Organocatalysis and Beyond: Activating Reactions with Two Catalytic Species

 ,
,  ,
, 
Abstract

{kind=link}
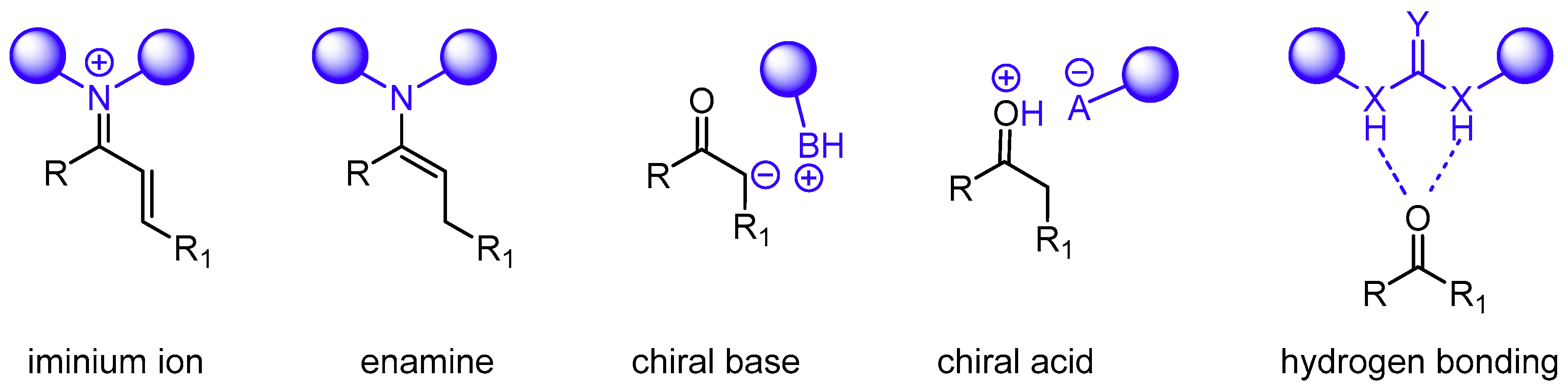{kind=link}
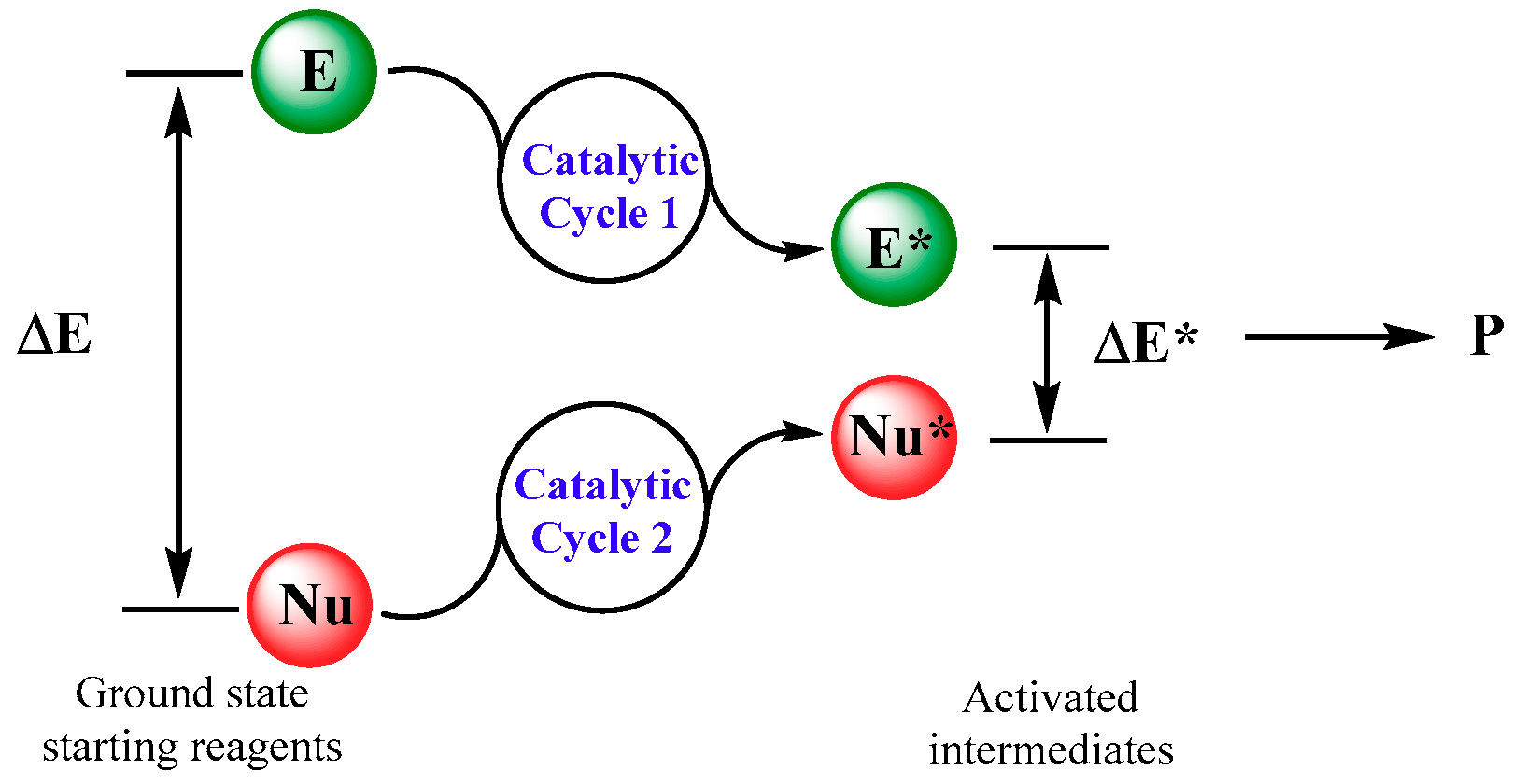{kind=link}
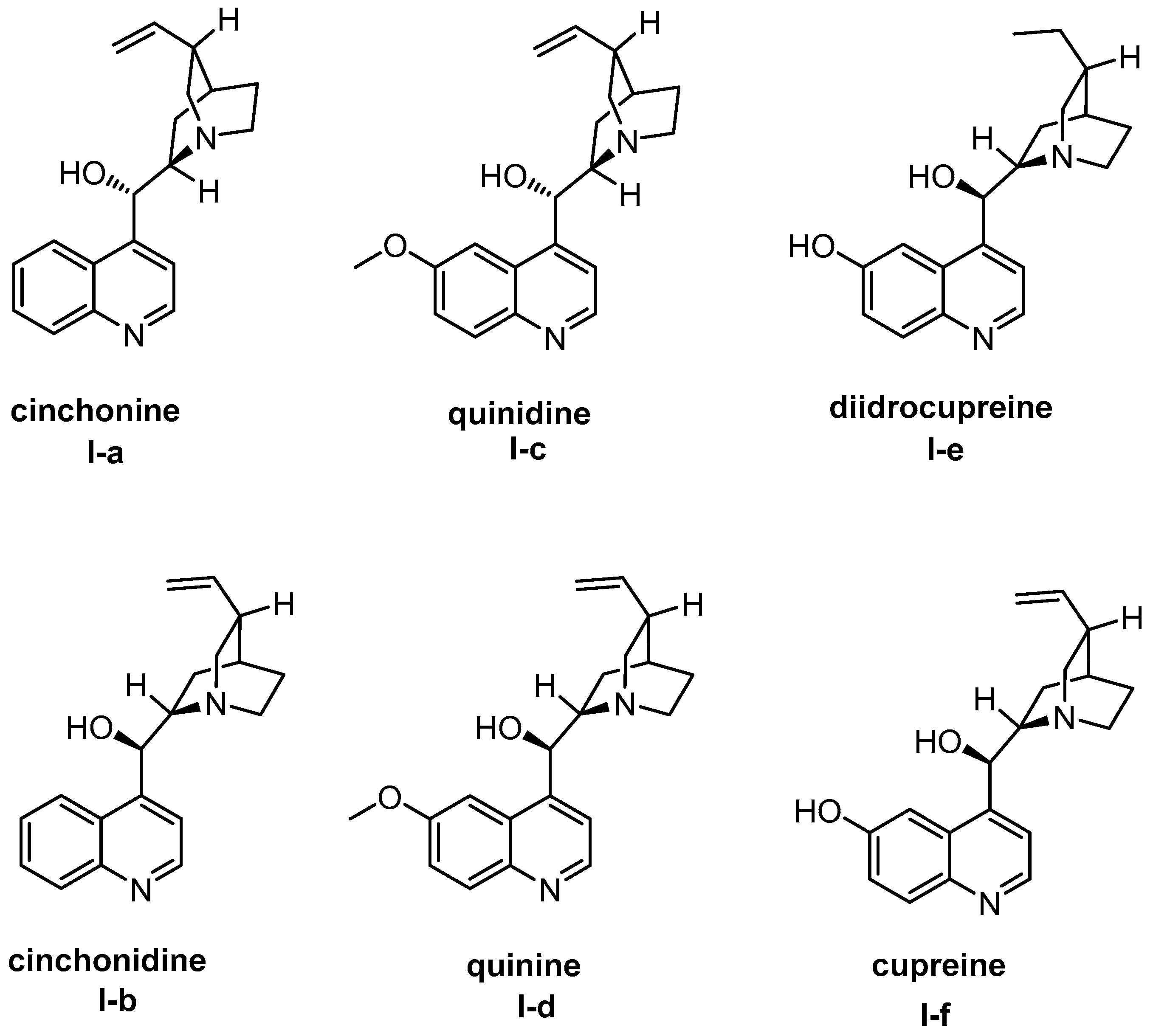{kind=link}
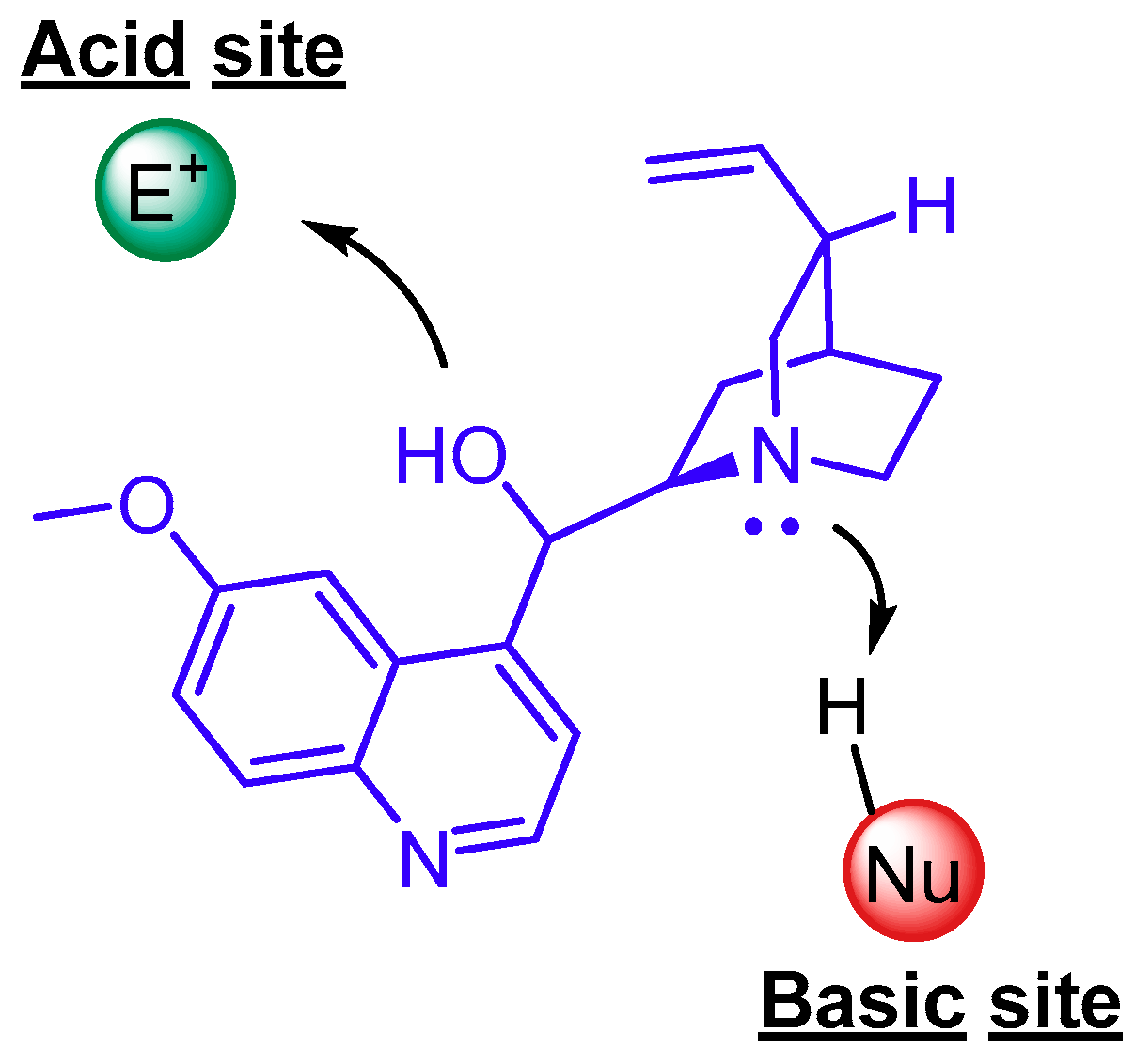{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
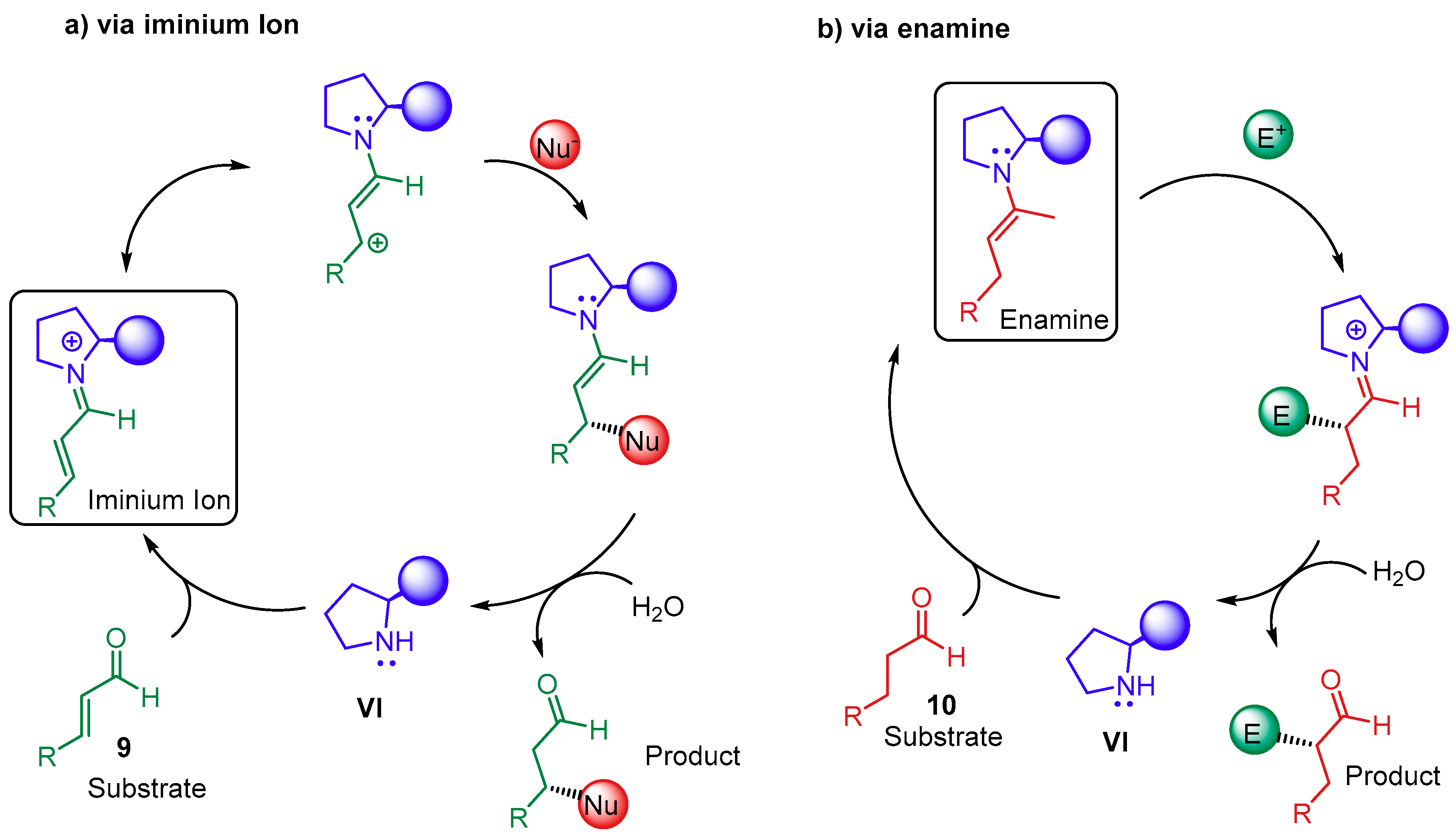{kind=link}
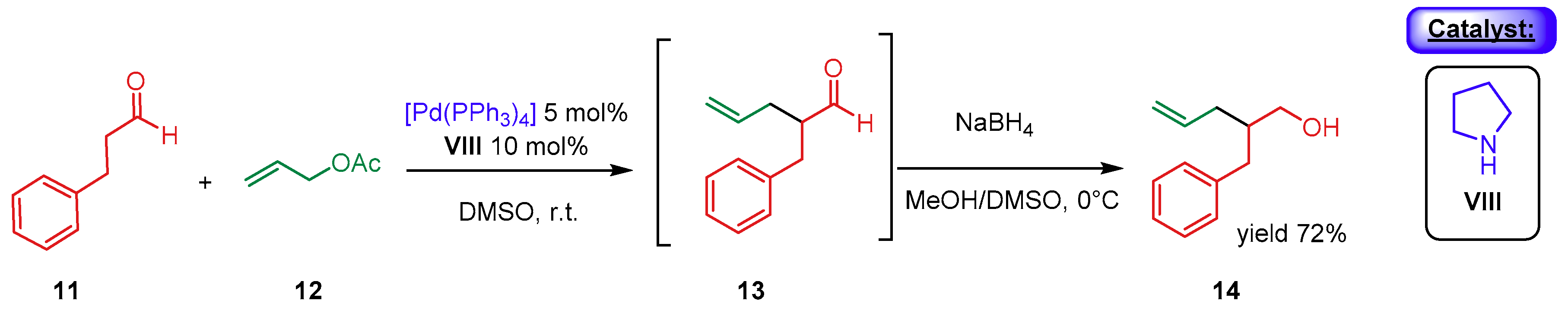{kind=link}
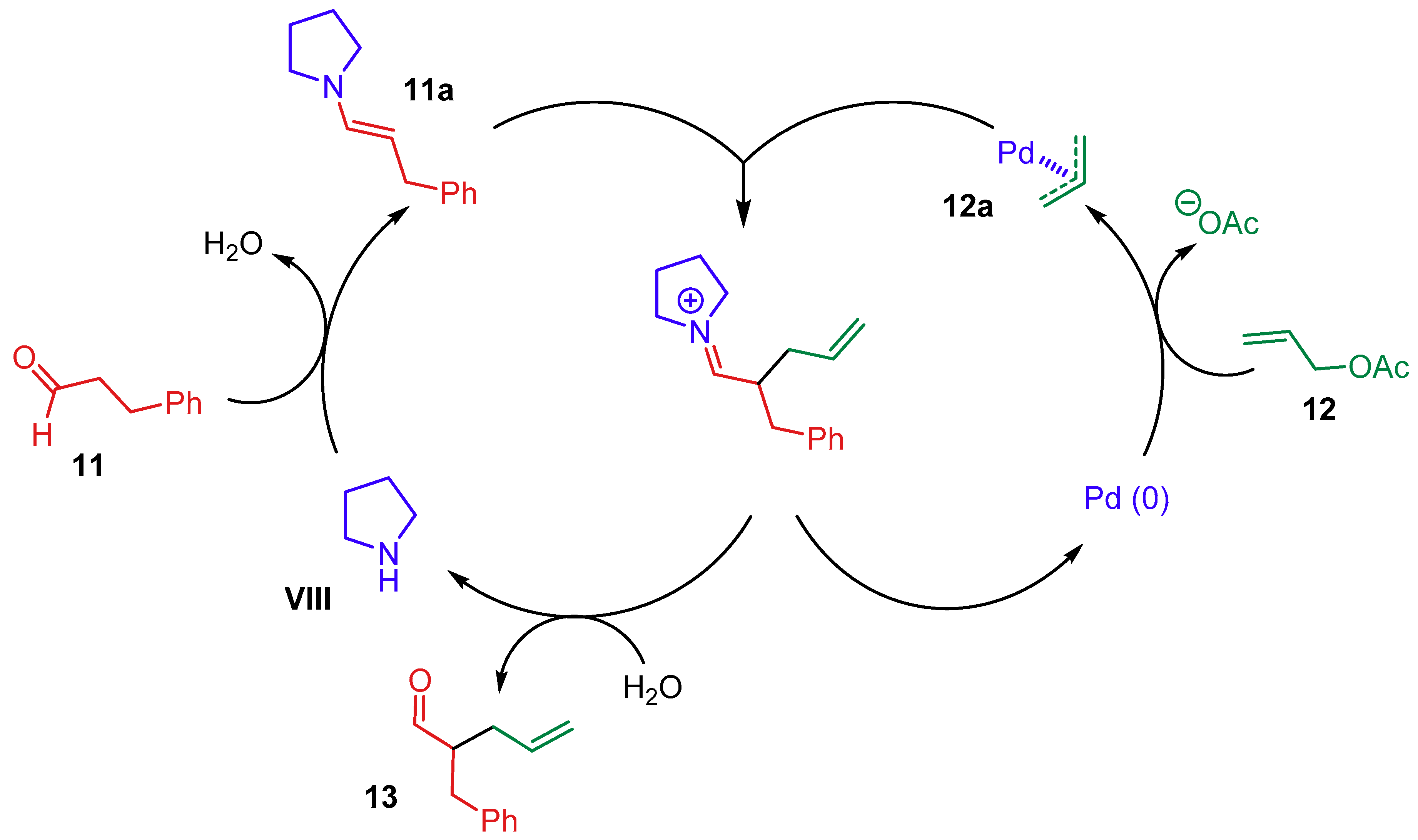{kind=link}
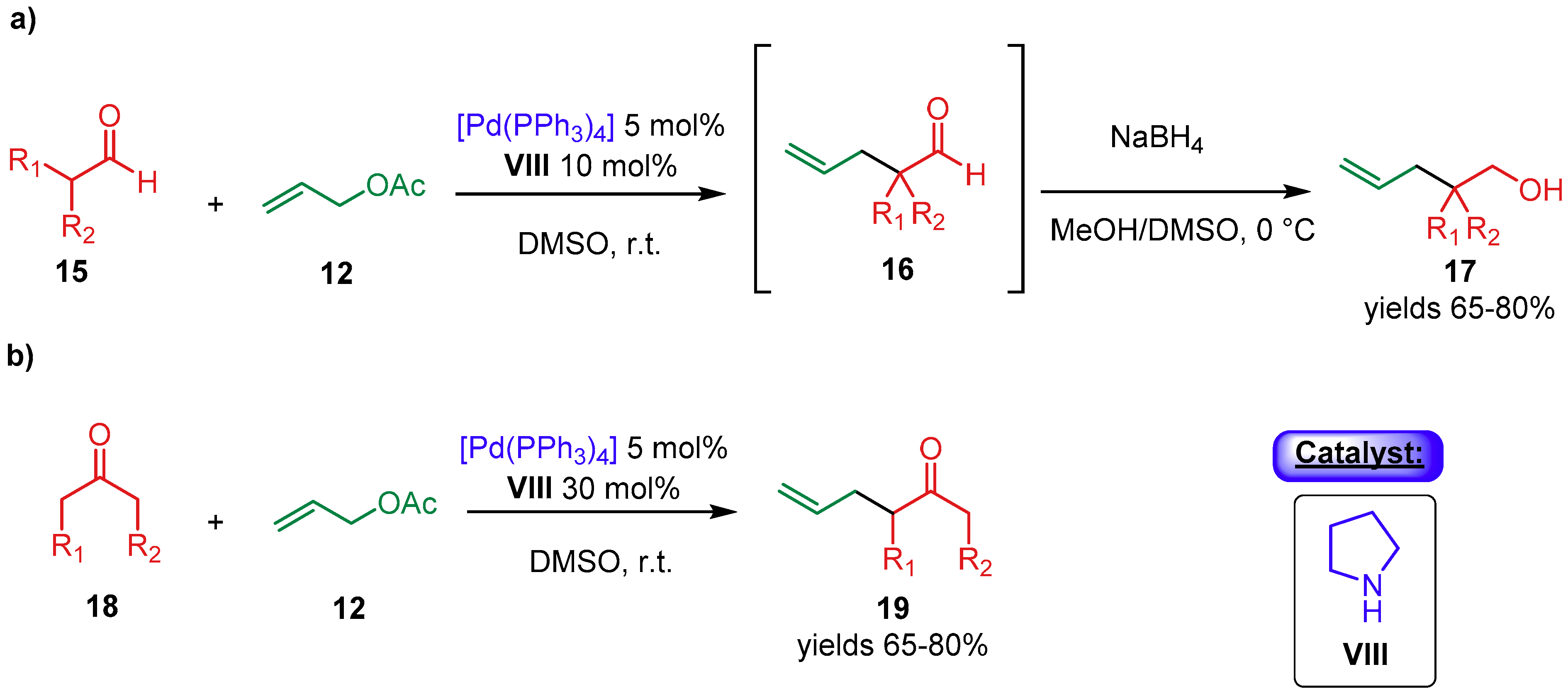{kind=link}
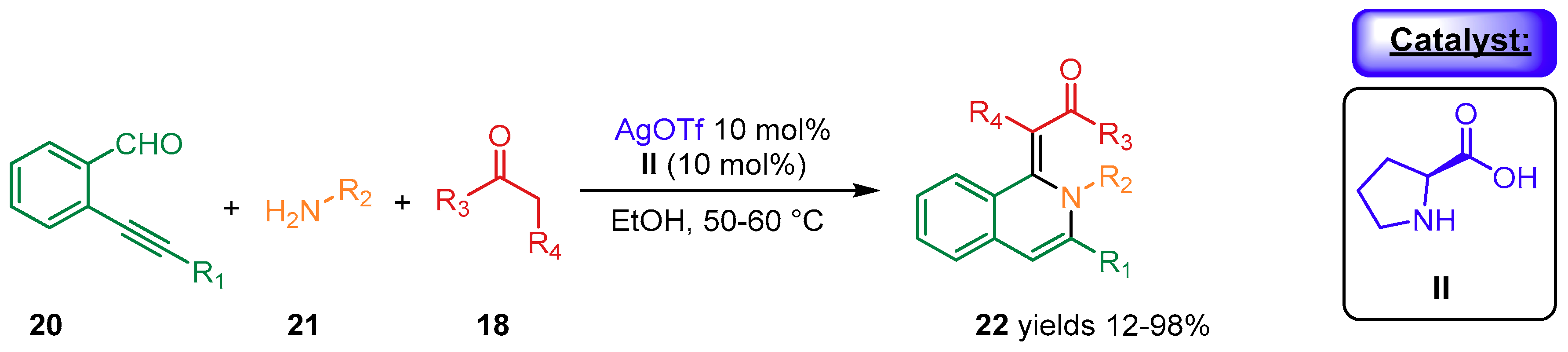{kind=link}
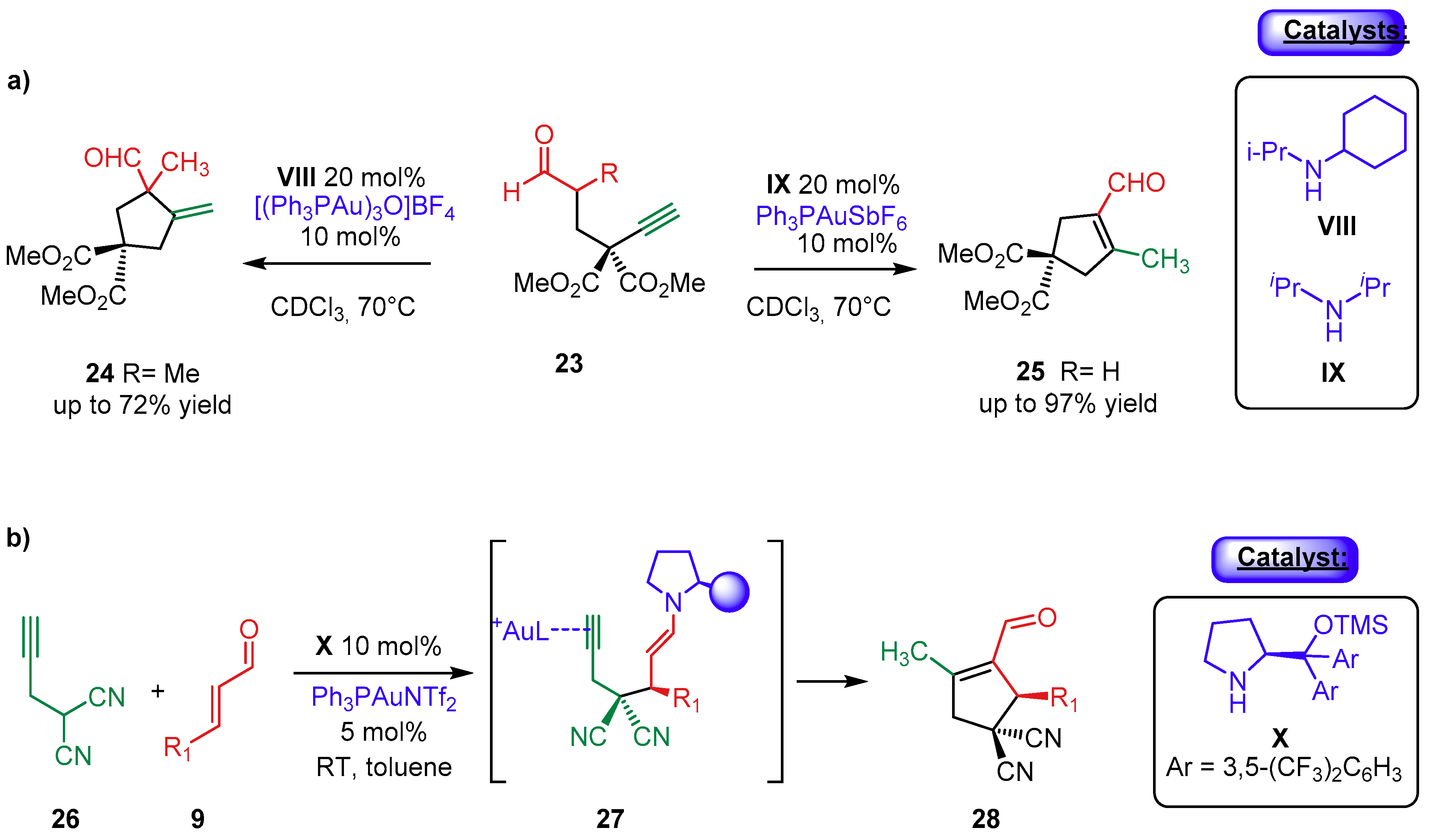{kind=link}
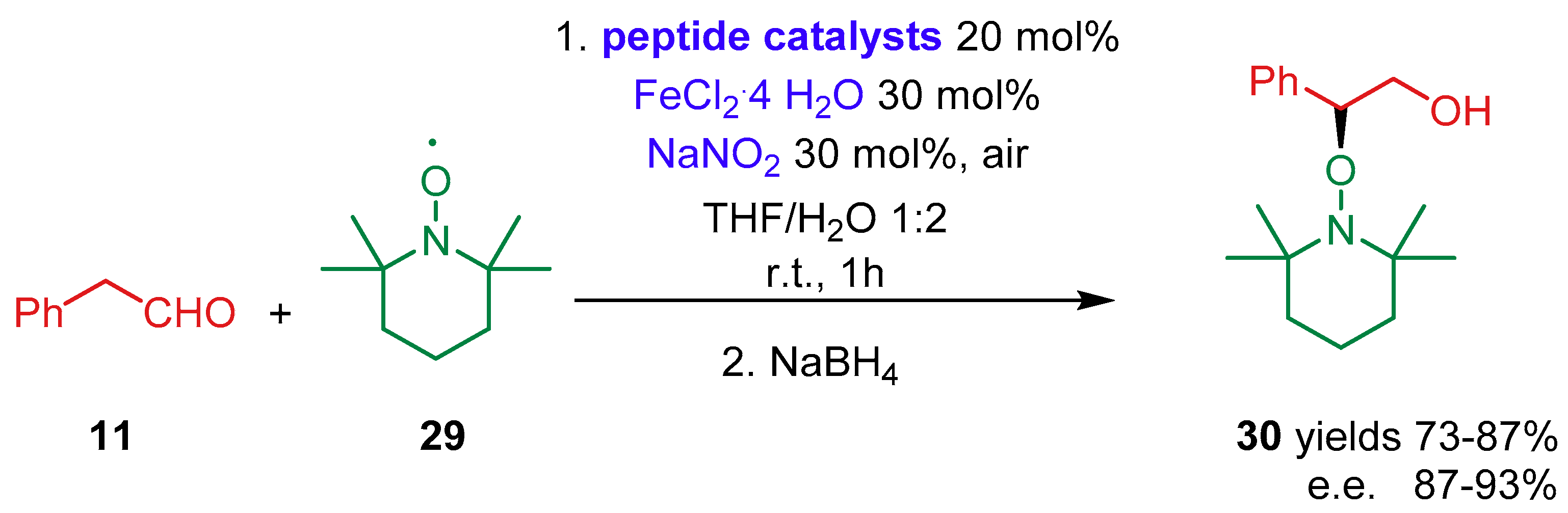{kind=link}
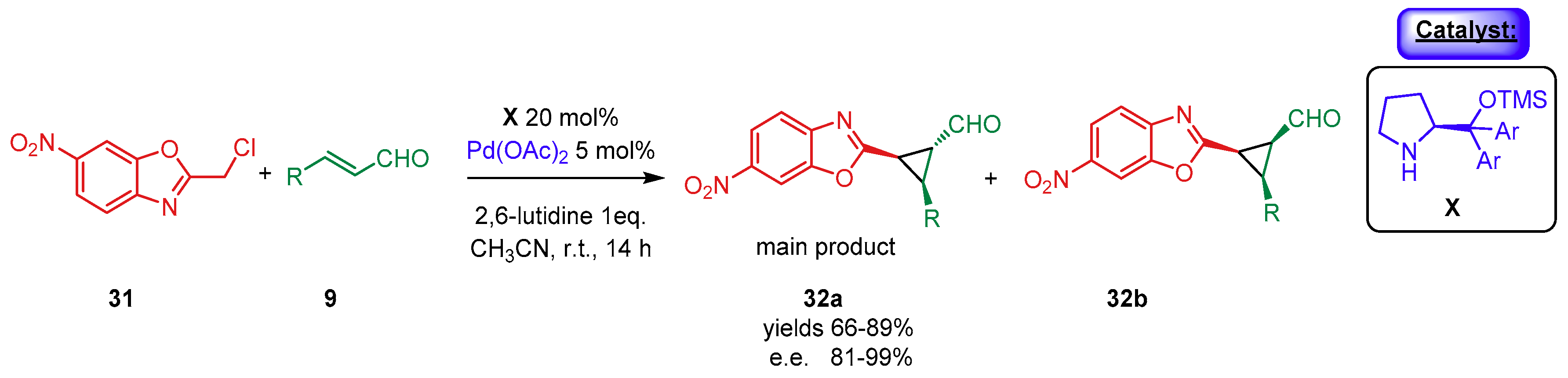{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
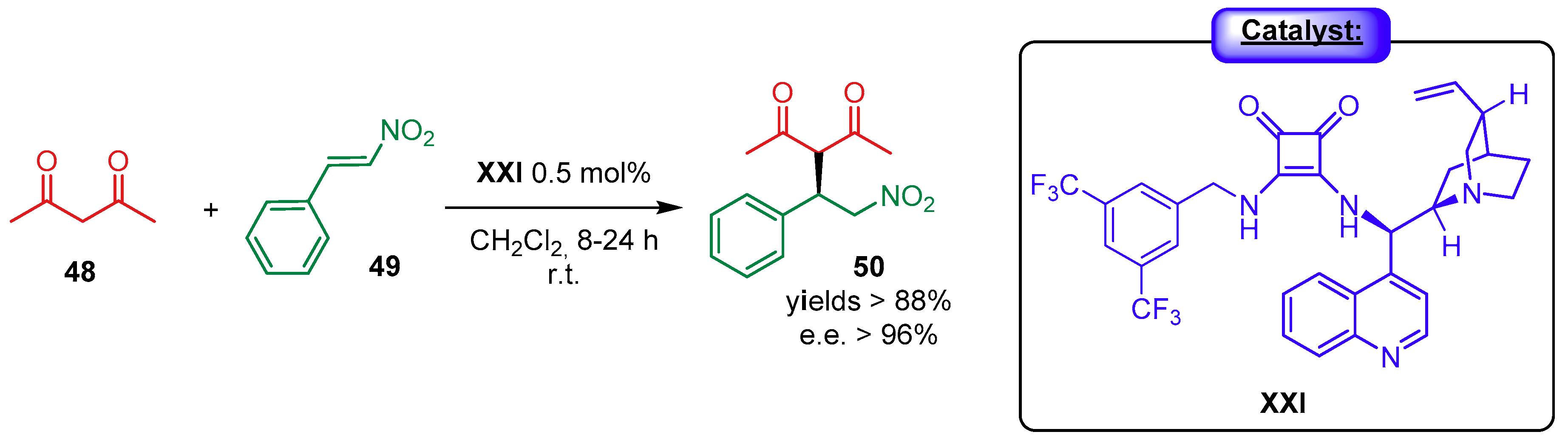{kind=link}
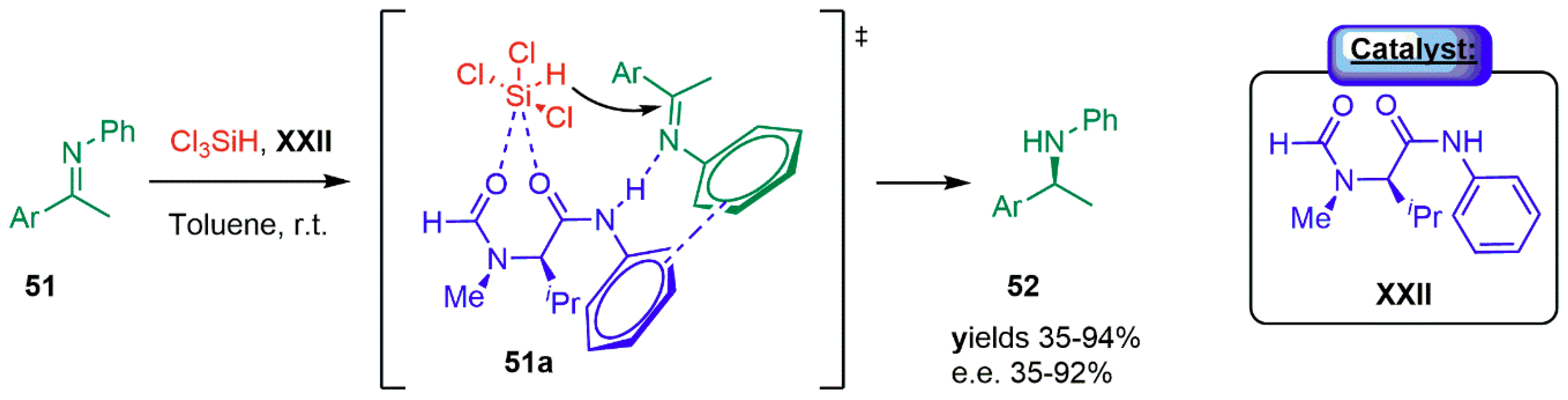{kind=link}
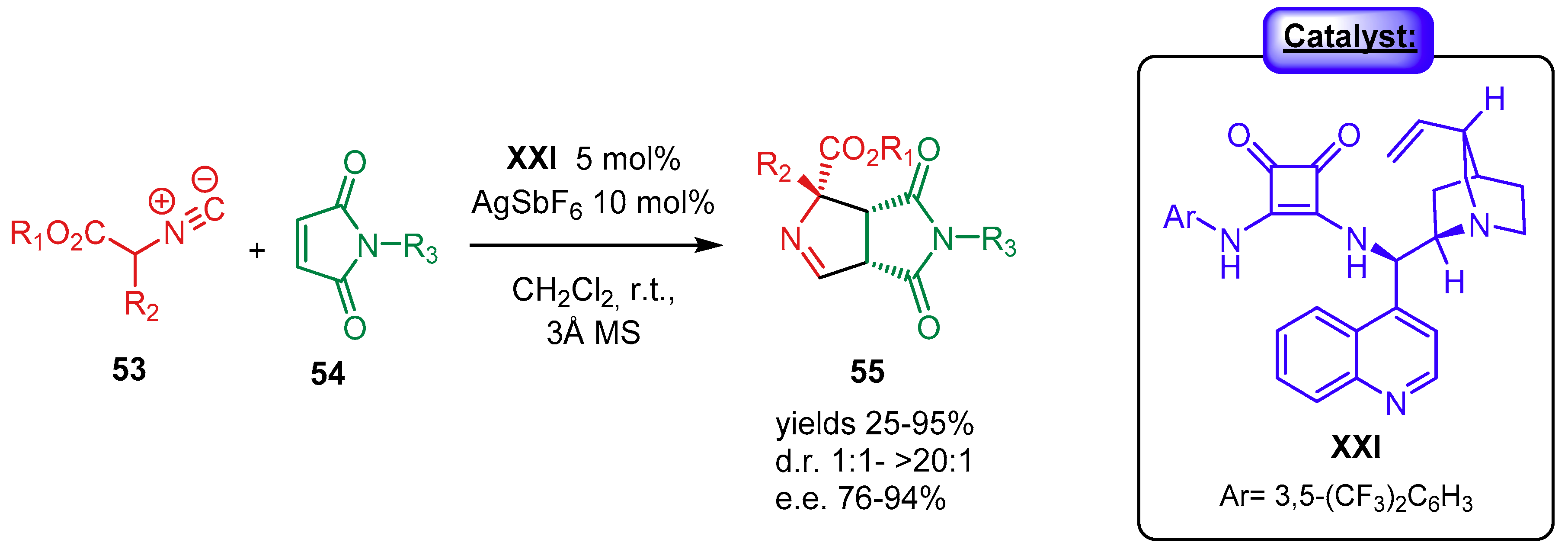{kind=link}
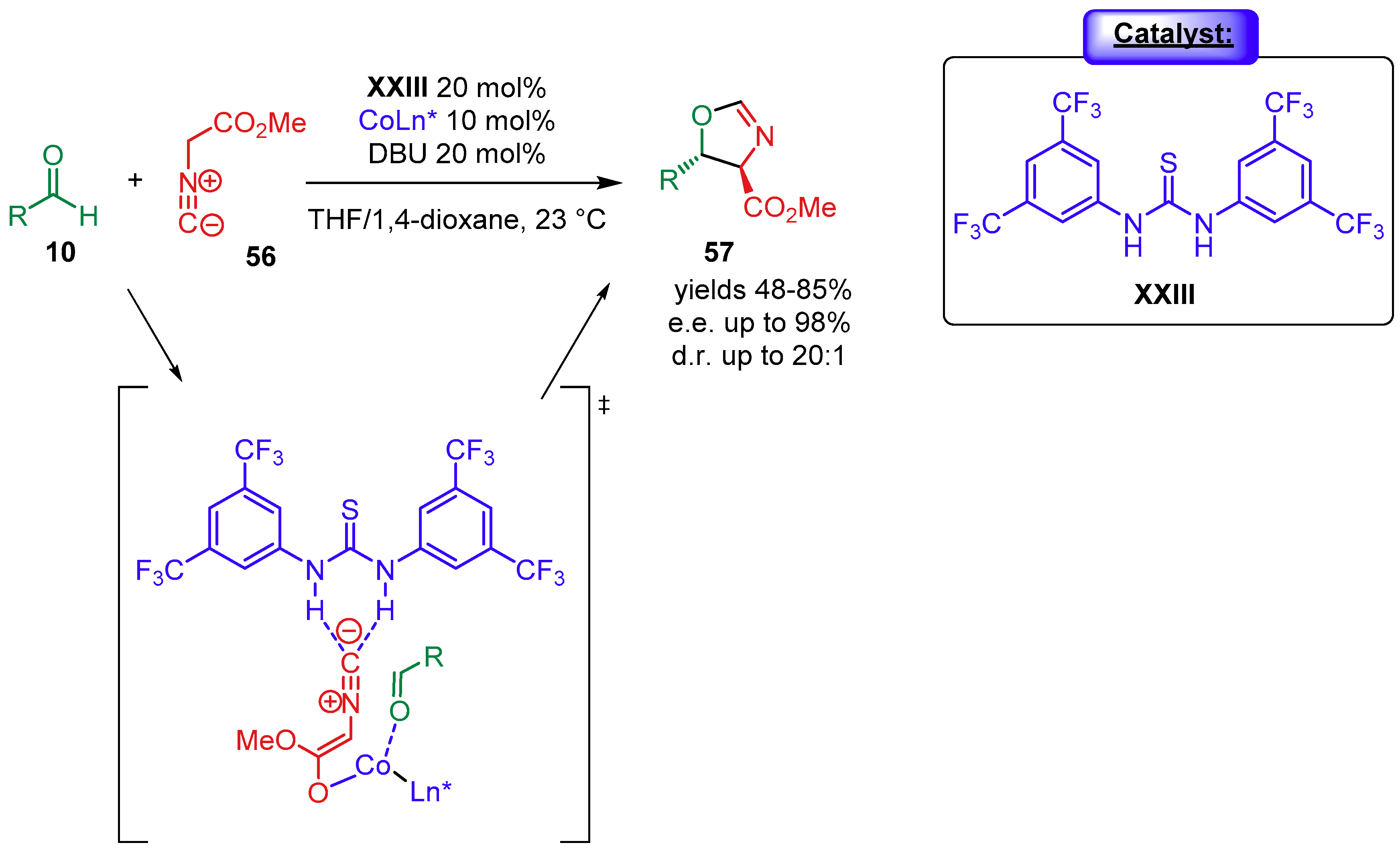{kind=link}
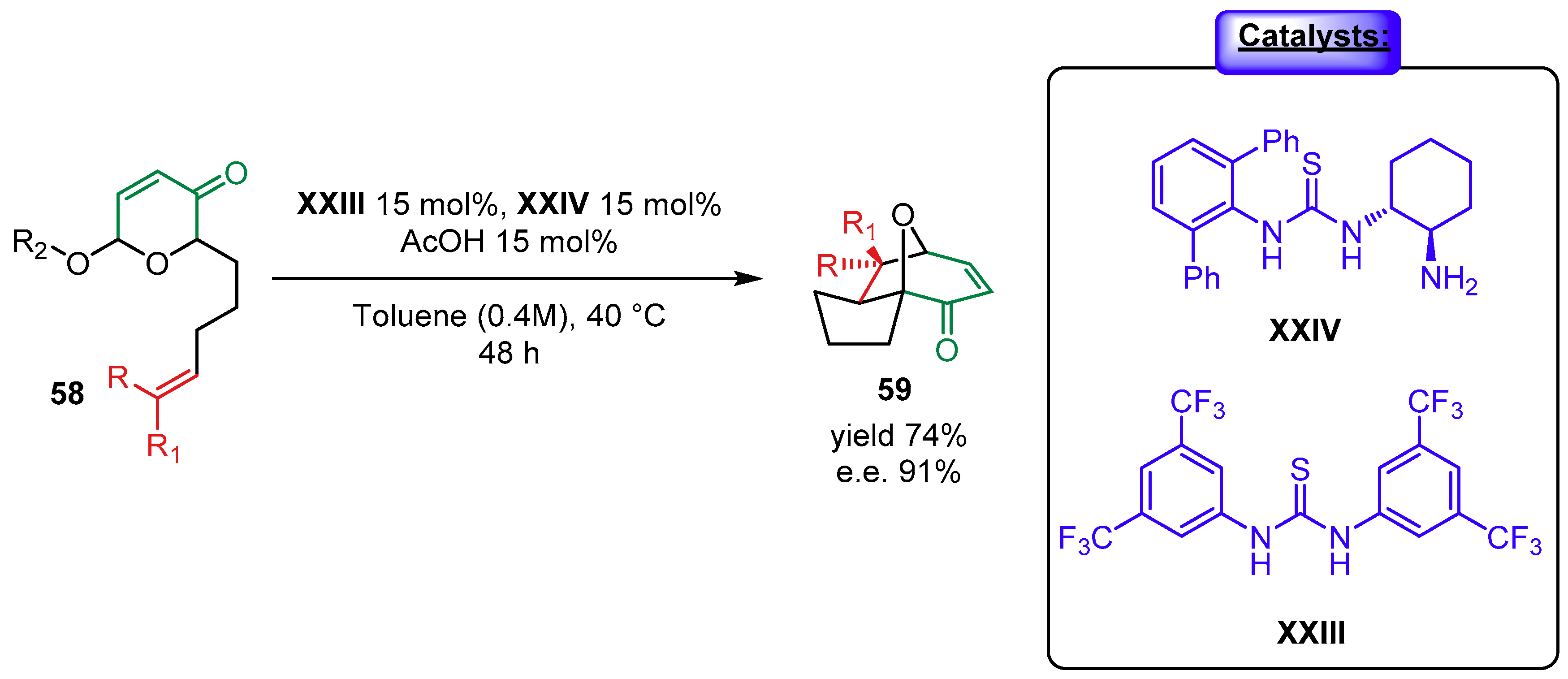{kind=link}
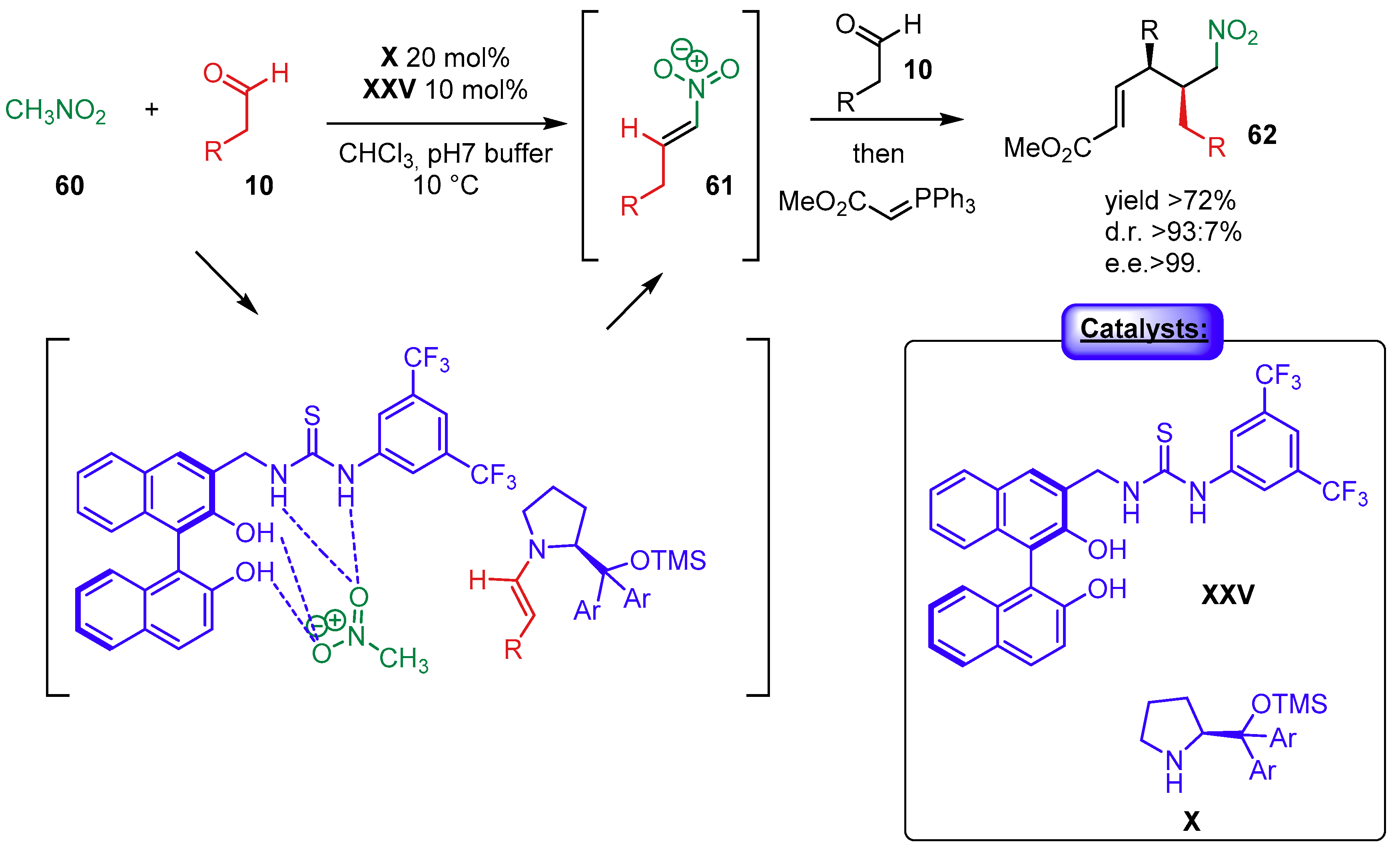{kind=link}
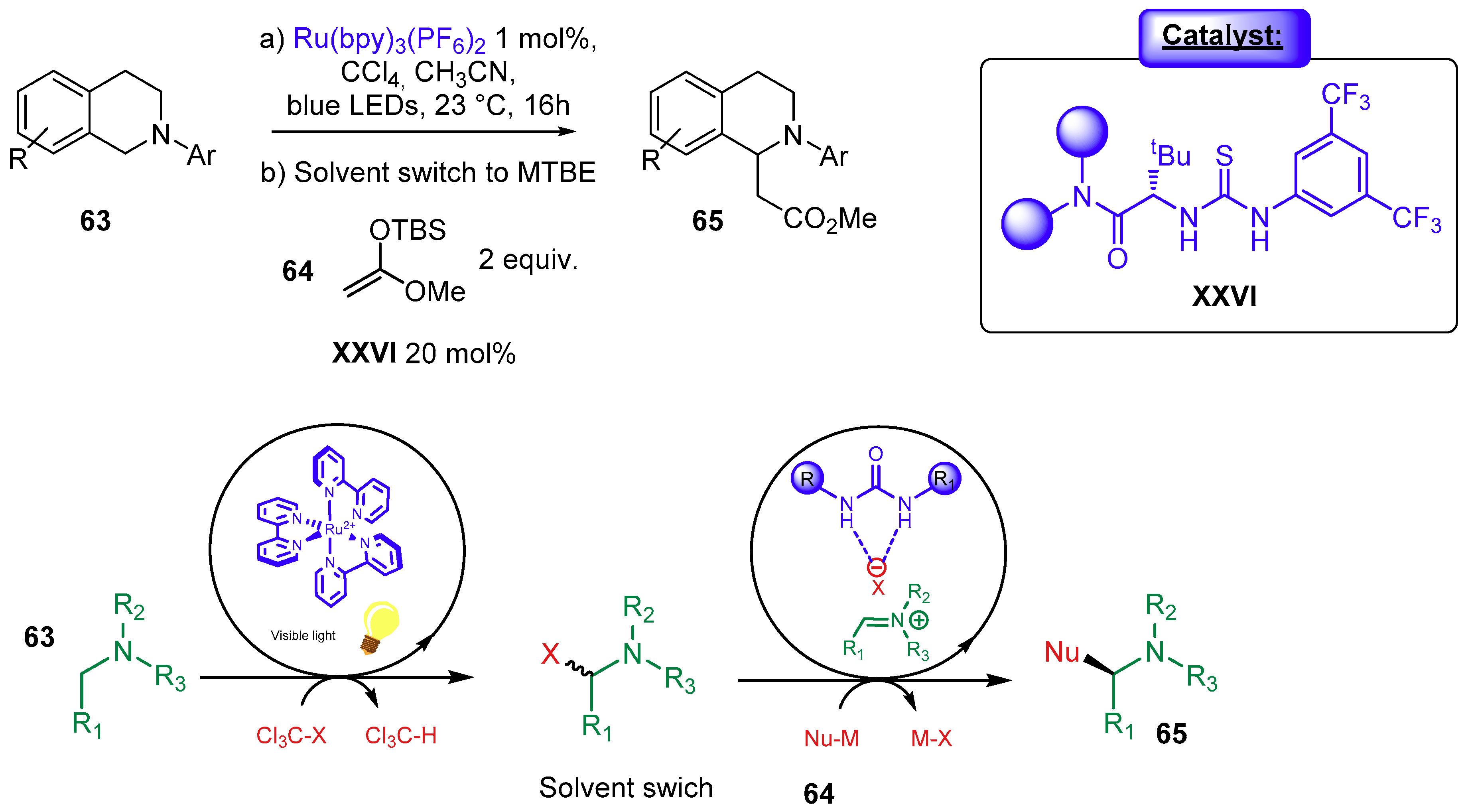{kind=link}
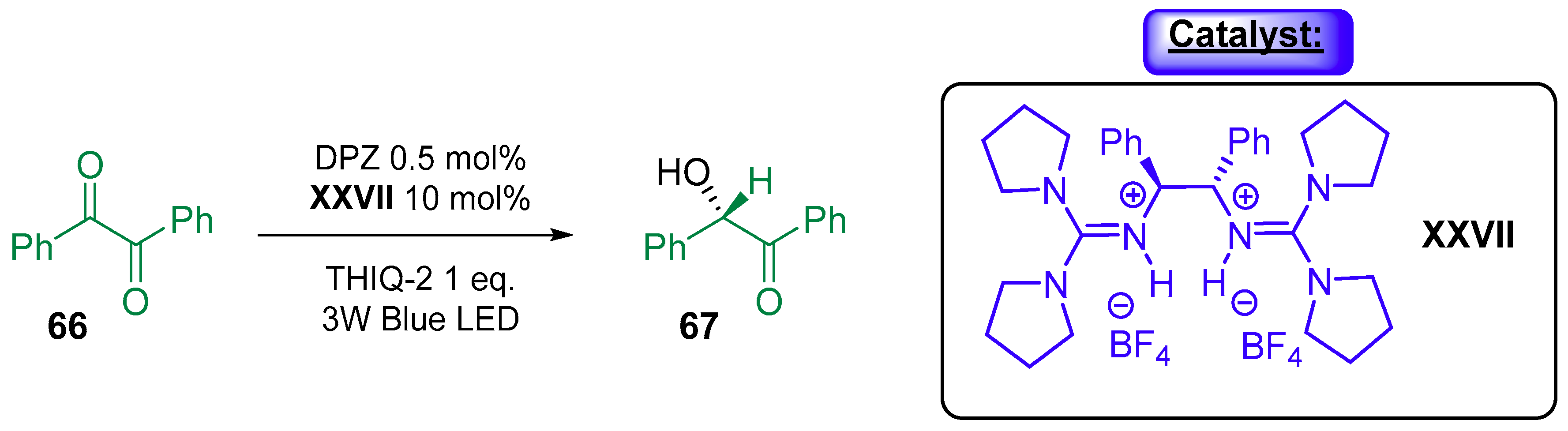{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
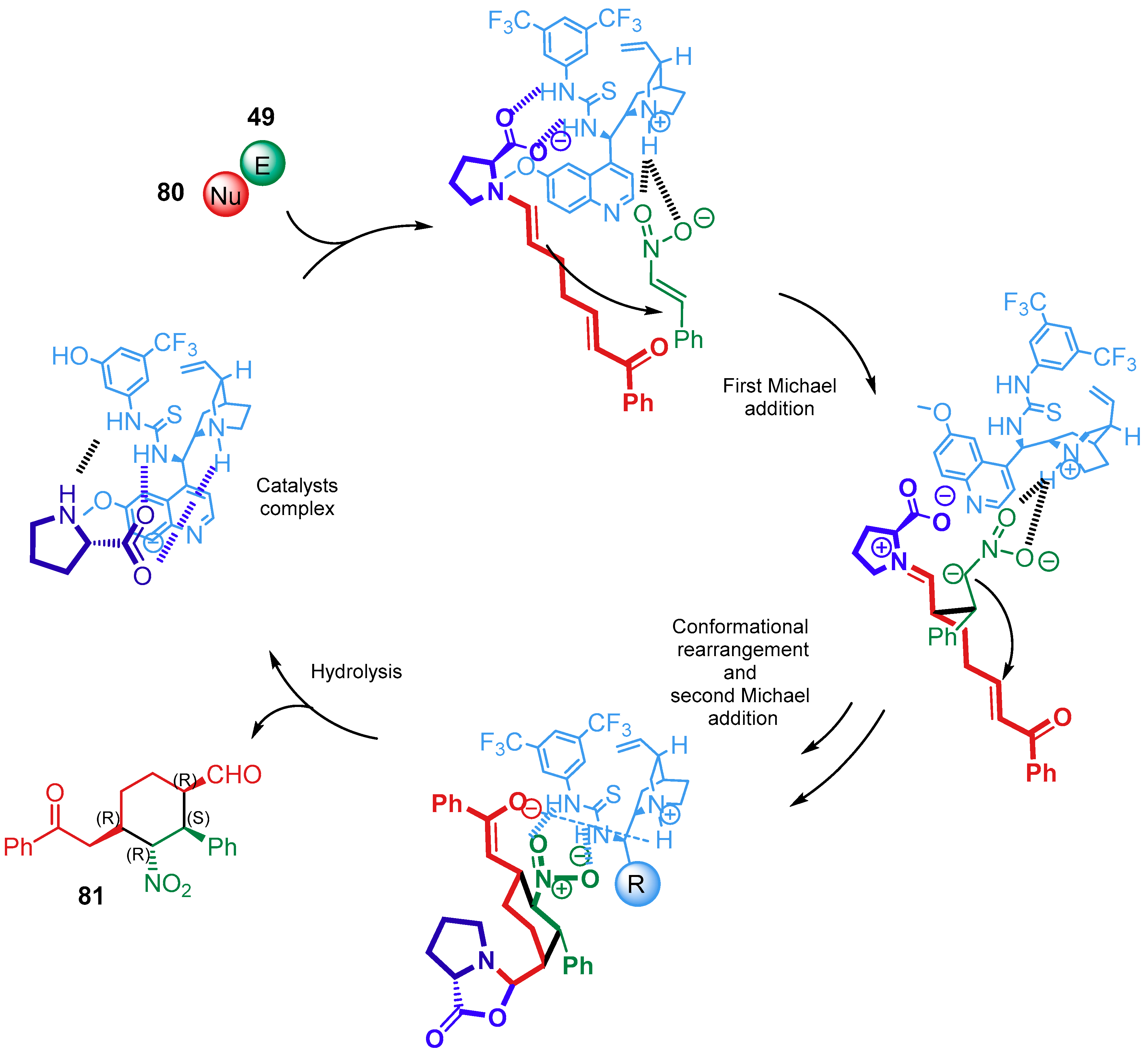{kind=link}
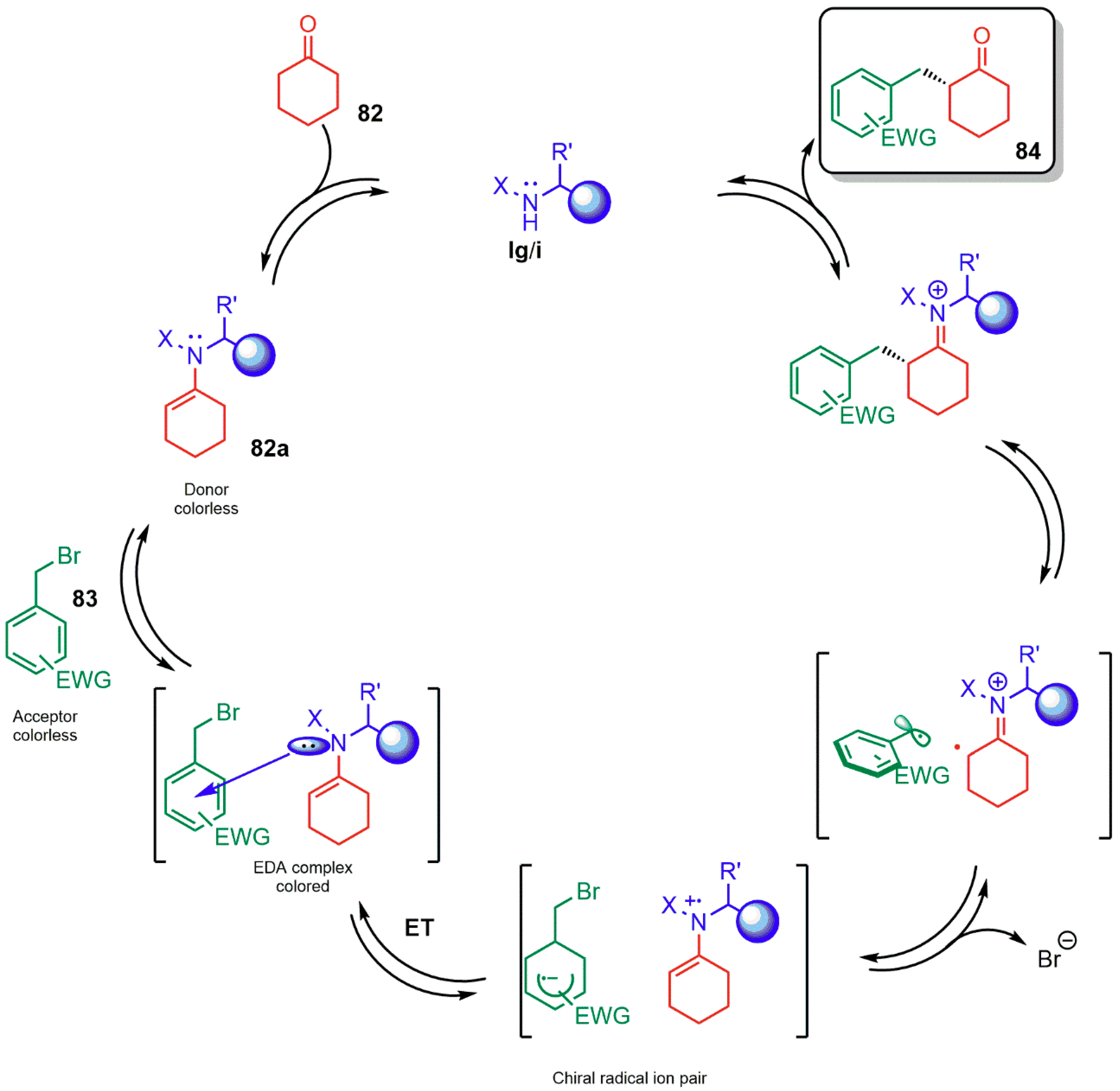{kind=link}
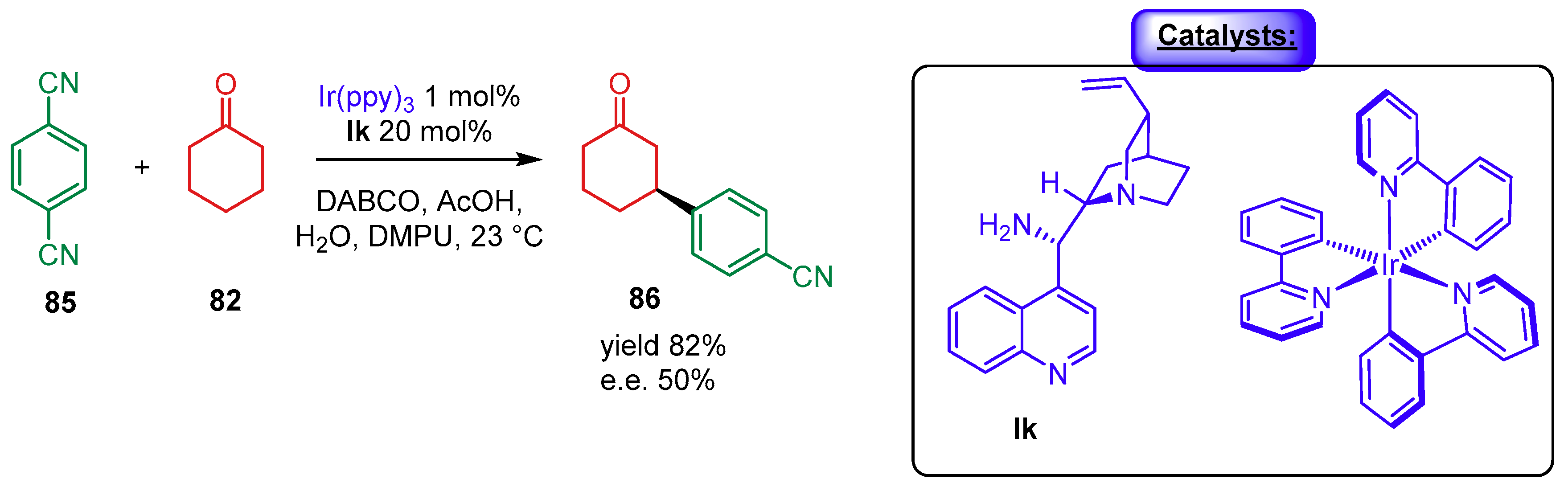{kind=link}
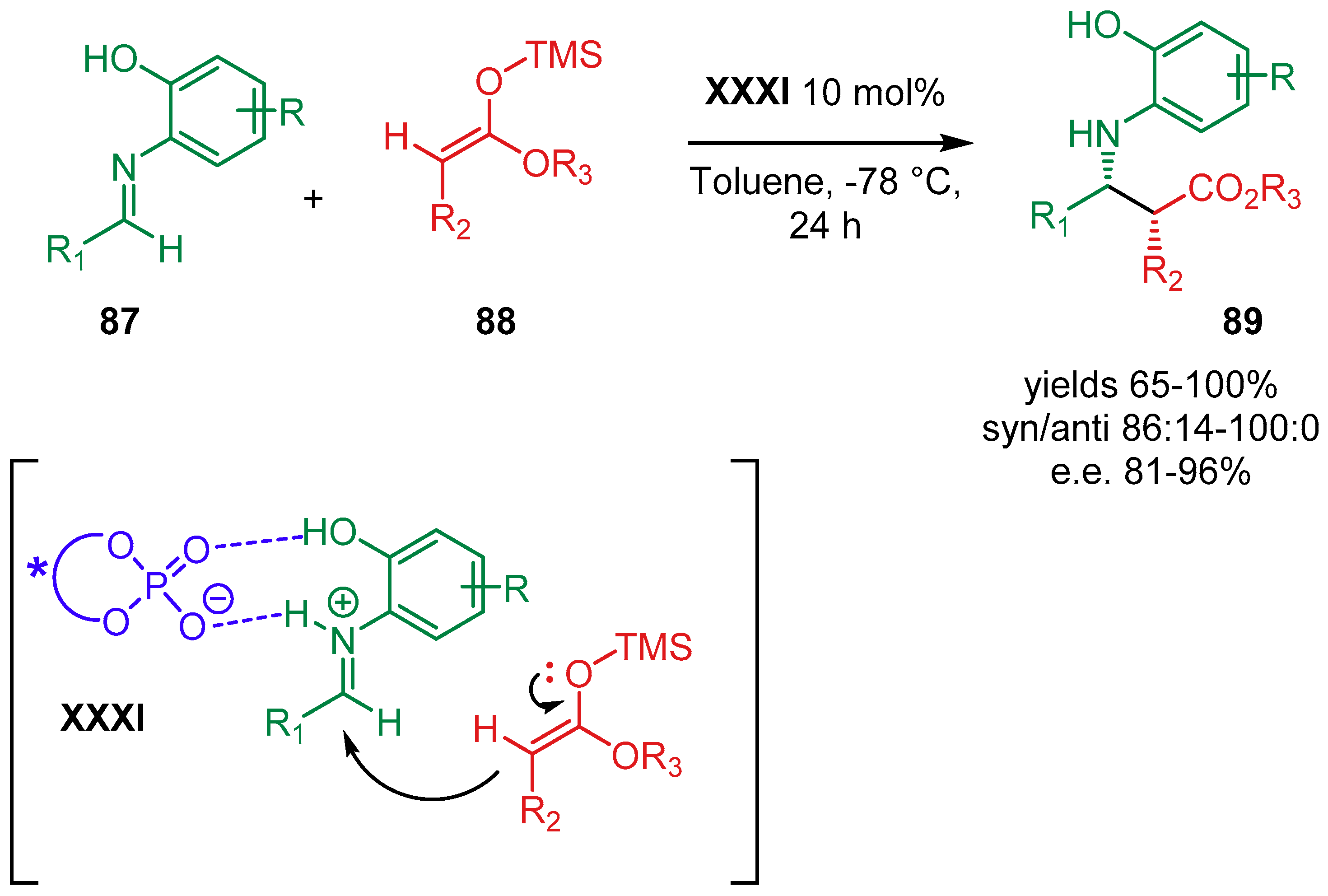{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
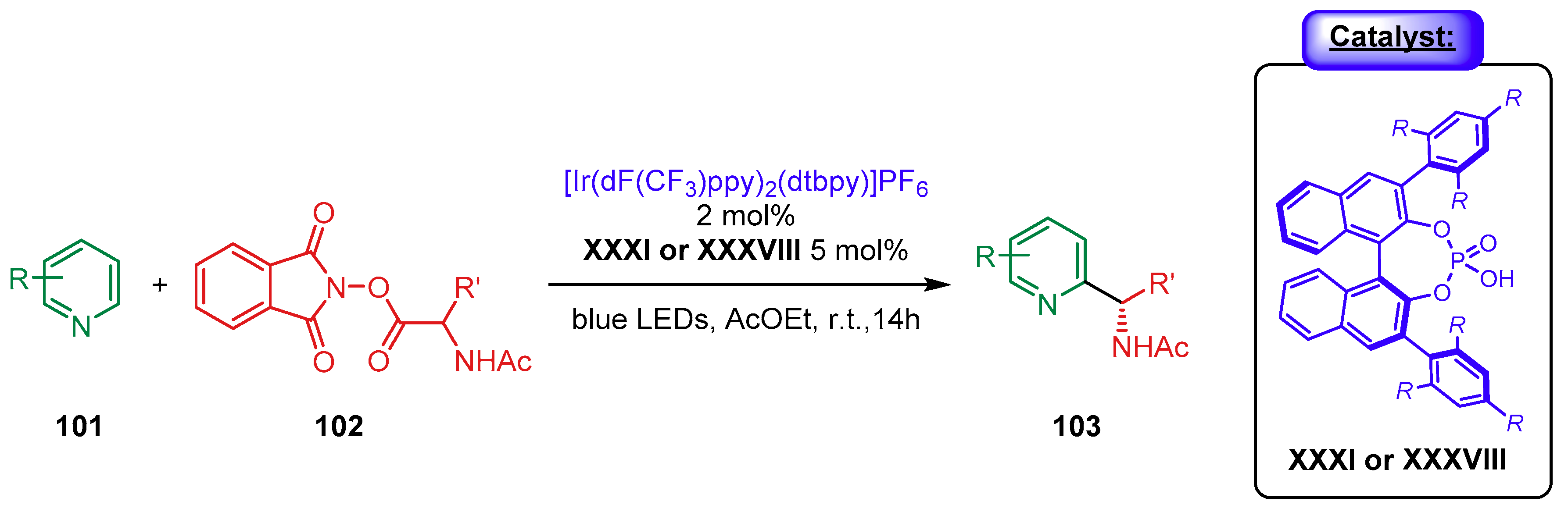{kind=link}
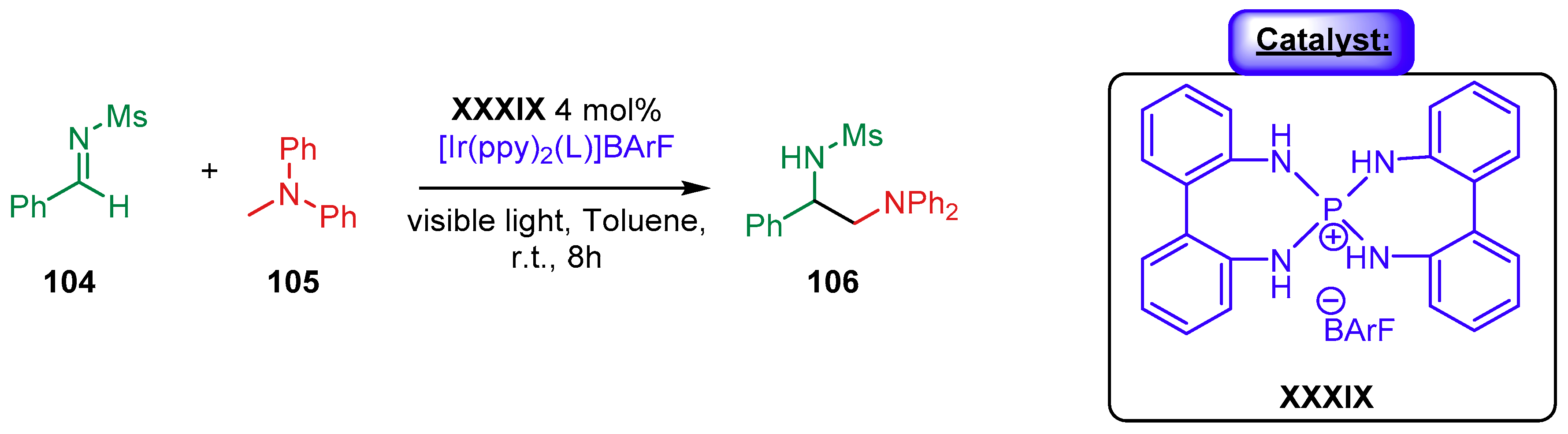{kind=link}
1. Introduction
2. Proline-Derivatives Catalysis
2.1. Synergistic Catalysis with Proline-Based Catalysts
2.1.1. Combination with Transition Metal-Based Catalysts
2.1.2. Combination with Other Organocatalysts
2.1.3. Combination with Photocatalysts
3. Hydrogen Bonding Catalysis
3.1. Synergistic Catalysis with Hydrogen Bonding Catalysts
3.1.1. Combination with Transition Metal-Based Catalysts
3.1.2. Combination with Other Organocatalysts
3.1.3. Combination with Photocatalyst
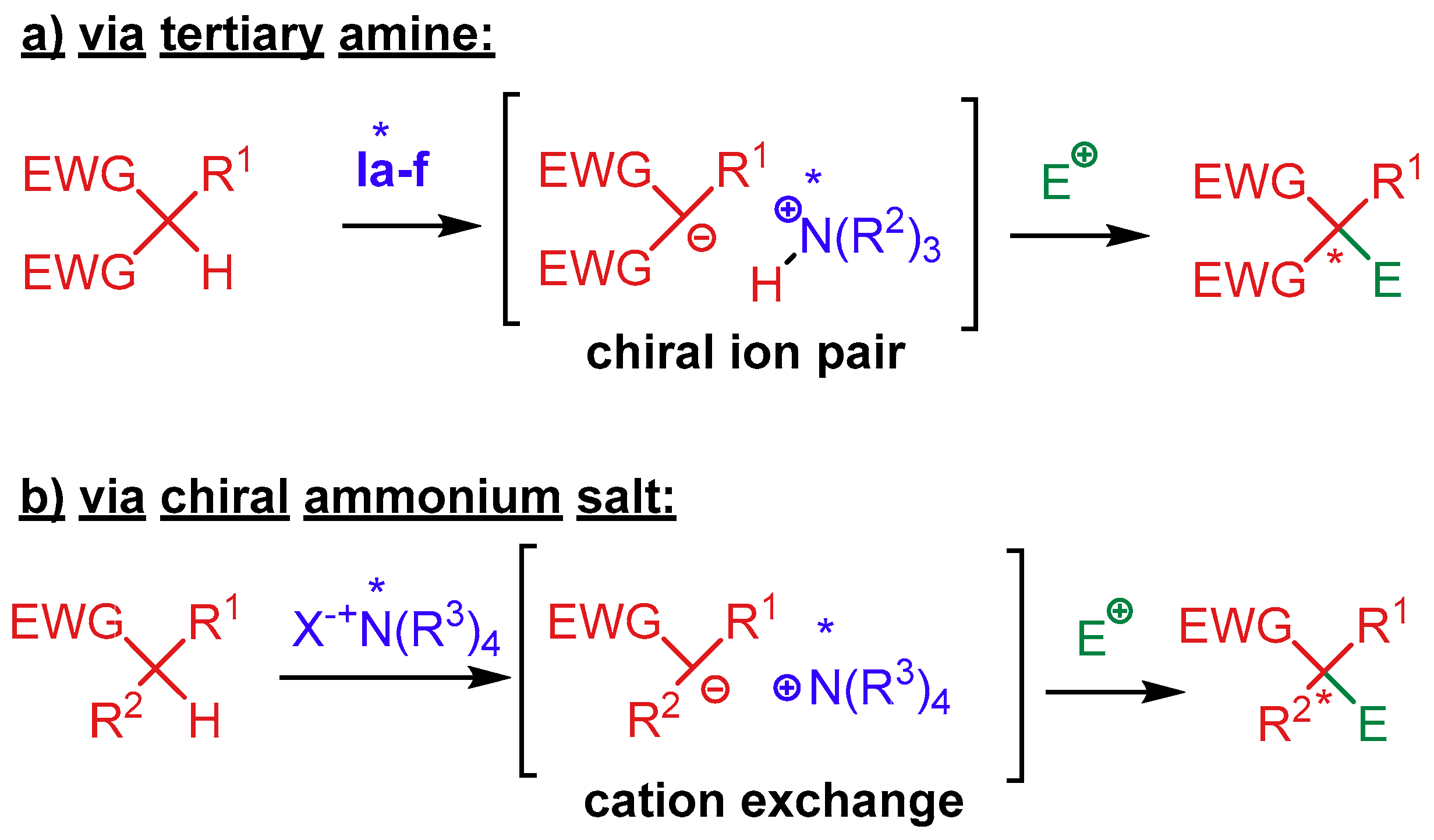
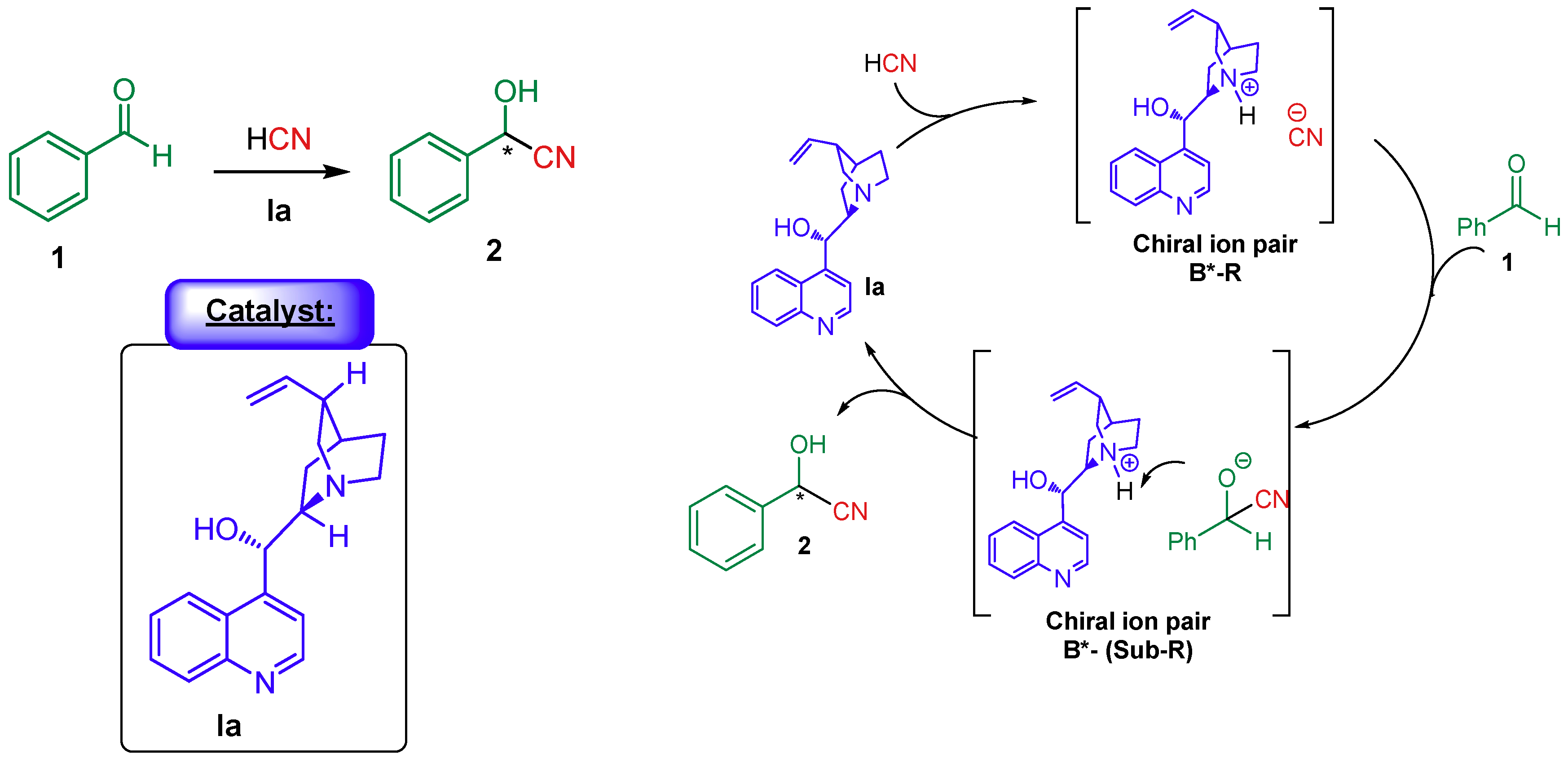
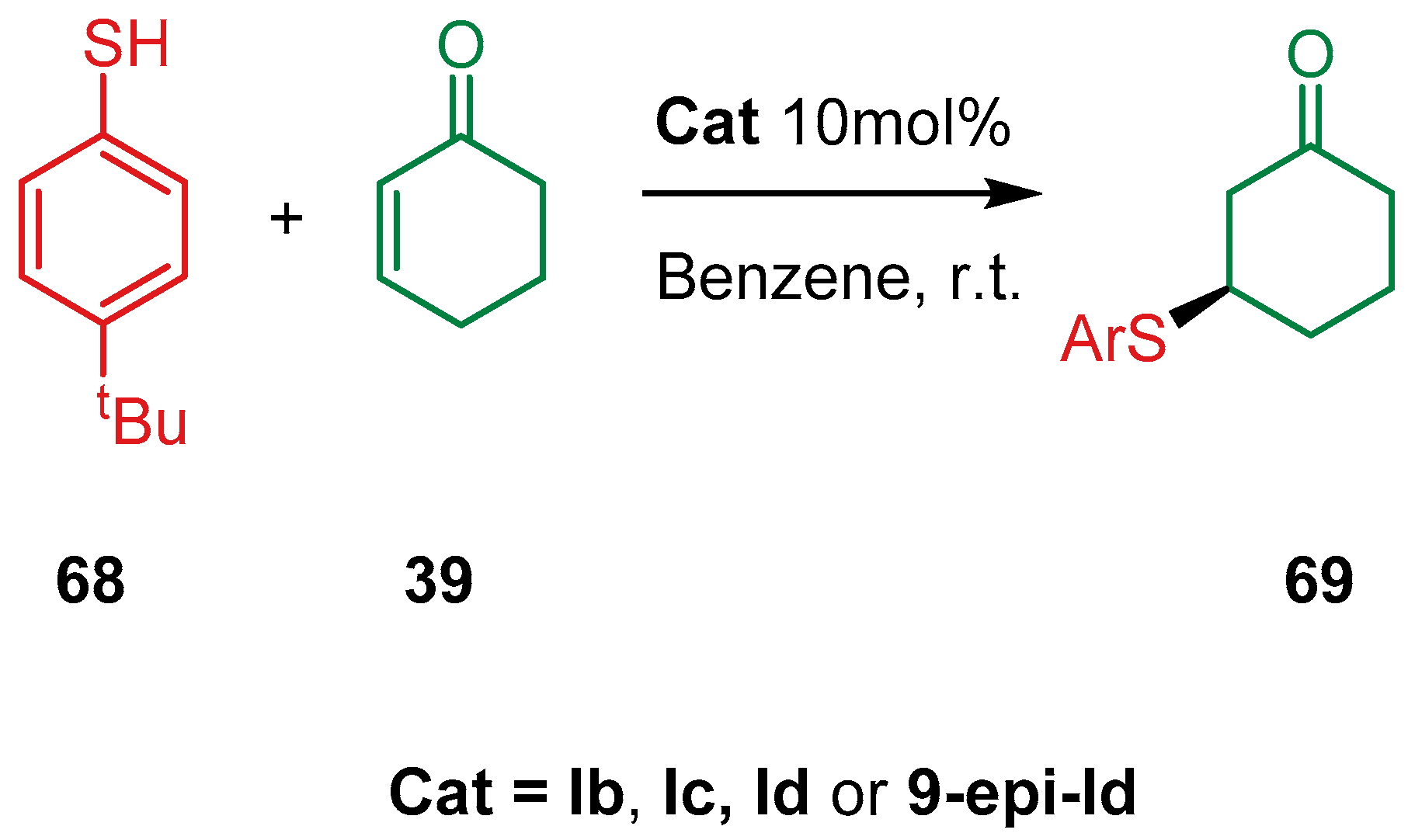
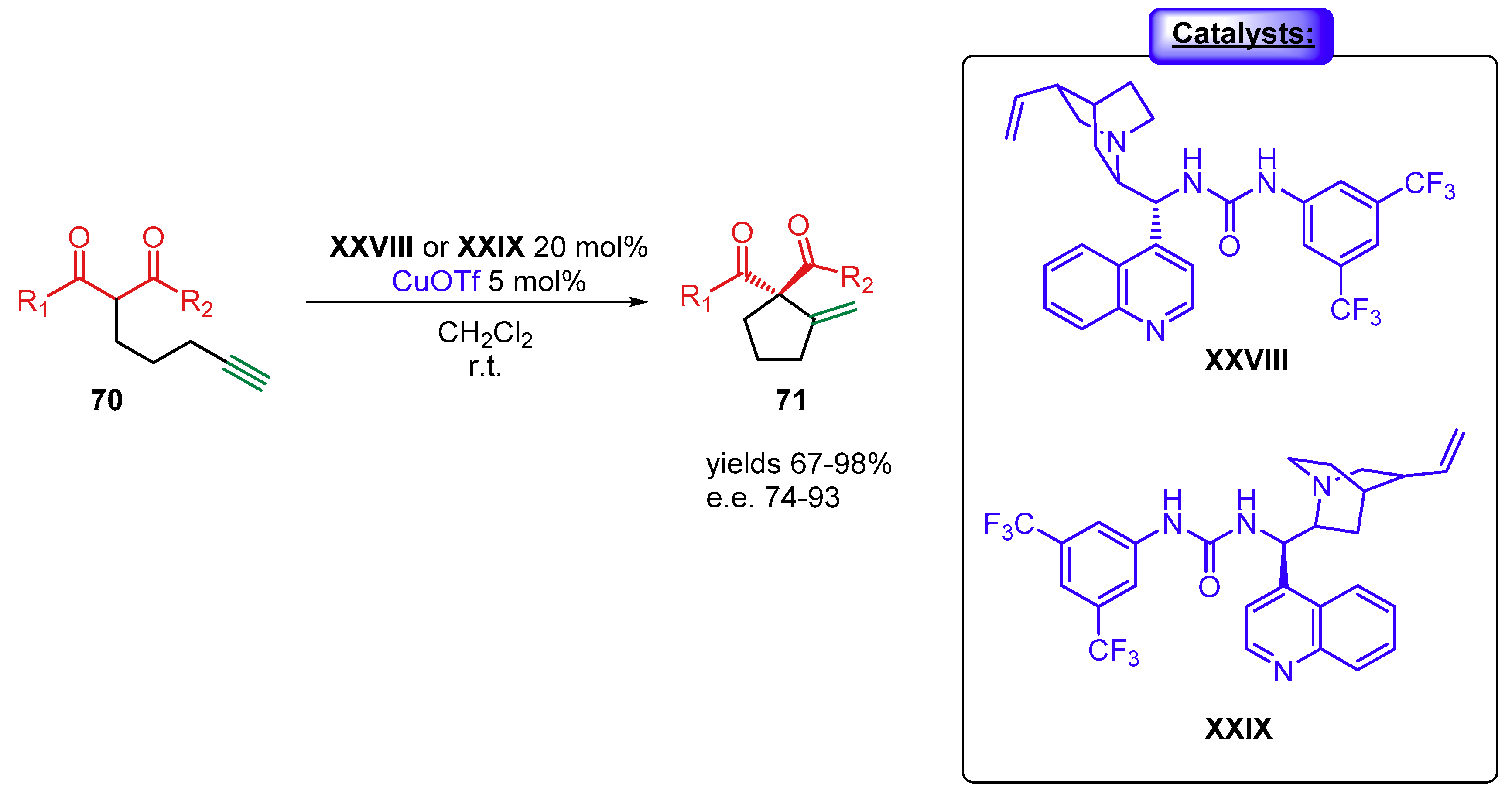
4. Cinchona Alkaloids Organocatalysis
4.1. Synergistic Catalysis with Cinchona Alkaloids
4.1.1. Combination with Transition Metal-Based Catalysts
4.1.2. Combination with Other Organocatalysts
4.1.3. Combination with Photocatalysts
5. Phosphoric Acids Catalysis
5.1. Synergistic Catalysis with Phosphoric Acids
5.1.1. Combination with Transition Metal-Based Catalysts
5.1.2. Combination with Other Organocatalysts
5.1.3. Combination with Photocatalysts
