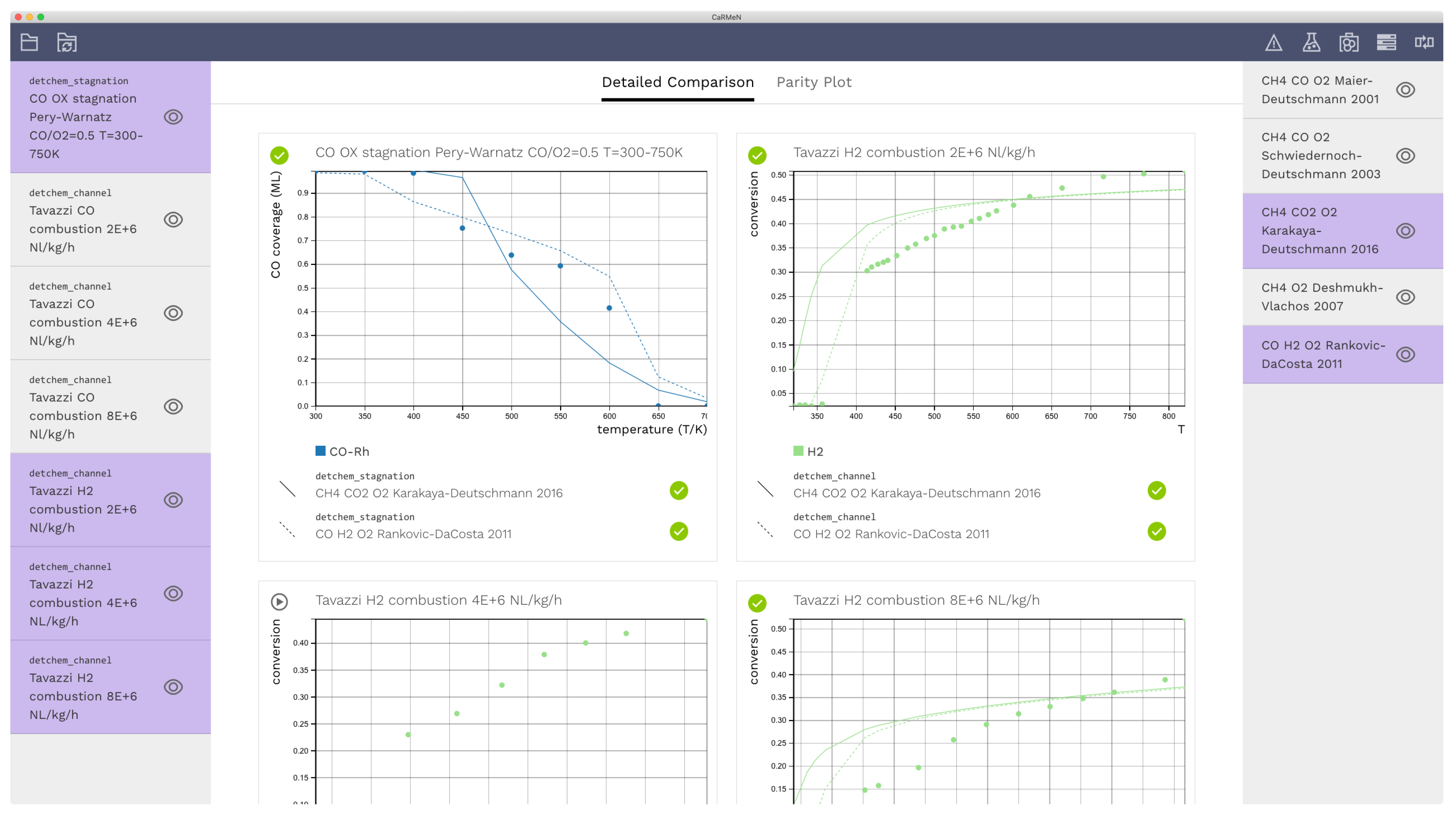
3. Illustrative Examples
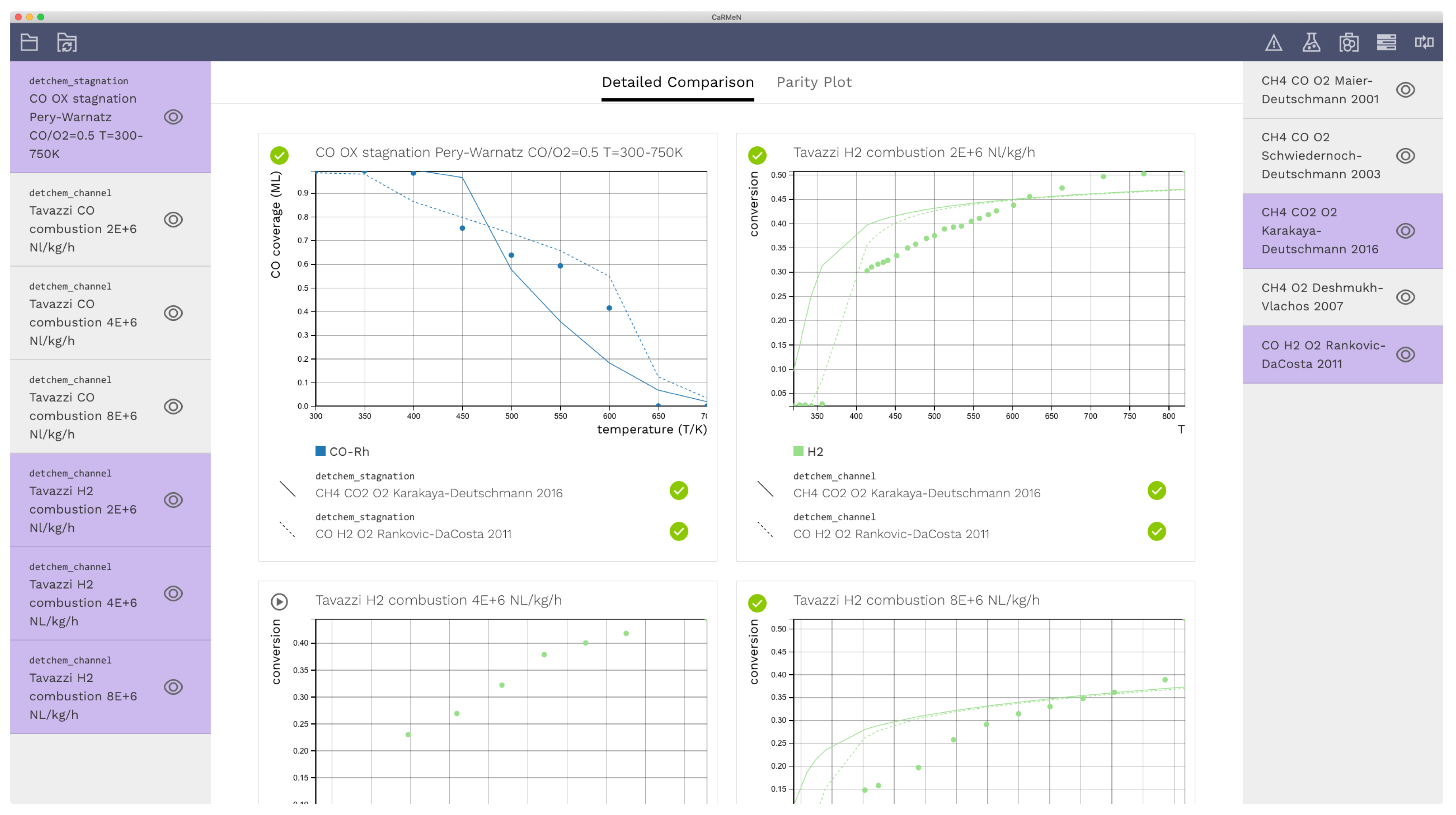
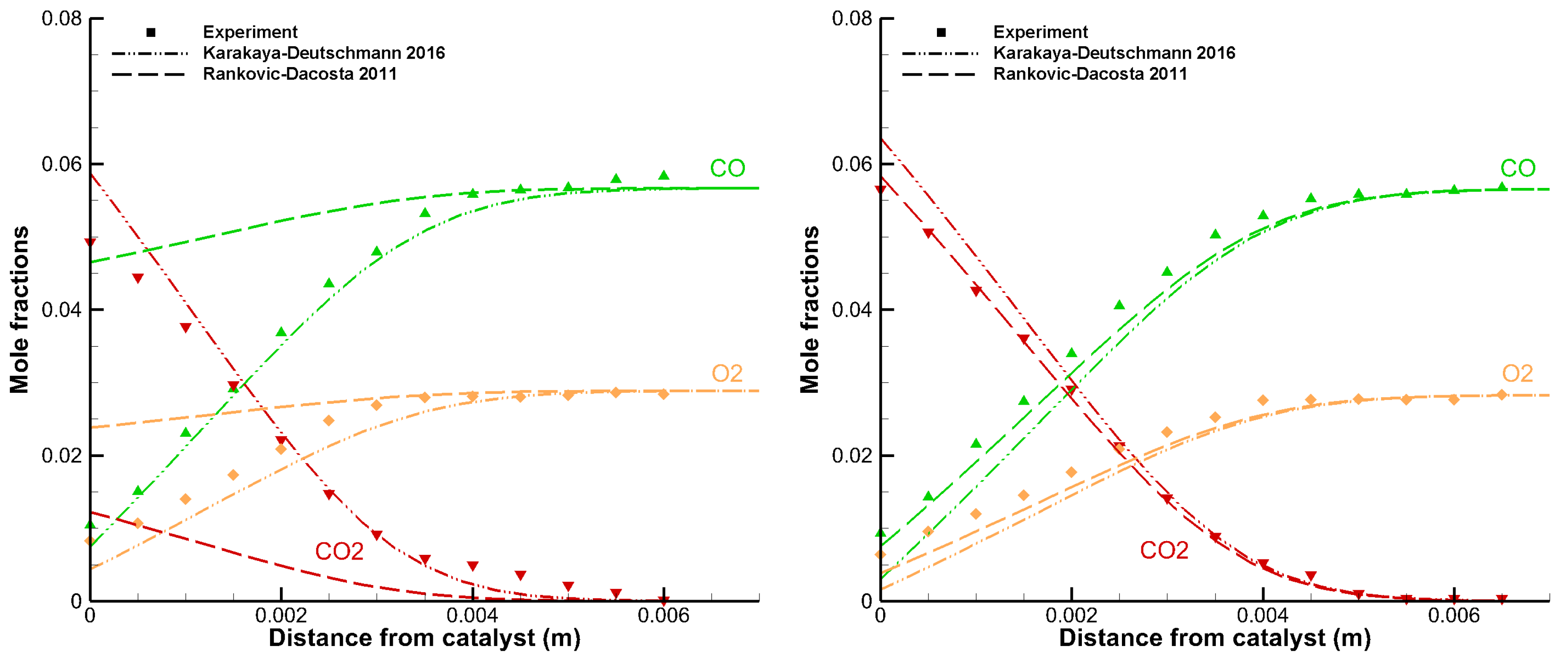