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bredig, G.; Fiske, P.S. Durch Katalysatoren Bewirkte Asymmetrische Synthese. Biochem. Z. 1912, 46, 7–23. [Google Scholar]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels-Alder reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Asymmetric aminocatalysis - Gold rush in organic chemistry. Angew. Chem. Int. Ed. 2008, 47, 6138–6171. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, S.; Scarpino Schietroma, D.M.; Bella, M. Multiple catalysis with two chiral units: An additional dimension for asymmetric synthesis. Angew. Chem. Int. Ed. 2011, 50, 6216–6232. [Google Scholar] [CrossRef]
- Franzén, J.; Marigo, M.; Fielenbach, D.; Wabnitz, T.C.; Kjærsgaard, A.; Jørgensen, K.A. A general organocatalyst for direct α-functionalization of aldehydes: Stereoselective C-C, C-N, C-F, C-Br, and C-S bond-forming reactions. Scope and mechanistic insights. J. Am. Chem. Soc. 2005, 127, 18296–18304. [Google Scholar] [CrossRef]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef]
- Sigman, M.S.; Jacobsen, E.N. Schiff base catalysts for the asymmetric Strecker reaction identified and optimized from parallel synthetic libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef]
- Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Diphenylprolinol silyl ethers as efficient organocatalysts for the asymmetric Michael reaction of aldehydes and nitroalkenes. Angew. Chem. Int. Ed. 2005, 44, 4212–4215. [Google Scholar] [CrossRef]
- Klussmann, M.; Ratjen, L.; Hoffmann, S.; Wakchaure, V.; Goddard, R.; List, B. Synthesis of TRIP and analysis of phosphate salt impurities. Synlett 2010, 14, 2189–2192. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef] [PubMed]
- Eder, U.; Sauer, G.; Wiechert, R. New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures. Chem. Int. Ed. 1971, 10, 496–497. [Google Scholar] [CrossRef]
- Hajos, Z.G.; Parrish, D.R. Asymmetric Synthesis of Bicyclic Intermediates of Natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Berkessel, A. Asymmetric Organocatalysis—From Biomimetic Concepts to Applications in Asymmetric Synthesis; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; p. 405. [Google Scholar]
- Cohen, N. Asymmetric Induction in 19-Norsteroid Total Synthesis. Acc. Chem. Res. 1976, 9, 412–417. [Google Scholar] [CrossRef]
- Woodward, R.B.; Logusch, E.; Nambiar, K.P.; Sakan, K.; Ward, D.E.; Au-Yeung, B.W.; Balaram, P.; Browne, L.J.; Card, P.J.; Chen, C.H. Asymmetric total synthesis of erythromcin. 1. Synthesis of an erythronolide A secoacid derivative via asymmetric induction. J. Am. Chem. Soc. 1981, 103, 3210–3213. [Google Scholar] [CrossRef]
- Agami, C.; Sevestre, H. Si-Enantioface Selectivity in (S)-Proline-catalysed Asymmetric Annelation. J. Chem. Soc. Chem. Commun. 1984, 21, 1385–1386. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Shiraishi, T.; Hirama, M. A Catalytic Enantioselective Michael Addition of a Simple Malonate to Prochiral α,β-Unsaturated Ketoses and Aldehydes. Angew. Chem. Int. Ed. Engl. 1993, 32, 1176–1178. [Google Scholar] [CrossRef]
- Kawara, A.; Taguchi, T. An enantioselective Michael addition of soft nucleophiles to prochiral enone catalyzed by (2-pyrrolidyl) alkyl ammonium hydroxide. Tetrahedron Lett. 1994, 35, 8805–8808. [Google Scholar] [CrossRef]
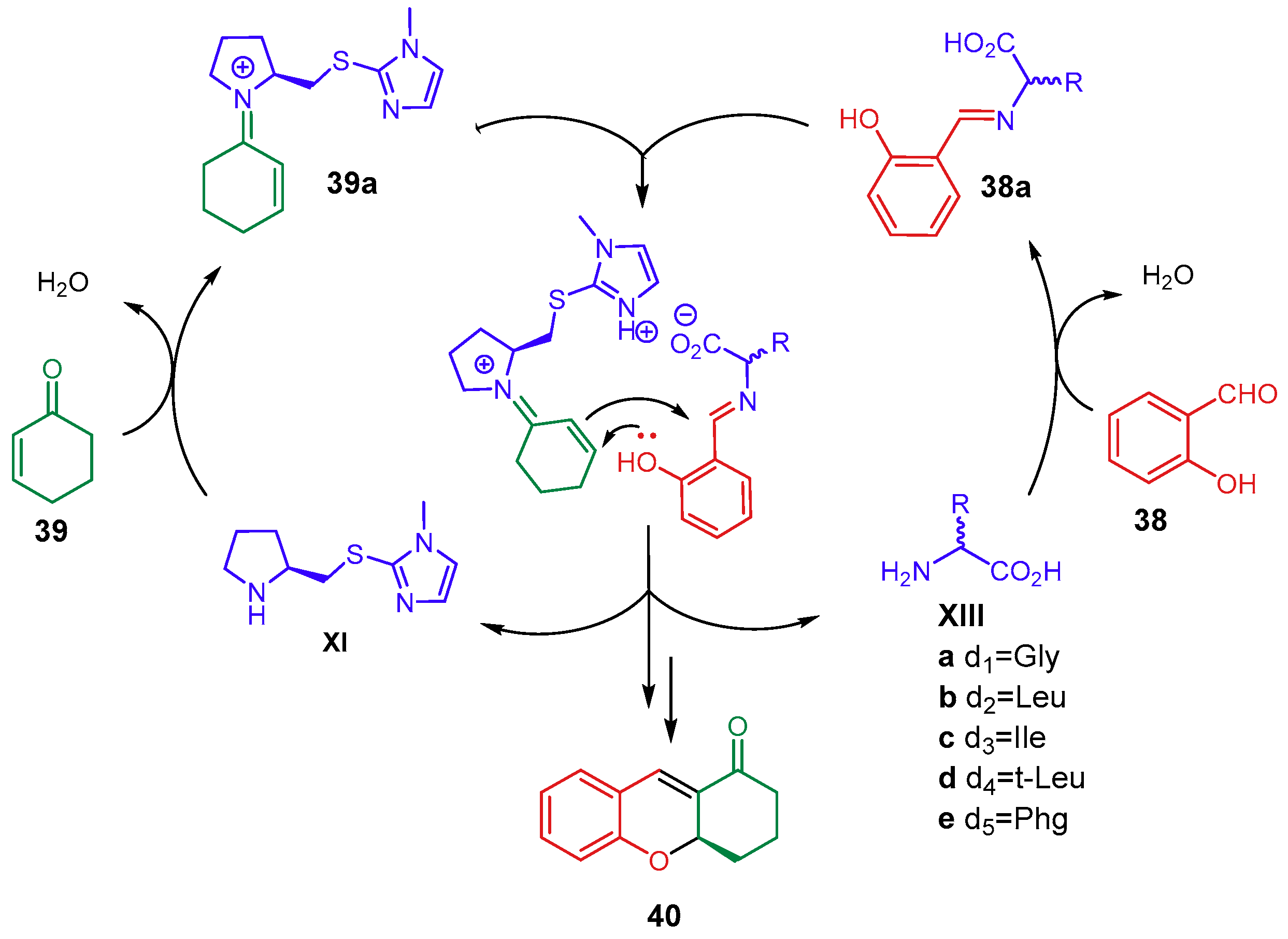
- Vega-Peñaloza, A.; Paria, S.; Bonchio, M.; Dell’Amico, L.; Companyó, X. Profiling the Privileges of Pyrrolidine-Based Catalysts in Asymmetric Synthesis: From Polar to Light-Driven Radical Chemistry. ACS Catal. 2019, 21, 6058–6072. [Google Scholar] [CrossRef]
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W. Mechanochemistry: Opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. [Google Scholar] [CrossRef] [PubMed]
- Ibrahem, I.; Córdova, A. Direct catalytic intermolecular α-allylic alkylation of aldehydes by combination of transition-metal and organocatalysis. Angew. Chem. Int. Ed. 2006, 45, 1952–1956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, J. Friedländer synthesis of quinolines using a lewis acid-surfactant- combined catalyst in water. Adv. Synth. Catal. 2007, 349, 1047–1051. [Google Scholar] [CrossRef]
- Sun, W.; Ding, Q.; Sun, X.; Fan, R.; Wu, J. AgOTf-catalyzed three-component reactions of 2-alkynylbenzaldehydes, amines, and diethylphosphite. An efficient route to 2,3-disubstituted-1,2- dihydroisoqumolin-1-ylphosphonates. J. Comb. Chem. 2007, 9, 690–694. [Google Scholar] [CrossRef]
- Ding, Q.; Ye, Y.; Fan, R.; Wu, J. Selective synthesis of 2,3-disubstituted-2H-isoindol-1-ylphosphonate and 2,3-disubstituted-1,2-dihydroisoquinolin-1-ylphosphonate via metal-tuned reaction of α-amino (2-alkynylphenyl) methylphosphonate. J. Org. Chem. 2007, 72, 5439–5442. [Google Scholar] [CrossRef]
- Ding, Q.; Wu, J. Lewis acid- and organocatalyst-cocatalyzed multicomponent reactions of 2-alkynylbenzaldehydes, amines, and ketones. Org. Lett. 2007, 9, 4959–4962. [Google Scholar] [CrossRef]
- Binder, J.T.; Crone, B.; Haug, T.T.; Menz, H.; Kirsch, S.F. Direct carbocyclization of aldehydes with alkynes: Combining gold catalysis with aminocatalysis. Org. Lett. 2008, 10, 1025–1028. [Google Scholar] [CrossRef]
- Jensen, K.L.; Franke, P.T.; Arróniz, C.; Kobbelgaard, S.; Jørgensen, K.A. Enantioselective synthesis of cyclopentene carbaldehydes by a direct multicatalytic cascade sequence: Carbocyclization of aldehydes with alkynes. Chem. Eur. J. 2010, 16, 1750–1753. [Google Scholar] [CrossRef]
- Akagawa, K.; Fujiwara, T.; Sakamoto, S.; Kudo, K. Efficient asymmetric α-oxyamination of aldehydes by resin-supported peptide catalyst in aqueous media. Org. Lett. 2010, 12, 1804–1807. [Google Scholar] [CrossRef]
- Itsuno, S.; Sakakura, M.; Koichi, I. Polymer-Supported Poly (amino acids) as New Asymmetric Epoxidation Catalyst of α,β-Unsaturated Ketones. J. Org. Chem. 1990, 55, 6047–6049. [Google Scholar] [CrossRef]
- Sibi, M.P.; Hasegawa, M. Organocatalysis in radical chemistry. Enantioselective α-oxyamination of aldehydes. J. Am. Chem. Soc. 2007, 129, 4124–4125. [Google Scholar] [CrossRef] [PubMed]
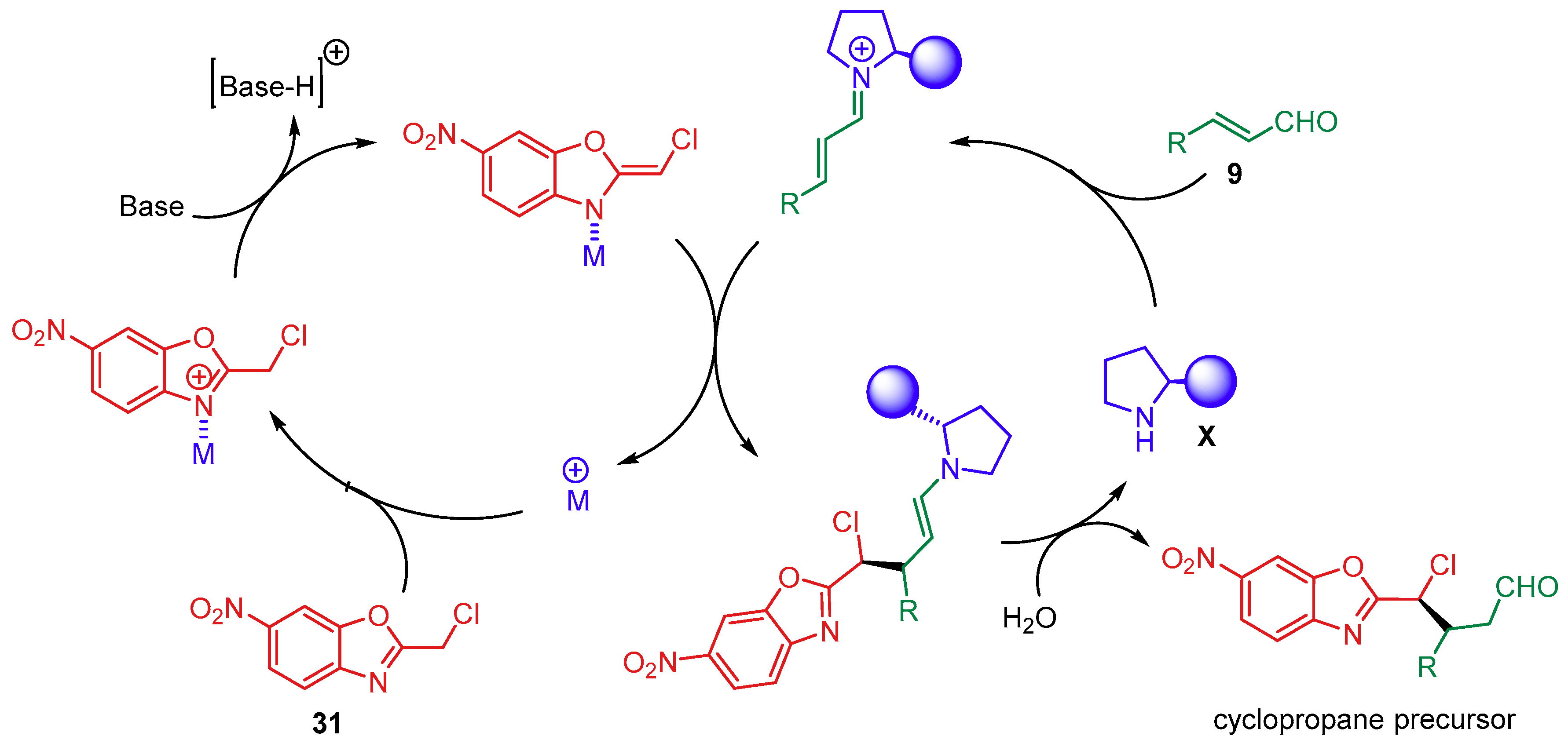
- Meazza, M.; Polo, V.; Merino, P.; Rios, R. Synergistic catalysis: Enantioselective cyclopropanation of alkylidene benzoxazoles by Pd(II) and secondary amine catalysis. Scope, limitations and mechanistic insightf. Org. Chem. Front. 2018, 5, 806–812. [Google Scholar] [CrossRef]