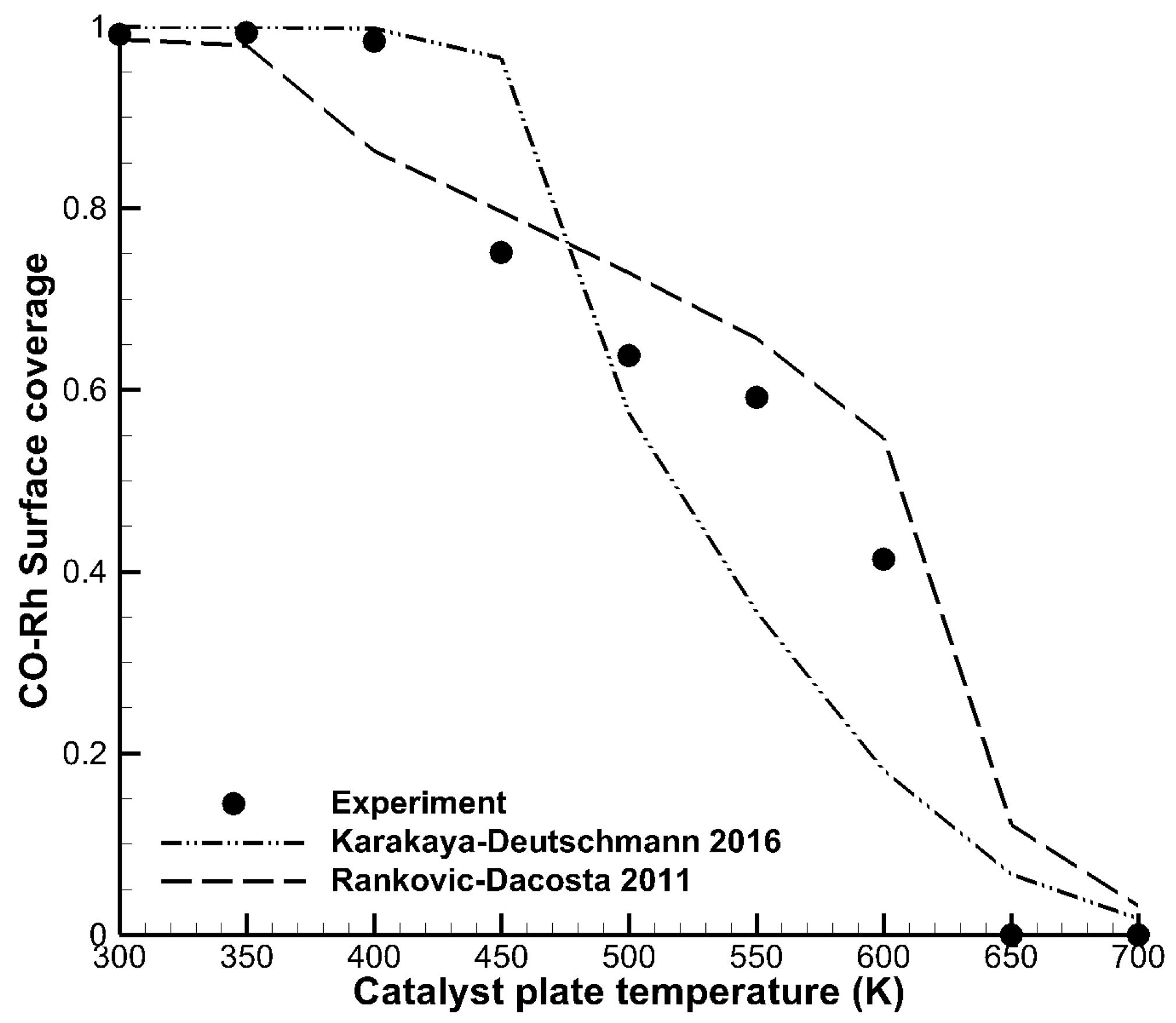
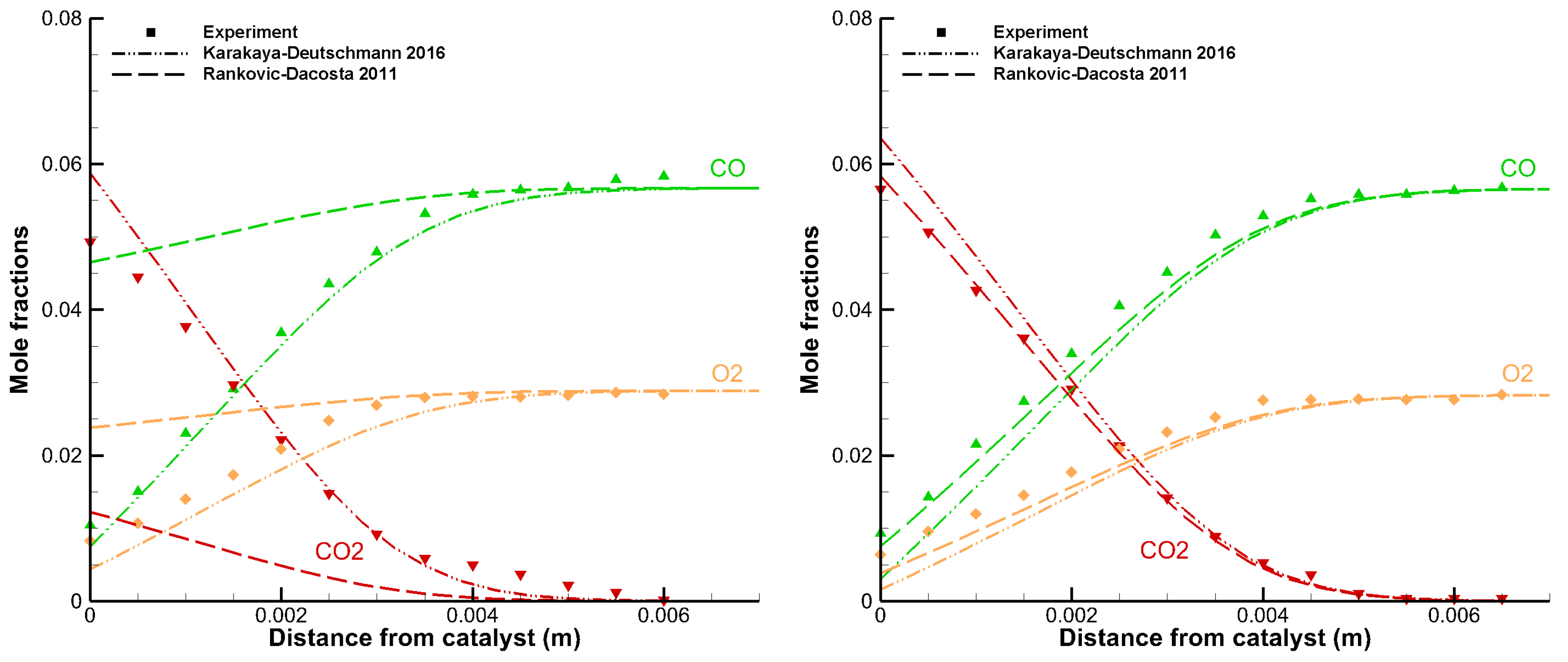
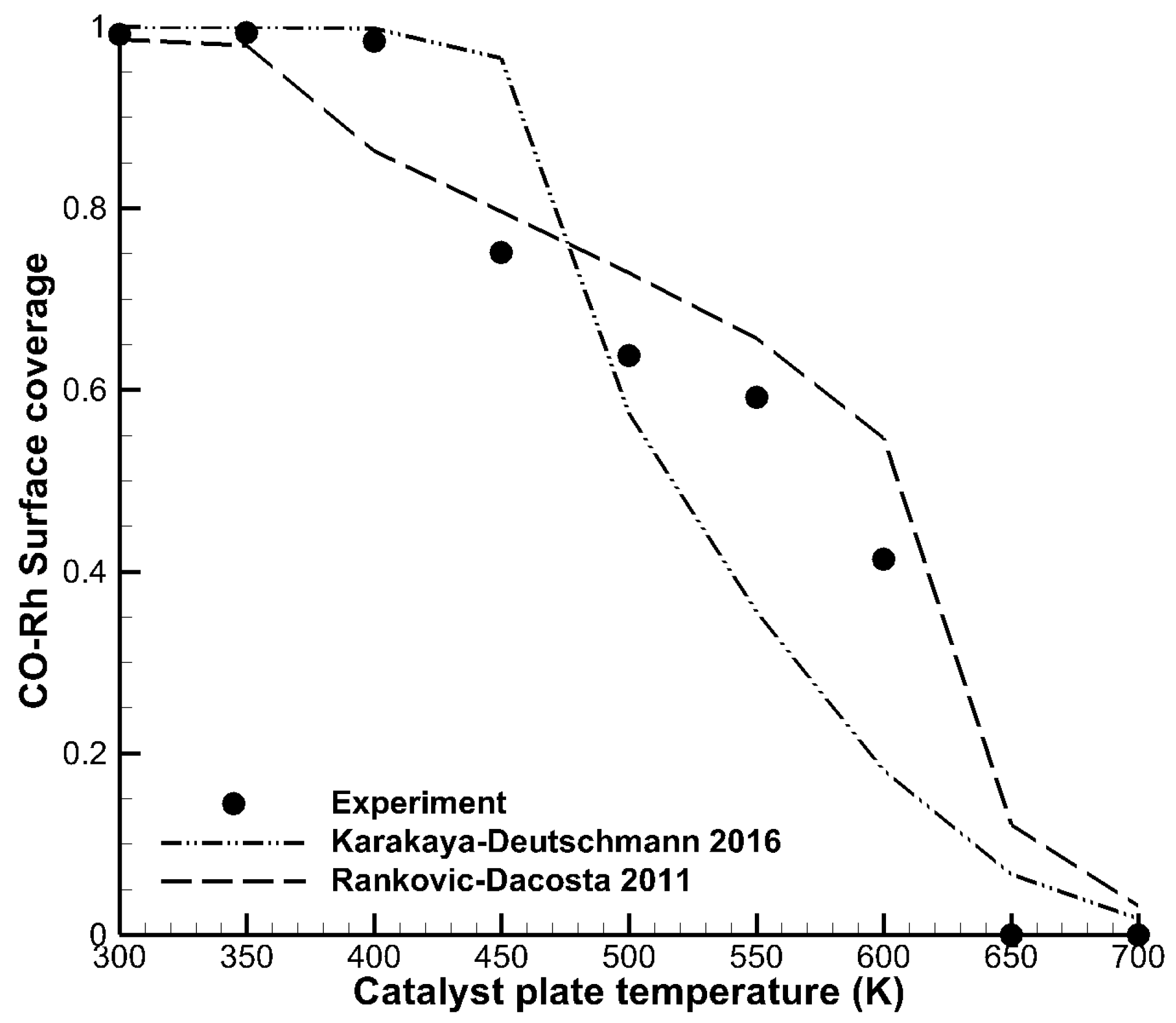
3.1. Detailed Comparison—CO Combustion