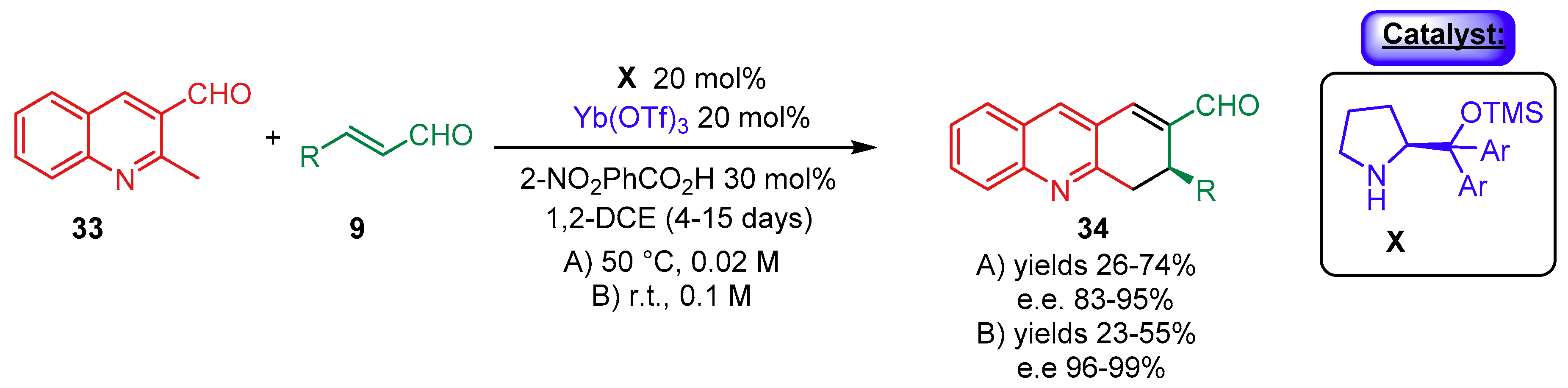
- Meninno, S.; Meazza, M.; Yang, J.W.; Tejero, T.; Merino-Gomez, P.; Rios, R. Synergistic Catalysis: Highly Enantioselective Cascade Reaction for the Synthesis of Dihydroacridines. Chem. Eur. J. 2019, 25, 7623–7627. [Google Scholar] [CrossRef] [PubMed]
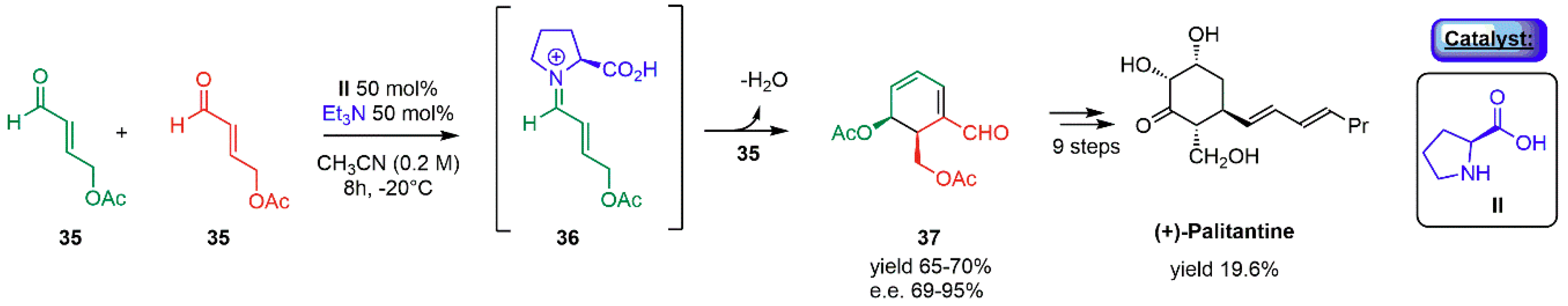
- Hong, B.C.; Wu, M.F.; Tseng, H.C.; Huang, G.F.; Su, C.F.; Liao, J.H. Organocatalytic asymmetric Robinson annulation of α,β-unsaturated aldehydes: Applications to the total synthesis of (+)-palitantin. J. Org. Chem. 2007, 72, 8459–8471. [Google Scholar] [CrossRef] [PubMed]
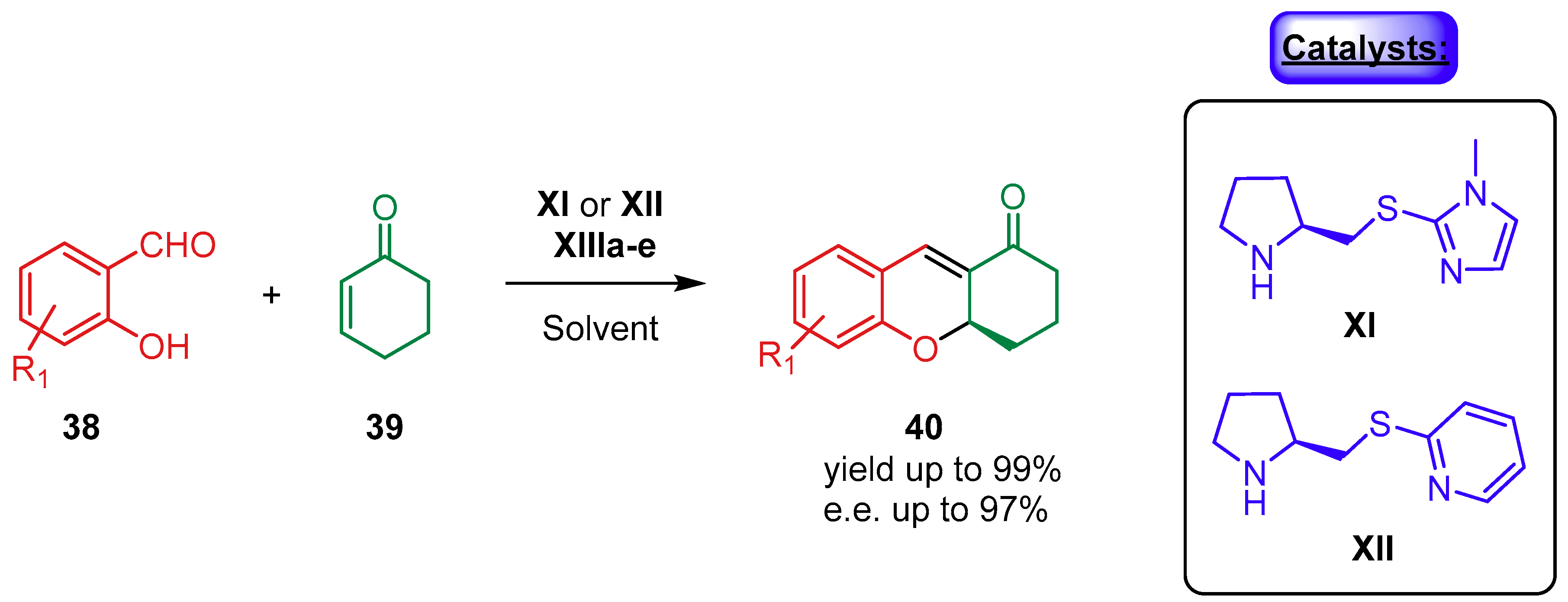
- Xia, A.B.; Xu, D.Q.; Luo, S.P.; Jiang, J.R.; Tang, J.; Wang, Y.F.; Xu, Z.Y. Dual organocatalytic ion-pair assemblies: A highly efficient approach for the enantioselective oxa-michael-mannich reaction of salicylic aldehydes with cyclohexenones. Chem. Eur. J. 2010, 16, 801–804. [Google Scholar] [CrossRef]
- Albini, A.; Fagnoni, M. Green chemistry and photochemistry were born at the same time. Green Chem. 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Hoffmann, N. Photochemical reactions of aromatic compounds and the concept of the photon as a traceless reagent. Photochem. Photobiol. Sci. 2012, 11, 1613–1641. [Google Scholar] [CrossRef]
- Ravelli, D.; Dondi, D.; Fagnoni, M.; Albini, A. Photocatalysis. A multi-faceted concept for green chemistry. Chem. Soc. Rev. 2009, 38, 1999–2011. [Google Scholar] [CrossRef]
- Condie, A.G.; González-Gómez, J.C.; Stephenson, C.R.J. Visible-light photoredox catalysis: Aza-Henry reactions via C-H functionalization. J. Am. Chem. Soc. 2010, 132, 1464–1465. [Google Scholar] [CrossRef]
- Nicewicz, D.A.; Macmillan, D.W.C. Merging Photoredox Catalysis with Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [Google Scholar] [CrossRef]
- Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Metal-free, cooperative asymmetric organophotoredox catalysis with visible light. Angew. Chem. Int. Ed. 2011, 50, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Nagib, D.A.; Scott, M.E.; MacMillan, D.W.C. Enantioselective a-Trifluoromethylation of aldehydes via photoredox organocatalysis. J. Am. Chem. Soc. 2009, 131, 10875–10877. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.W.; Wal, M.N.V.; Grange, R.L.; MacMillan, D.W.C. Enantioselective α-benzylation of aldehydes via photoredox organocatalysis. J. Am. Chem. Soc. 2010, 132, 13600–13603. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.R.; Meghani, P.; Ekkundi, V.S. Diels-alder reactions: Rate acceleration promoted by a biphenylenediol. Tetrahedron Lett. 1990, 31, 3381–3384. [Google Scholar] [CrossRef]
- Schreiner, P.R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Wheeler, S.E. Origin of the superior performance of (thio)squaramides over (thio)ureasin organocatalysis. Chem. Eur. J. 2013, 19, 15141–15147. [Google Scholar] [CrossRef] [PubMed]
- Malkov, A.V.; Mariani, A.; MacDougall, K.N.; Kočovský, P. Role of noncovalent interactions in the enantioselective reduction of aromatic ketimines with trichlorosilane. Org. Lett. 2004, 6, 2253–2256. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-X.; Ji, F.-H.; Wei, D.-K.; Shi, M. Chiral squaramides catalyzed diastereo- and enantioselective Michael addition of α-substituted isocyanoacetates to N-aryl maleimides. Tetrahedron 2013, 69, 10763–10771. [Google Scholar] [CrossRef]
- Kim, H.Y.; Oh, K. Highly diastereo- and enantioselective aldol reaction of methyl α-isocyanoacetate: A cooperative catalysis approach. Org. Lett. 2011, 13, 1306–1309. [Google Scholar] [CrossRef]
- Burns, N.Z.; Witten, M.R.; Jacobsen, E.N. Dual catalysis in enantioselective oxidopyrylium-based [5 + 2] cycloadditions. J. Am. Chem. Soc. 2011, 133, 14578–14581. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, H.; Madarász, Ú.; Pápai, I.; Pihko, P.M. Dual hydrogen-bond/enamine catalysis enables a direct enantioselective three-component domino reaction. Angew. Chem. Int. Ed. 2011, 50, 6123–6127. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.W.; Stephenson, C.R.J. Tandem visible light-mediated radical cyclization-divinylcyclopropane rearrangement to tricyclic pyrrolidinones. Org. Lett. 2011, 13, 5468–5471. [Google Scholar] [CrossRef]
- Ischay, M.A.; Yoon, T.P. Accessing the synthetic chemistry of radical ions. Eur. J. Org. Chem. 2012, 2012, 3359–3372. [Google Scholar] [CrossRef]
- Lin, S.; Ischay, M.A.; Fry, C.G.; Yoon, T.P. Radical cation Diels-Alder cycloadditions by visible light photocatalysis. J. Am. Chem. Soc. 2011, 133, 19350–19353. [Google Scholar] [CrossRef] [PubMed]
- Brak, K.; Jacobsen, E.N. Asymmetric ion-pairing catalysis. Angew. Chem. Int. Ed. 2013, 52, 534–561. [Google Scholar] [CrossRef] [PubMed]
- Bergonzini, G.; Schindler, C.S.; Wallentin, C.J.; Jacobsen, E.N.; Stephenson, C.R.J. Photoredox activation and anion binding catalysis in the dual catalytic enantioselective synthesis of β-amino esters. Chem. Sci. 2014, 5, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.A.; Chen, B.C. Asymmetric Hydroxylation of Enolates with N-Sulfonyloxaziridines. Chem. Rev. 1992, 92, 919–934. [Google Scholar] [CrossRef]
- Rono, L.J.; Yayla, H.G.; Wang, D.Y.; Armstrong, M.F.; Knowles, R.R. Enantioselective photoredox catalysis enabled by proton-coupled electron transfer: Development of an asymmetric aza-pinacol cyclization. J. Am. Chem. Soc. 2013, 135, 17735–17738. [Google Scholar] [CrossRef]
- Lin, L.; Bai, X.; Ye, X.; Zhao, X.; Tan, C.H.; Jiang, Z. Organocatalytic Enantioselective Protonation for Photoreduction of Activated Ketones and Ketimines Induced by Visible Light. Angew. Chem. Int. Ed. 2017, 56, 13842–13846. [Google Scholar] [CrossRef]
- Bella, M.; Gasperi, T. Organocatalytic formation of quaternary stereocenters. Synthesis 2009, 10, 1583–1614. [Google Scholar] [CrossRef]
- Brandes, S.; Niess, B.; Bella, M.; Prieto, A.; Overgaard, J.; Jørgensen, K.A. Non-biaryl atropisomers in organocatalysis. Chem. Eur. J. 2006, 12, 6039–6052. [Google Scholar] [CrossRef] [PubMed]