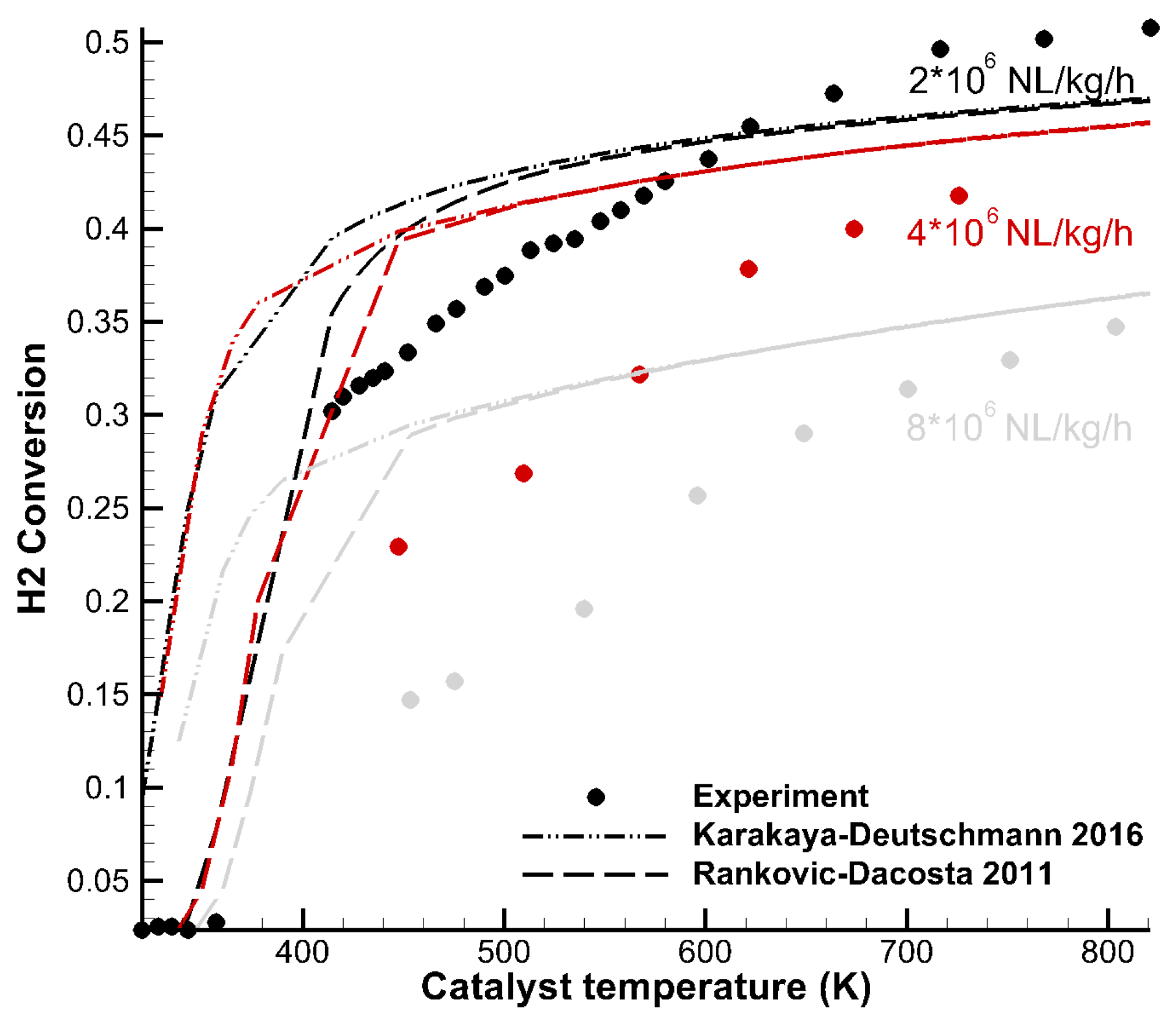
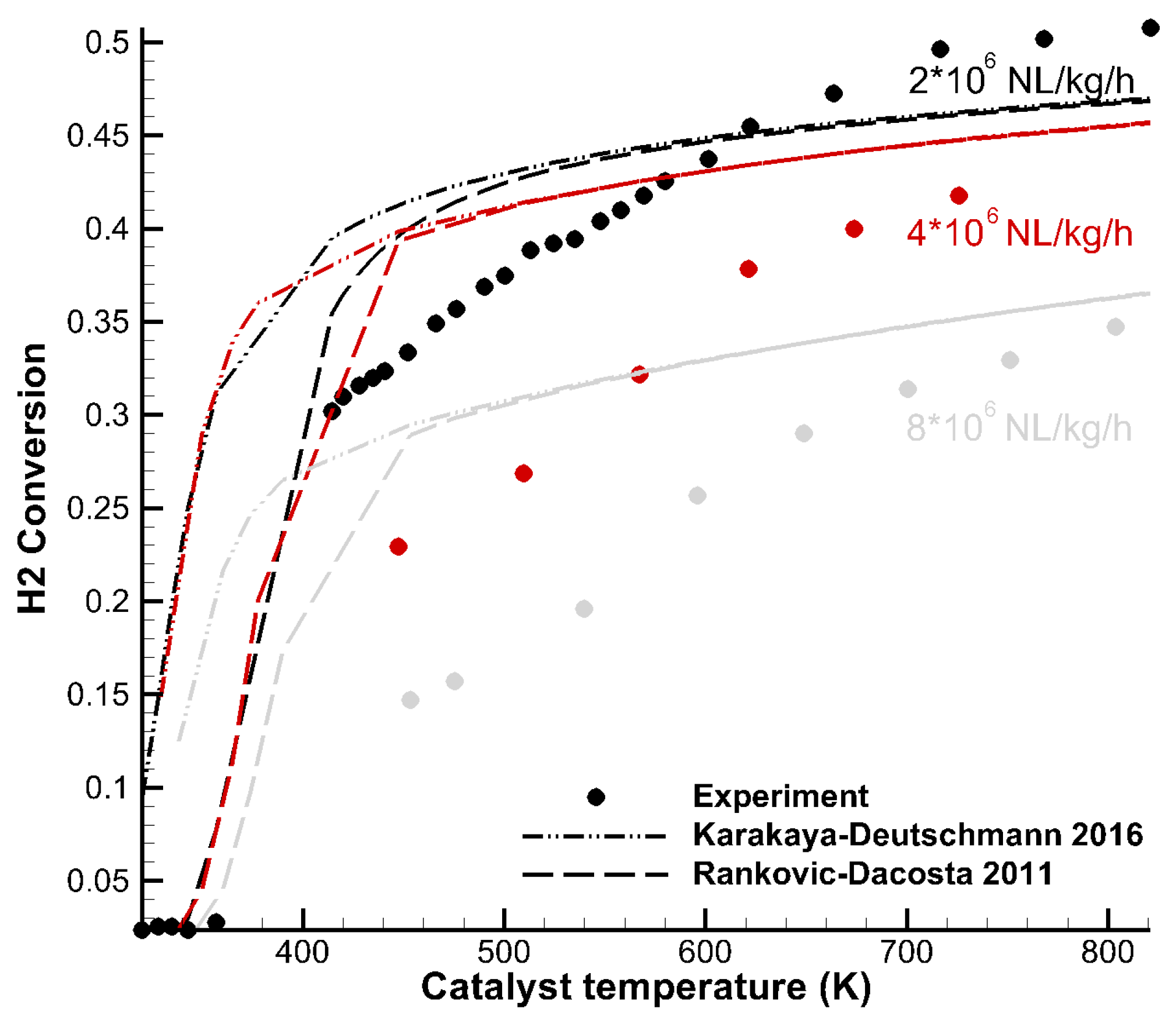
3.2. Light-Off Curves—H2 Combustion
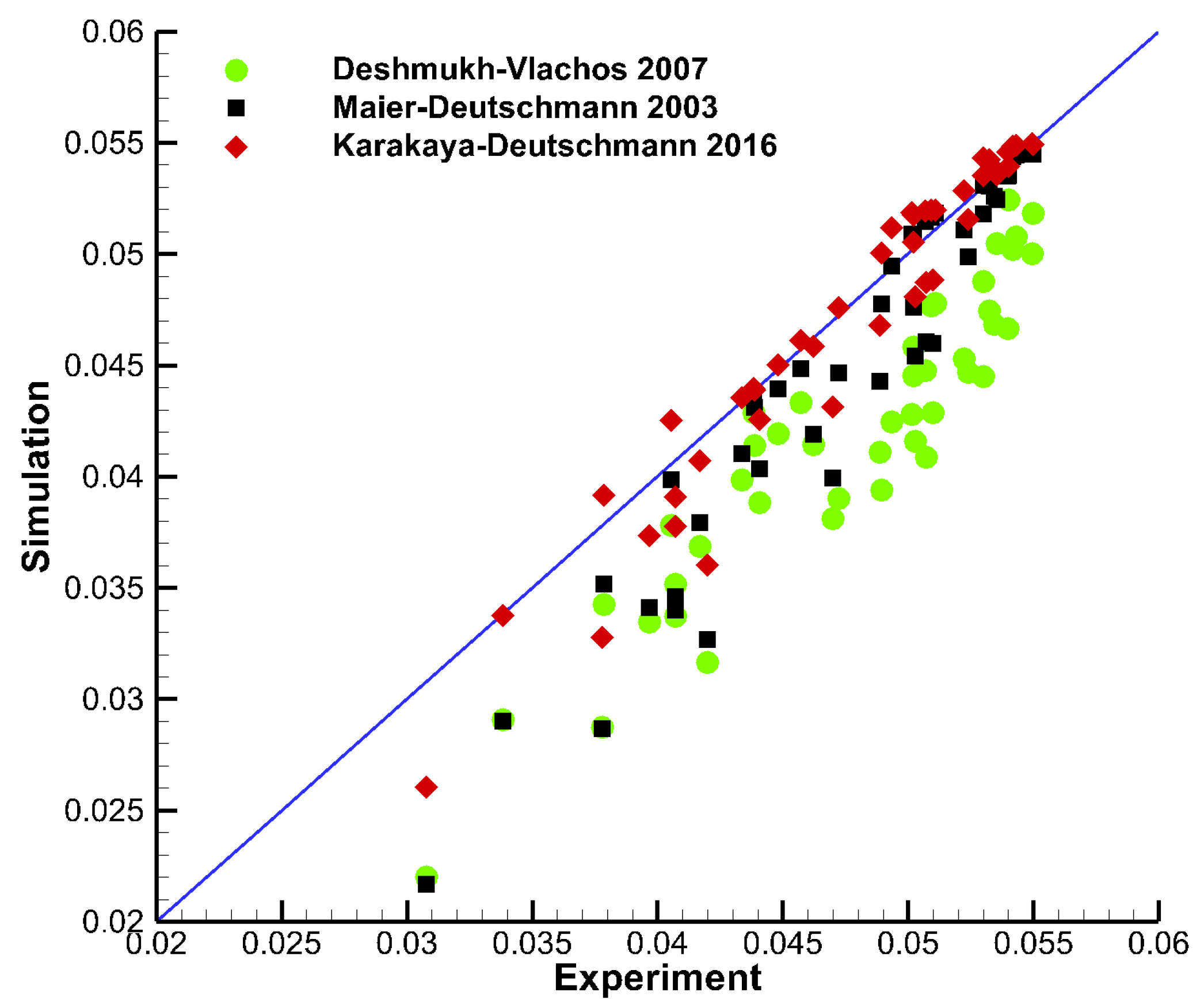
3.3. Parity Plot—CH4 Combustion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gossler, H.; Maier, L.; Angeli, S.; Tischer, S.; Deutschmann, O. CaRMeN: A Tool for Analysing and Deriving Kinetics in the Real World. Phys. Chem. Chem. Phys. 2018, 20, 10857–10876. [Google Scholar] [CrossRef] [PubMed]
- PrIMe. Available online: http://primekinetics.org (accessed on 9 December 2018).
- RESPECTH. Available online: http://respecth.chem.elte.hu/respecth/ (accessed on 9 December 2018).