- Pasteur, L. Umwandlung der Weinsäure in Traubensäure. Entdeckung von unwirksamer Weinsäure. Neue Methode der Zerlegung von Traubensäure in Rechts- und in Linksweinsäure. Ann. der Phys. und Chem. 1853, 166, 504–509. [Google Scholar] [CrossRef]
- Hiemstra, H.; Wynberg, H. Addition of Aromatic Thiols to Conjugated Cycloalkenones, Catalyzed by Chiral β-Hydroxy Amines. A Mechanistic Study on Homogeneous Catalytic Asymmetric Synthesis. J. Am. Chem. Soc. 1981, 103, 417–430. [Google Scholar] [CrossRef]
- Helder, R.; Arends, R.; Bolt, W.; Hiemstra, H.; Wynberg, H. Alkaloid catalyzed asymmetric synthesis III the addition of mercaptans to 2-cyclohexene-1-one; determination of enantiomeric excess using 13C NMR. Tetrahedron Lett. 1977, 18, 2181–2182. [Google Scholar] [CrossRef]
- Xie, J.W.; Chen, W.; Li, R.; Zeng, M.; Du, W.; Yue, L.; Chen, Y.C.; Wu, Y.; Zhu, J.; Deng, J.G. Highly asymmetric Michael addition to α,β-unsaturated ketones catalyzed by 9-amino-9-deoxyepiquinine. Angew. Chem. Int. Ed. 2007, 46, 389–392. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Carlone, A.; Pesciaioli, F.; Sambri, L.; Melchiorre, P. Organocatalytic asymmetric friedel-crafts alkylation of indoles with simple α,β-unsaturated ketones. Org. Lett. 2007, 9, 1403–1405. [Google Scholar] [CrossRef]
- Yang, T.; Ferrali, A.; Sladojevich, F.; Campbell, L.; Dixon, D.J. Brønsted base/Lewis acid cooperative catalysis in the enantioselective Conia-ene reaction. J. Am. Chem. Soc. 2009, 131, 9140–9141. [Google Scholar] [CrossRef]
- Kennedy-Smith, J.J.; Staben, S.T.; Toste, F.D. Gold(I)-Catalyzed Conia-Ene Reaction of β-Ketoesters with Alkynes. J. Am. Chem. Soc. 2004, 126, 4526–4527. [Google Scholar] [CrossRef]
- Gao, Q.; Zheng, B.F.; Li, J.H.; Yang, D. Ni(II)-catalyzed Conia-ene reaction of 1,3-dicarbonyl compounds with alkynes. Org. Lett. 2005, 7, 2185–2188. [Google Scholar] [CrossRef]
- Bouyssi, D.; Monteiro, N.; Balme, G. Intramolecular carbocupration reaction of unactivated alkynes bearing a stabilized nucleophile: Application to the synthesis of iridoid monoterpenes. Tetrahedron Lett. 1999, 40, 1297–1300. [Google Scholar] [CrossRef]
- Wack, H.; France, S.; Hafez, A.M.; Drury, W.J.; Weatherwax, A.; Lectka, T. Development of a new dimeric cyclophane ligand: Application to enhanced diastereo- and enantioselectivity in the catalytic synthesis of β-lactams. J. Org. Chem. 2004, 69, 4531–4533. [Google Scholar] [CrossRef]
- Taggi, A.E.; Wack, H.; Hafez, A.M.; France, S.; Lectka, T. Generation of ketenes from acid chlorides using NaH/crown ether shuttle-deprotonation for use in asymmetric catalysis. Org. Lett. 2002, 4, 627–629. [Google Scholar] [CrossRef]
- Dudding, T.; Hafez, A.M.; Taggi, A.E.; Wagerle, T.R.; Lectka, T. A catalyst that plays multiple roles: Asymmetric synthesis of β-substituted aspartic acid derivatives through a four-stage, one-pot procedure. Org. Lett. 2002, 4, 387–390. [Google Scholar] [CrossRef]