- Goteng, G.; Speight, M.; Nettyam, N.; Farooq, A.; Franklach, M.; Sarathy, S. A Hybrid Cloud System for Combustion Kinetics Simulation. In Proceedings of the 23rd International Symposium on Gas Kinetics and Related Phenomena, Szeged, Hungary, 20–24 July 2014. [Google Scholar]
- Lunsford, J.H. Catalytic Conversion of Methane to More Useful Chemicals and Fuels: A Challenge for the 21st Century. Catal. Today 2000, 63, 165–174. [Google Scholar] [CrossRef]
- Abbasi, R.; Wu, L.; Wanke, S.E.; Hayes, R.E. Kinetics of Methane Combustion over Pt and Pt–Pd Catalysts. Chem. Eng. Res. Des. 2012, 90, 1930–1942. [Google Scholar] [CrossRef]
- Chen, D.; Lødeng, R.; Svendsen, H.; Holmen, A. Hierarchical Multiscale Modeling of Methane Steam Reforming Reactions. Ind. Eng. Chem. Res. 2011, 50, 2600–2612. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; Laffir, F.; Curtin, T.; Thompson, J.M.; Rooney, D.W. A Bimetallic Catalyst on a Dual Component Support for Low Temperature Total Methane Oxidation. Appl. Catal. B Environ. 2016, 187, 408–418. [Google Scholar] [CrossRef]
- Osman, A.I.; Meudal, J.; Laffir, F.; Thompson, J.; Rooney, D. Enhanced Catalytic Activity of Ni on η-Al2O3 and ZSM-5 on Addition of Ceria Zirconia for the Partial Oxidation of Methane. Appl. Catal. B Environ. 2017, 212, 68–79. [Google Scholar] [CrossRef]
- Maier, L.; Schädel, B.; Delgado, K.H.; Tischer, S.; Deutschmann, O. Steam Reforming of Methane Over Nickel: Development of a Multi-Step Surface Reaction Mechanism. Top. Catal. 2011, 54, 845. [Google Scholar] [CrossRef]
- Deutschmann, O.; Schwiedemoch, R.; Maier, L.I.; Chatterjee, D. Natural Gas Conversion in Monolithic Catalysts: Interaction of Chemical Reactions and Transport Phenomena. In Studies in Surface Science and Catalysis; Iglesia, E., Spivey, J.J., Fleisch, T.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; Volume 136, Natural Gas Conversion VI; pp. 251–258. [Google Scholar]
- Schwiedernoch, R.; Tischer, S.; Correa, C.; Deutschmann, O. Experimental and Numerical Study on the Transient Behavior of Partial Oxidation of Methane in a Catalytic Monolith. Chem. Eng. Sci. 2003, 58, 633–642. [Google Scholar] [CrossRef]
- Williams, K.A.; Leclerc, C.A.; Schmidt, L.D. Rapid Lightoff of Syngas Production from Methane: A Transient Product Analysis. AIChE J. 2005, 51, 247–260. [Google Scholar] [CrossRef]
- Karakaya, C.; Maier, L.; Deutschmann, O. Surface Reaction Kinetics of the Oxidation and Reforming of CH4 over Rh/Al2O3 Catalysts. Int. J. Chem. Kinet. 2016, 48, 144–160. [Google Scholar] [CrossRef]
- Deshmukh, S.R.; Vlachos, D.G. A Reduced Mechanism for Methane and One-Step Rate Expressions for Fuel-Lean Catalytic Combustion of Small Alkanes on Noble Metals. Combust. Flame 2007, 149, 366–383. [Google Scholar] [CrossRef]
- Mhadeshwar, A.B.; Vlachos, D.G. Hierarchical Multiscale Mechanism Development for Methane Partial Oxidation and Reforming and for Thermal Decomposition of Oxygenates on Rh. J. Phys. Chem. B 2005, 109, 16819–16835. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, N.; Nicolle, A.; Berthout, D.; Da Costa, P. Kinetic Modeling Study of the Oxidation of Carbon Monoxide–Hydrogen Mixtures over Pt/Al2O3 and Rh/Al2O3 Catalysts. J. Phys. Chem. C 2011, 115, 20225–20236. [Google Scholar] [CrossRef]
- Boll, W. Korrelation Zwischen Umsatzverhalten Und Katalytischer Oberfläche von Dieseloxidationskatalysatoren Unter Variation von Beladung Und Alterungszustand. Ph.D. Thesis, Karlsruhe Institute of Technology, Karlsruhe, Germany, 2011. [Google Scholar]
- Cai, Y.; Stenger, H.G., Jr.; Lyman, C.E. Catalytic CO Oxidation over Pt–Rh/γ-Al2O3Catalysts. J. Catal. 1996, 161, 123–131. [Google Scholar] [CrossRef]
- Ito, S.I.; Fujimori, T.; Nagashima, K.; Yuzaki, K.; Kunimori, K. Strong Rhodium–Niobia Interaction in Rh/Nb2O5, Nb2O5–Rh/SiO2 and RhNbO4/SiO2 Catalysts: Application to Selective CO Oxidation and CO Hydrogenation. Catal. Today 2000, 57, 247–254. [Google Scholar] [CrossRef]
- Zum Mallen, M.P.; Williams, W.R.; Schmidt, L.D. Steps in Hydrogen Oxidation on Rhodium: Hydroxyl Desorption at High Temperatures. J. Phys. Chem. 1993, 97, 625–632. [Google Scholar] [CrossRef]
- Hickman, D.A.; Schmidt, L.D. Steps in CH4 Oxidation on Pt and Rh Surfaces: High-Temperature Reactor Simulations. AIChE J. 1993, 39, 1164–1177. [Google Scholar] [CrossRef]
- Appel, C.; Mantzaras, J.; Schaeren, R.; Bombach, R.; Inauen, A.; Tylli, N.; Wolf, M.; Griffin, T.; Winkler, D.; Carroni, R. Partial Catalytic Oxidation of Methane to Synthesis Gas over Rhodium: In Situ Raman Experiments and Detailed Simulations. Proc. Combust. Inst. 2005, 30, 2509–2517. [Google Scholar] [CrossRef]
- Sui, R.; Mantzaras, J.; Bombach, R. A Comparative Experimental and Numerical Investigation of the Heterogeneous and Homogeneous Combustion Characteristics of Fuel-Rich Methane Mixtures over Rhodium and Platinum. Proc. Combust. Inst. 2017, 36, 4313–4320. [Google Scholar] [CrossRef]