- Hafez, A.M.; Taggi, A.E.; Dudding, T.; Lectka, T. Asymmetric catalysis on sequentially-linked columns. J. Am. Chem. Soc. 2001, 123, 10853–10859. [Google Scholar] [CrossRef]
- Hafez, A.M.; Taggi, A.E.; Wack, H.; Drury, W.J.; Lectka, T. Column asymmetric catalysis for β-lactam synthesis. Org. Lett. 2000, 25, 3963–3965. [Google Scholar] [CrossRef]
- Taggi, A.E.; Hafez, A.M.; Wack, H.; Young, B.; Drury, W.J.; Lectka, T. Catalytic, asymmetric synthesis of β-lactams. J. Am. Chem. Soc. 2000, 122, 7831–7832. [Google Scholar] [CrossRef]
- Ramachary, D.B.; Shruthi, K.S. Asymmetric synthesis of tetrahydroquinolines through supramolecular organocatalysis. Org. Biomol. Chem. 2014, 12, 4300–4304. [Google Scholar] [CrossRef]
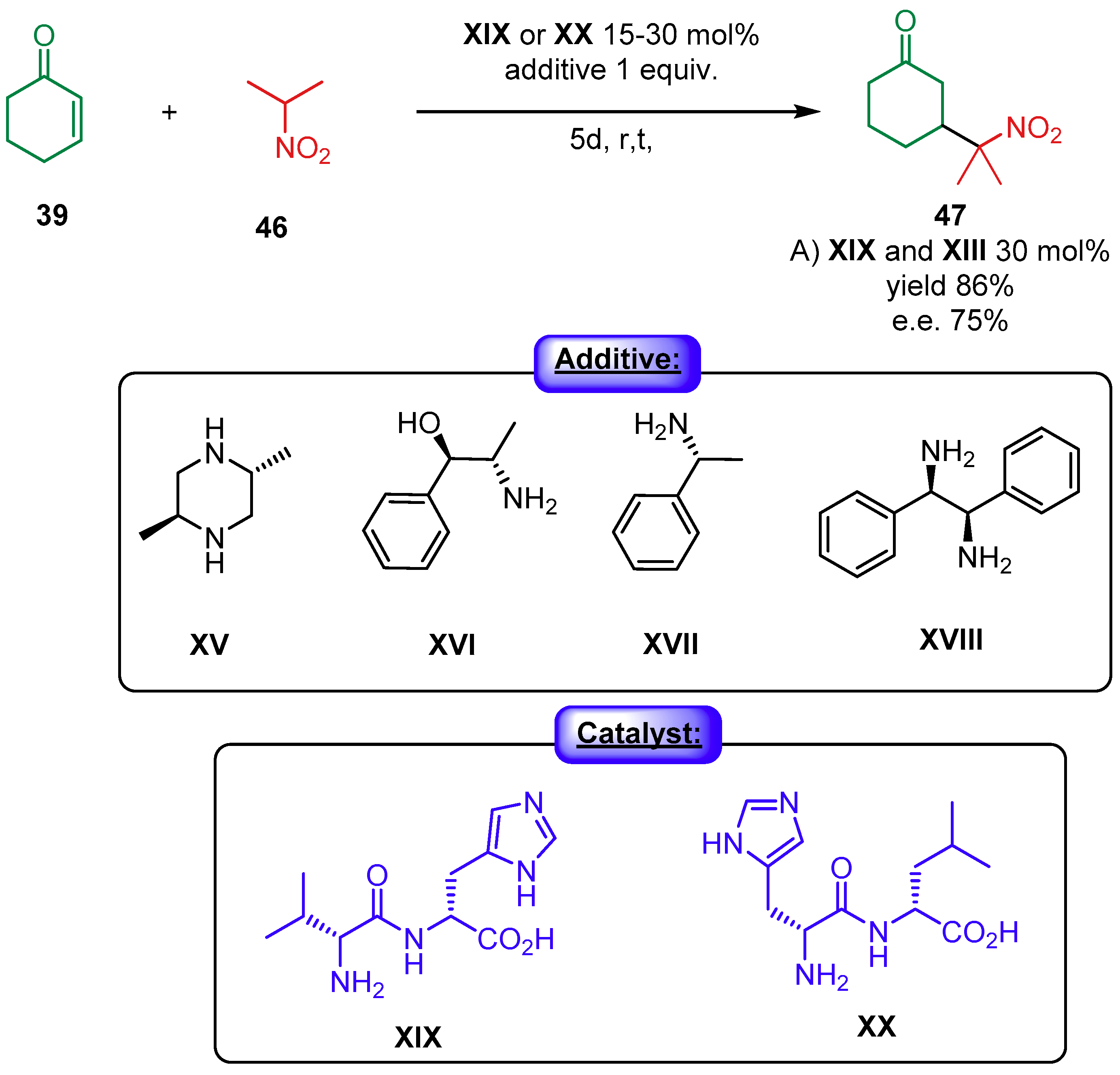
- Mandal, T.; Zhao, C.G. Modularly designed organocatalytic assemblies for direct nitro-Michael addition reactions. Angew. Chem. Int. Ed. 2008, 47, 7714–7717. [Google Scholar] [CrossRef]
- Rana, N.K.; Huang, H.; Zhao, J.C.G. Highly diastereodivergent synthesis of tetrasubstituted cyclohexanes catalyzed by modularly designed organocatalysts. Angew. Chem. Int. Ed. 2014, 53, 7619–7623. [Google Scholar] [CrossRef]
- Bhaskararao, B.; Sunoj, R.B. Two chiral catalysts in action: Insights into cooperativity and stereoselectivity in proline and cinchona-thiourea dual organocatalysis. Chem. Sci. 2018, 9, 8738–8747. [Google Scholar] [CrossRef]
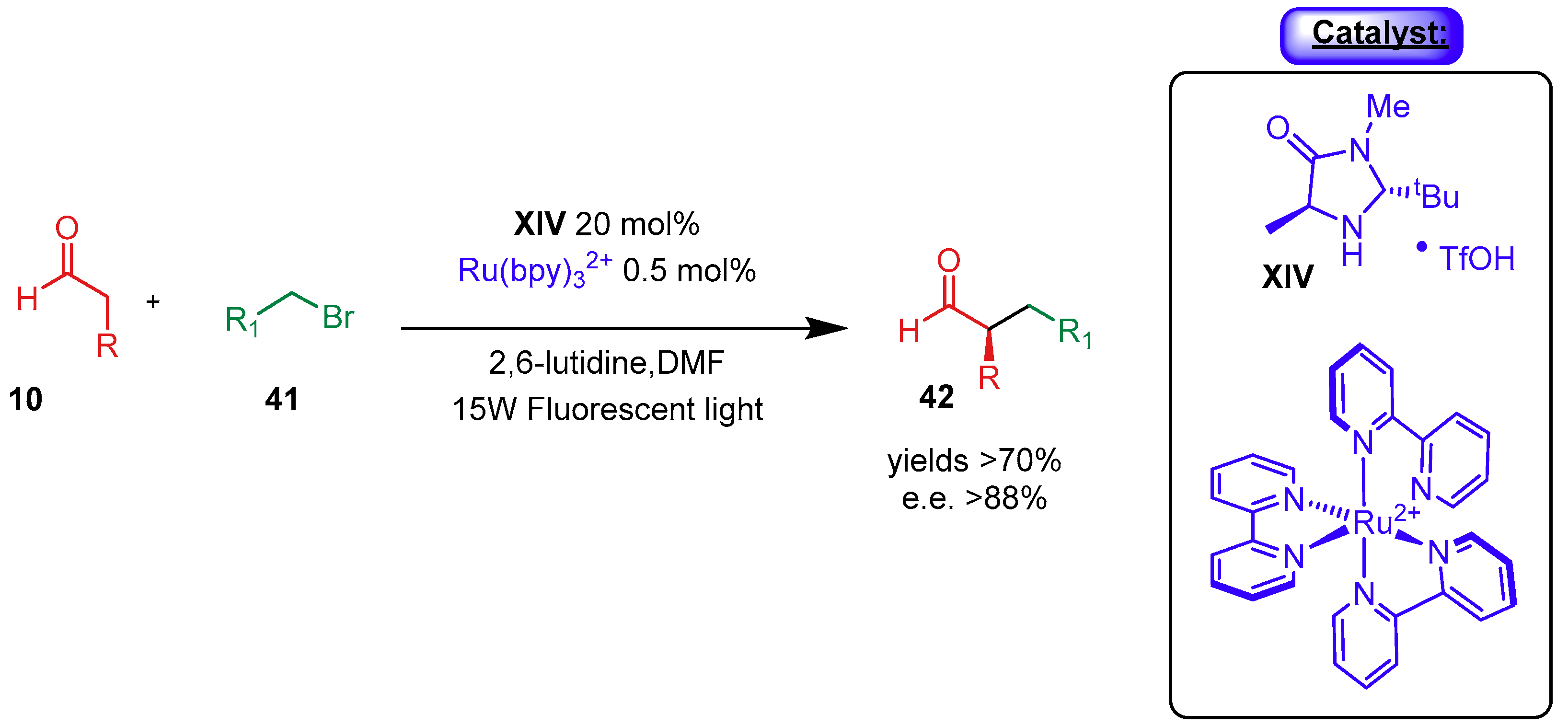
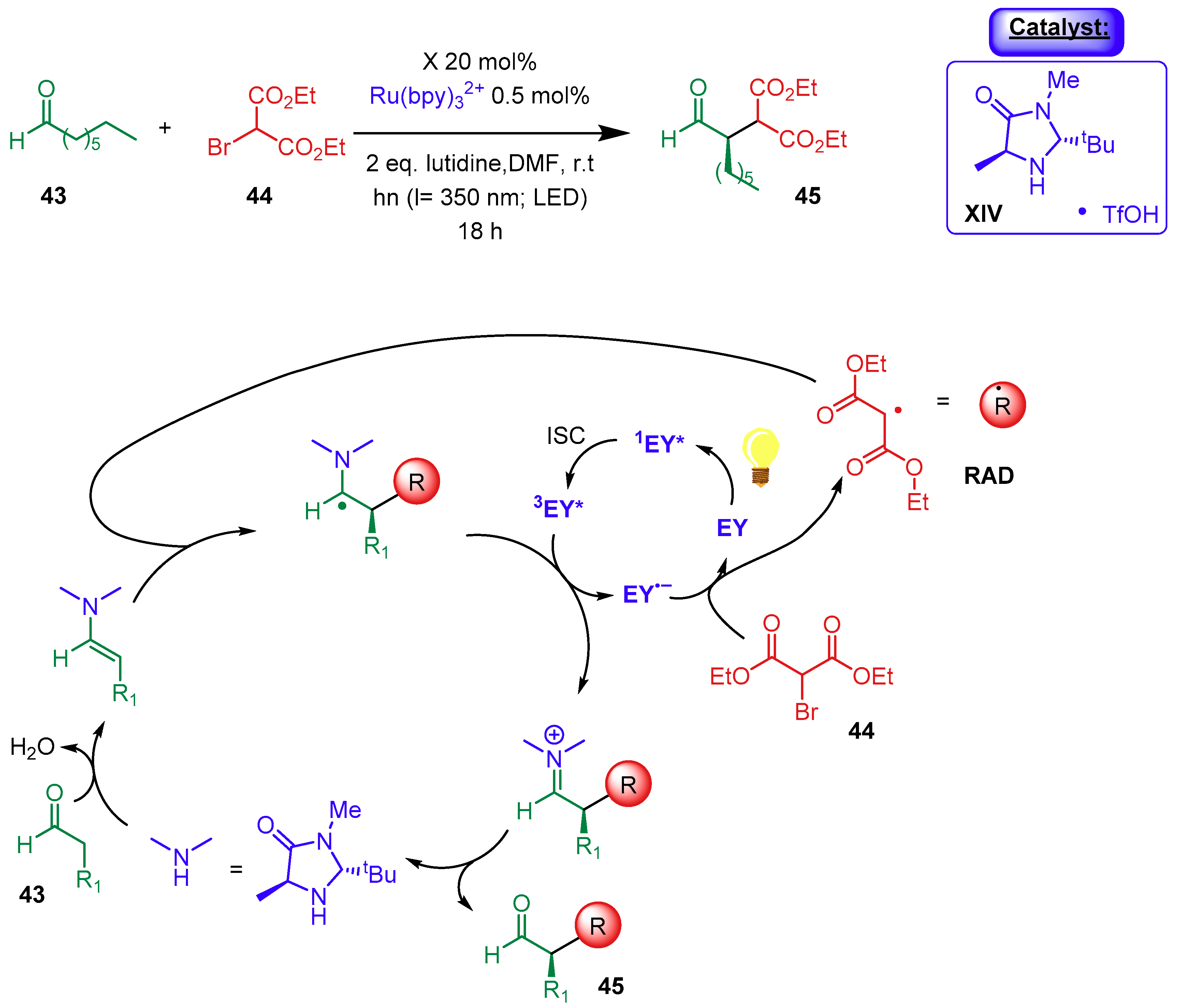
- Arceo, E.; Bahamonde, A.; Bergonzini, G.; Melchiorre, P. Enantioselective direct α-alkylation of cyclic ketones by means of photo-organocatalysis. Chem. Sci. 2014, 5, 2438–2442. [Google Scholar] [CrossRef]
- Arceo, E.; Jurberg, I.D.; Álvarez-Fernández, A.; Melchiorre, P. Photochemical activity of a key donor-acceptor complex can drive stereoselective catalytic α-alkylation of aldehydes. Nat. Chem. 2013, 5, 750–756. [Google Scholar] [CrossRef]
- Pirnot, M.T.; Rankic, D.A.; Martin, D.B.C.; MacMillan, D.W.C. Photoredox activation for the direct β-arylation of ketones and aldehydes. Science 2013, 339, 1593–1596. [Google Scholar] [CrossRef]
- Cornforth, J. The imitation of enzymic catalysis. Proc. R. Soc. Lond. Biol. Sci. 1978, 203, 101–117. [Google Scholar]
- Cornforth, J. Synthesis of substituted dibenzophospholes. Part 9. Preparation of two water-soluble phosphinic-polyphosphonic acids. J. Chem. Soc. Perkin Trans. 1996, 24, 2889–2893. [Google Scholar] [CrossRef]
- Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Enantioselective Mannich-type reaction catalyzed by a chiral Brønsted acid. Angew. Chem. Int. Ed. 2004, 43, 1566–1568. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Kouchi, M.; Sorimachi, K.; Terada, M. On the mechanism of stereoselection in direct Mannich reaction catalyzed by BINOL-derived phosphoric acids. Tetrahedron Lett. 2007, 48, 497–500. [Google Scholar] [CrossRef]
- Kaupmees, K.; Tolstoluzhsky, N.; Raja, S.; Rueping, M.; Leito, I. On the acidity and reactivity of highly effective chiral Brønsted acid catalysts: Establishment of an acidity scale. Angew. Chem. Int. Ed. 2013, 52, 11569–11572. [Google Scholar] [CrossRef]
- Zhou, S.; Fleischer, S.; Junge, K.; Beller, M. Cooperative transition-metal and chiral Brønsted acid catalysis: Enantioselective hydrogenation of imines to form amines. Angew. Chem. Int. Ed. 2011, 50, 5120–5124. [Google Scholar] [CrossRef]
- Shima, S.; Pilak, O.; Vogt, S.; Schick, M.; Stagni, M.S.; Meyer-Klaucke, W.; Warkentin, E.; Thauer, R.K.; Ermler, U. The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site. Science 2008, 321, 572–575. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, L.; Hu, S.; Cheng, J.P.; Luo, S. Asymmetric binary-acid catalysis with InBr3 in the inverse-electron-demanding hetero-diels-alder reaction of mono- and bis-substituted cyclopentadienes: Remote fluoro-effect on stereocontrol. Chem. Eur. J. 2012, 18, 799–803. [Google Scholar] [CrossRef]
- Yang, X.; Phipps, R.J.; Toste, F.D. Asymmetric fluorination of α-branched cyclohexanones enabled by a combination of chiral anion phase-transfer catalysis and enamine catalysis using protected amino acids. J. Am. Chem. Soc. 2014, 136, 5225–5228. [Google Scholar] [CrossRef]
- Bergonzini, G.; Vera, S.; Melchiorre, P. Cooperative organocatalysis for the asymmetric γ alkylation of α-branched enals. Angew. Chem. Int. Ed. 2010, 49, 9685–9688. [Google Scholar] [CrossRef]
- Inoue, Y. Asymmetric Photochemical Reactions in Solution. Chem. Rev. 1992, 92, 741–770. [Google Scholar] [CrossRef]
- Du, J.; Skubi, K.L.; Schultz, D.M.; Yoon, T.P. A dual-catalysis approach to enantioselective [2 + 2] photocycloadditions using visible light. Science 2014, 344, 392–396. [Google Scholar] [CrossRef]
- Proctor, R.S.J.; Davis, H.J.; Phipps, R.J. Catalytic enantioselective Minisci-Type addition to heteroarenes. Science 2018, 360, 419–422. [Google Scholar] [CrossRef]
- Uraguchi, D.; Kinoshita, N.; Kizu, T.; Ooi, T. Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J. Am. Chem. Soc. 2015, 137, 13768–13771. [Google Scholar] [CrossRef]


















































© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinibaldi, A.; Nori, V.; Baschieri, A.; Fini, F.; Arcadi, A.; Carlone, A. Organocatalysis and Beyond: Activating Reactions with Two Catalytic Species. Catalysts 2019, 9, 928. https://doi.org/10.3390/catal9110928
Sinibaldi A, Nori V, Baschieri A, Fini F, Arcadi A, Carlone A. Organocatalysis and Beyond: Activating Reactions with Two Catalytic Species. Catalysts. 2019; 9(11):928. https://doi.org/10.3390/catal9110928
Chicago/Turabian StyleSinibaldi, Arianna, Valeria Nori, Andrea Baschieri, Francesco Fini, Antonio Arcadi, and Armando Carlone. 2019. "Organocatalysis and Beyond: Activating Reactions with Two Catalytic Species" Catalysts 9, no. 11: 928. https://doi.org/10.3390/catal9110928
APA StyleSinibaldi, A., Nori, V., Baschieri, A., Fini, F., Arcadi, A., & Carlone, A. (2019). Organocatalysis and Beyond: Activating Reactions with Two Catalytic Species. Catalysts, 9(11), 928. https://doi.org/10.3390/catal9110928