- Sui, R.; Mantzaras, J.; Bombach, R.; Denisov, A. Hetero-/Homogeneous Combustion of Fuel-Lean Methane/Oxygen/Nitrogen Mixtures over Rhodium at Pressures up to 12 bar. Proc. Combust. Inst. 2017, 36, 4321–4328. [Google Scholar] [CrossRef]
- Maestri, M.; Beretta, A.; Faravelli, T.; Groppi, G.; Tronconi, E.; Vlachos, D.G. Two-Dimensional Detailed Modeling of Fuel-Rich H2 Combustion over Rh/Al2O3 Catalyst. Chem. Eng. Sci. 2008, 63, 2657–2669. [Google Scholar] [CrossRef]
- Karakaya, C.; Deutschmann, O. Kinetics of Hydrogen Oxidation on Rh/Al2O3 Catalysts Studied in a Stagnation-Flow Reactor. Chem. Eng. Sci. 2013, 89, 171–184. [Google Scholar] [CrossRef]
- Karadeniz, H.; Karakaya, C.; Tischer, S.; Deutschmann, O. Numerical Modeling of Stagnation-Flows on Porous Catalytic Surfaces: CO Oxidation on Rh/Al2O3. Chem. Eng. Sci. 2013, 104, 899–907. [Google Scholar] [CrossRef]
- Pery, T.; Schweitzer, M.G.; Volpp, H.R.; Wolfrum, J.; Ciossu, L.; Deutschmann, O.; Warnatz, J. Sum-Frequency Generation in Situ Study of CO Adsorption and Catalytic CO Oxidation on Rhodium at Elevated Pressures. Proc. Combust. Inst. 2002, 29, 973–980. [Google Scholar] [CrossRef]
- Karakaya, C.; Otterstätter, R.; Maier, L.; Deutschmann, O. Kinetics of the Water-Gas Shift Reaction over Rh/Al2O3 Catalysts. Appl. Catal. A Gen. 2014, 470, 31–44. [Google Scholar] [CrossRef]
- McGuire, N.E.; Sullivan, N.P.; Deutschmann, O.; Zhu, H.; Kee, R.J. Dry Reforming of Methane in a Stagnation-Flow Reactor Using Rh Supported on Strontium-Substituted Hexaaluminate. Appl. Catal. A Gen. 2011, 394, 257–265. [Google Scholar] [CrossRef]
- Behrendt, F.; Deutschmann, O.; Maas, U.; Warnatz, J. Simulation and Sensitivity Analysis of the Heterogeneous Oxidation of Methane on a Platinum Foil. J. Vac. Sci. Technol. A 1995, 13, 1373–1377. [Google Scholar] [CrossRef]
- Kee, R.J.; Coltrin, M.E.; Glarborg, P. Chemically Reacting Flow: Theory and Practice; Wiley Interscience: Hoboken, NJ, USA, 2003. [Google Scholar]
- Tischer, S.; Correa, C.; Deutschmann, O. Transient Three-Dimensional Simulations of a Catalytic Combustion Monolith Using Detailed Models for Heterogeneous and Homogeneous Reactions and Transport Phenomena. Catal. Today 2001, 69, 57–62. [Google Scholar] [CrossRef]
- Tavazzi, I.; Beretta, A.; Groppi, G.; Forzatti, P. Development of a Molecular Kinetic Scheme for Methane Partial Oxidation over a Rh/α-Al2O3 Catalyst. J. Catal. 2006, 241, 1–13. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism | R | Features | Used in | |
|---|---|---|---|---|
| 1 | Zum Mallen–Schmidt 1993 [21] H2/O2; H2/H2O | 12 | high temperature H2-oxidation with partially noncompetitive adsorption of O2 | [21] |
| 2 | Hickman–Schmidt (1993) [22] CO-H2/O2; CH4/O2 | 19 | high temperature CO, H2-oxidation, CO-H2 coupling/ pyrolysis CH4 mechanism including a desorption of OH radical | [22] |
| 3 | Maier–Deutschmann (2001) [11] CO-H2/O2; CH4/O2/H2O | 38 | CO, H2-oxidation, CO-H2 coupling, WGS, CH4 oxidation and reforming [11]. Including coverage-dependent desorption energies for CO and O2: [12] | [11,12,23,24,25] |
| 4 | Karakaya–Deutschmann (2016) [14] CO-H2/O2; CH4/O2/H2O/CO2 | 48 | CO, H2-oxidation, CO-H2 coupling, WGS, CH4 oxidation and reforming / CO-H2 coupling via COOH | [14,25] |
| 5 | Mhadeshwar–Vlachos (2005) [16] CO-H2/O2 | 44 | CO, H2-oxidation, CO-H2 coupling/WGS via COOH and HCOO; activation energies are coverage-dependent and temperature-dependent | [15,16] |
| 6 | Deshmukh–Vlachos (2007) [15] CH4/O2 | 15 | CH4 oxidation/reduced Mhadeshwar et al. [16] model for fuel-lean methane catalytic combustion (no CO and H2 in products) | [15,25] |
| 7 | Maestri–Vlachos (2008) [26] H2/O2 | 18 | H2-oxidation/H2 sub-mechanism from Mhadeshwar et al. [16], activation energies are coverage- and temperature-dependent | [26] |
| 8 | Rankovic–Da Costa (2011) [17] CO-H2/O2 | 28 | CO, H2-oxidation, CO-H2 coupling, WGS/mechanism includes N2 and NOx chemistry | [17] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gossler, H.; Maier, L.; Angeli, S.; Tischer, S.; Deutschmann, O. CaRMeN: An Improved Computer-Aided Method for Developing Catalytic Reaction Mechanisms. Catalysts 2019, 9, 227. https://doi.org/10.3390/catal9030227
Gossler H, Maier L, Angeli S, Tischer S, Deutschmann O. CaRMeN: An Improved Computer-Aided Method for Developing Catalytic Reaction Mechanisms. Catalysts. 2019; 9(3):227. https://doi.org/10.3390/catal9030227
Chicago/Turabian StyleGossler, Hendrik, Lubow Maier, Sofia Angeli, Steffen Tischer, and Olaf Deutschmann. 2019. "CaRMeN: An Improved Computer-Aided Method for Developing Catalytic Reaction Mechanisms" Catalysts 9, no. 3: 227. https://doi.org/10.3390/catal9030227
APA StyleGossler, H., Maier, L., Angeli, S., Tischer, S., & Deutschmann, O. (2019). CaRMeN: An Improved Computer-Aided Method for Developing Catalytic Reaction Mechanisms. Catalysts, 9(3), 227. https://doi.org/10.3390/catal9030227